 Open Journal of Physical Chemistry, 2013, 3, 127-137 Published Online November 2013 (http://www.scirp.org/journal/ojpc) http://dx.doi.org/10.4236/ojpc.2013.34016 Open Access OJPC Computational Study of the Alkylation Reaction of the Nitrogen Mustard Mechlorethamine Using NBO Model and the QTAIM Theory Michell O. Almeida1,2, Sérgio Henrique D. M. Faria1,3* 1Universidade Paulista, Limeira, Brazil 2CCNH—Universidade Federal do ABC, Santo André, Brazil 3Instituto de Química, Universidade Estadual de Campinas, Campinas, Brazil Email: *sehenrique@gmail.com Received July 26, 2013; revised August 24, 2013; accepted September 1, 2013 Copyright © 2013 Michell O. Almeida, Sérgio Henrique D. M. Faria. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Substances known as nitrogen mustards turn into aziridinium ion through the intramolecular cyclization SN1. This ion reacts with the DNA preferably at the N7 position of the guanine, and because of this, it is an important antineoplastic agent. Based on this, the objective of this study is to quantify the interaction between the nitrogen mustard mechlore- thamine and the guanine, using the NBO analysis and the QTAIM theory. The results of the NBO analysis showed that when the triangular cycle C4-N1-C5 is formed, there is some resonance among these atoms. This analysis also showed that the electronic transition at the sigma antibondingorbital σ* N1-C4 presents higher perturbation energy of second order, indicating that this bond is broken at the nucleophilic attack of the N7 nitrogen of guanine. The analysis that re- fers to the electron density obtained by the QTAIM theory indicates that the guanine proximity enables an electron den- sity polarization of the BCPs aziridinium ion of mechlorethamine making that the frontal part of the ion becomes elec- tron deficient. Finally, the relative results to the Laplacian of the electron density obtained by the QTAIM theory showed that the guanine approximation increases the “hole” factor at the C4, proving that the nucleophilic attack based on the “lump-hole” concept causes the region of that atom is the site of alkylation reaction. Keywords: NBO; QTAIM; Aziridinium; Ion; Nucleophilic Attack; Electron Density; Laplacian of the Electron Density 1. Introduction Antitumor alkylating agents are classified as Celular- Cycle non specific [1] and form crosslinked bonds with DNA. These crosslinked bonds cause DNA lesions re- quiring complex repair mechanisms, including replica- tion inhibition. Because of it, in 1942, mechlorethamine was successfully used to induce transient tumor remis- sion in a patient with lymphoma, this event marked the beginning of the modern era cancer chemotherapy [1]. Among the alkylating agents, the mechlorethamine was the first anti-cancer drug effectively used for clinic pur- pose, and today it is the most common one used against tumor cells [2]. The alkylating agents efficiency, like the mechlore- thamine, was studied previously using molecular model [3,4] and also by rational planning [5,6] that highlighted the correlation between the intramolecular distance of the electrophilic centers of these agents and the nucleophilic nitrogen of nucleotide [1]. As the traditional QSAR tech- niques are laborious and require a long investigation time and high cost [7], the computational analysis of the alky- lating agents is becoming more attractive. The mustard nitrogen compounds are among the most popular agents studied [2,8-11] by the theoretical com- putational chemistry. These compounds form the azirid- inium ion, being this ion highly reactive through the in- tramolecular cyclization SN1. The ion reacts with the DNA, preferably at the guanine N7 position [1], however experiments have been noticing alkylation at the posi- tions N1, N3, N6 e O6. [1,2]. The physical fundamental understanding of this alkyalting family reaction can be interesting for the development on new drugs. *Corresponding author.  M. O. ALMEIDA, S. H. D. M. FARIA 128 With this objective, one of the ways to theoretically quantify the interaction intensity between the nitrogen mustards and the guanine, besides the analysis of the highest occupied molecular orbital (HOMO) [12], lowest unoccupied molecular orbital (LUMO) [13] and its dif- ference [14-16], it is through the NBO model [17]. A distinguishing feature of localized NBO functions is the simultaneous requirement of orthonormality and maxi- mum occupancy, leading to compact expressions for atomic and bond properties [18]. This way, ab initio wavefunctions transformed to NBO form are found to be in good agreement with the Lewis concept and with the classic bond form with hybridization and polarization from Pauling-Slater-Coulson [18-21]. Another way to quantify the nitrogen mustards inter- action with the guanines is using the QTAIM theory (Quantum Theory: Atoms in Molecules) [22] from Bader. At this theory, the Laplacian of the electron density 2 is shown at the local form of the virial theorem giving the mechanics of an atom inside of a molecule [23]. This way, the Laplacian can identify the reactivity sites of the molecule [23,24]. So, the objective of the present study is to quantify the interactions of the mechlorethamine molecule with the guanine using the Natural Bond Orbital (NBO) Analysis from Donor-Acceptor Viewpoint, the electron density, and the Laplacian of the electron density obtained by the QTAIM theory. 2. Computationaldetails The tridimensional molecule structures were built using the software GaussView 3.0 [25]. The structures of the mechlorethamine molecule were drawn in several steps of thealkylation reaction with the guanine. These struc- tures were built in four states: isolated mechlorethamine, isolated aziridinium ion of mechlorethamine, aziridinium ion in the presence of guanine and the transition state of the aziridinium ion. All these structures were optimized to the lower energy state with the program Gaussian03 [26] at the level B3LYP with the basis set 6-31G (d, p). All the post calculations were made with the same wavefunction, B3LYP/6-31G (d, p). The transition state of the aziridinium ion was obtained with the QST3 algo- rithm. With the optimized geometry, using the same software [26], the Natural Bond Orbitals (NBO) were obtained and also the electronic transitions determined by this theory. The electronic transitions chosen for the dis- cussion were the ones with the second order perturbation energy E2 ≥ 5 kcal·mol−1. All these calculations were made in a machine SGI Altix 1350/Altix 450 with 174 CPU’s Intel Itanium2, 866GB of RAM memory, tech- nology NUMAFlexGeração 4, interconnection Infiniband and storage system SGI TP9300 with 43 TB. This system is installed at CENAPAD-SP [27]. Using personal computers, continuing from the opti- mized geometries obtained, the critical points position (3,−1) and (3,−3) of the gradient of the electron density, were determined. Later, the electron density of these critical points were calculated and also the Laplacian of these densities, using the QTAIM theory. These quanti- ties were calculated using the software AI M2000 [28]. This software was also used to generate the relief maps of Laplacian of the electron density. 3. Results and Discussion 3.1. Construction of the Mechlorethamine Molecule in Different States of the Alkylation Reaction The different steps of the mechlorethamine molecule alkylation with the guanine have been widely discussed at the previous art [1,2,8-11,29,30]. It is know that this molecule reacts with aintramolecular cyclization SN1 releasing a chloride ion, forming the aziridinium ion. Right after the aziridinium ion gets close to the guanine, it goes to a transition state and lately suffers a nucleo- philic attack from the N7 of these nitrogenous base. This way, the molecules used in this study, according to the reaction steps, were built using the software Gaussview03 and they were optimized to the lowest en- ergy state using the program Gaussian03. The molecules were built in the following order: State (1)—isolated mechlorethamine molecule State (2)—isolated aziridinium ion of mechlore- thamine molecule State (3)—molecular cluster formed by the aziridinium ion of mechlorethamine + guanine State (4)—mechlorethamine molecule at the transition state + guanine (obtained with the algorithm QST3 from Gaussian03) These geometries obtained for the four states of the reaction, shown at Figure 1, were used at the NBO analy- sis and also to obtain the electron density of the QTAIM theory. 3.2. NBO Analysis of the Intermolecular Interaction between Donor and Acceptor at the Four Reactions States Thedata obtained with the NBO analysis represent the electronic transition within only one reaction state. The NBO analysis of the mechlorethamine molecule (state 1) did not show any electronic transition that satisfies the minimum stabilization energy condition E2 ≥ 5 kcal·mol−1. However, Table 1 shows that when the mechlorethamine becomes the aziridinium ion, forming the triangle C4-N1-C5 (state 2), there is a kind of reso- nance between the electrons of the sigma bond N1-C4 and N1-C5. However, a slight asymmetry can be noticed at Open Access OJPC 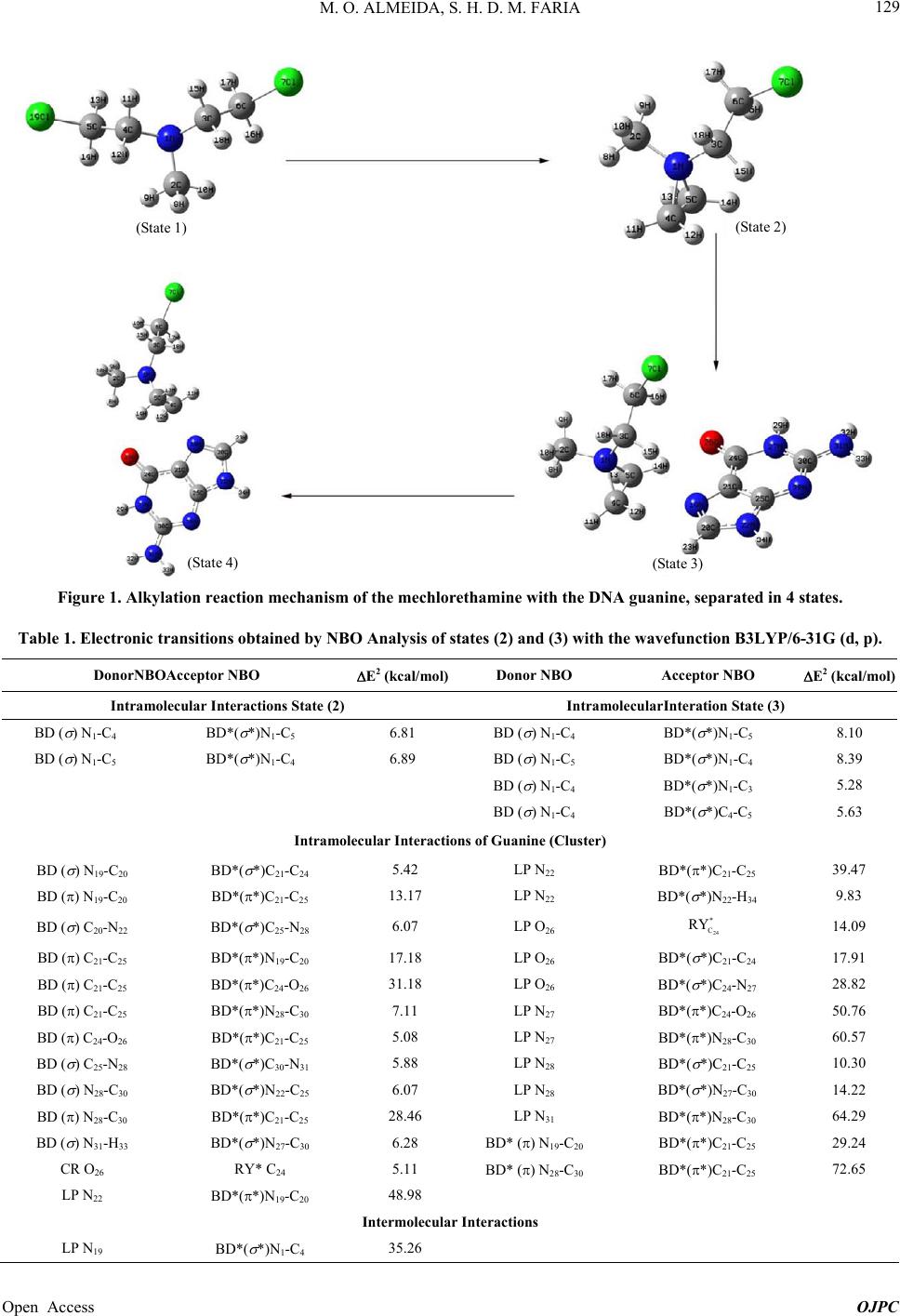 M. O. ALMEIDA, S. H. D. M. FARIA Open Access OJPC 129 (State 1) (State 2) (State 4)(State 3) Figure 1. Alkylation reaction mechanism of the mechlorethamine with the DNA guanine, separated in 4 states. Table 1. Electronic transitions obtained by NBO Analysis of states (2) and (3) with the wavefunction B3LYP/6-31G (d, p). DonorNBOAcceptor NBO E2 (kcal/mol)Donor NBO Acceptor NBO E2 (kcal/mol) Intramolecular Interactions State (2) IntramolecularInteration State (3) BD ( ) N1-C4 BD*( *)N1-C5 6.81 BD ( ) N1-C4 BD*( *)N1-C5 8.10 BD ( ) N1-C5 BD*( *)N1-C4 6.89 BD ( ) N1-C5 BD*( *)N1-C4 8.39 BD ( ) N1-C4 BD*( *)N1-C3 5.28 BD ( ) N1-C4 BD*( *)C4-C5 5.63 Intramolecular Interactions of Guanine (Cluster) BD ( ) N19-C20 BD*( *)C21-C24 5.42 LP N22 BD*(*)C21-C25 39.47 BD () N19-C20 BD*(*)C21-C25 13.17 LP N22 BD*( *)N22-H34 9.83 BD ( ) C20-N22 BD*( *)C25-N28 6.07 LP O26 24 * C RY 14.09 BD () C21-C25 BD*(*)N19-C20 17.18 LP O26 BD*( *)C21-C24 17.91 BD () C21-C25 BD*(*)C24-O26 31.18 LP O26 BD*( *)C24-N27 28.82 BD () C21-C25 BD*(*)N28-C30 7.11 LP N27 BD*(*)C24-O26 50.76 BD () C24-O26 BD*(*)C21-C25 5.08 LP N27 BD*(*)N28-C30 60.57 BD ( ) C25-N28 BD*( *)C30-N31 5.88 LP N28 BD*( *)C21-C25 10.30 BD ( ) N28-C30 BD*( *)N22-C25 6.07 LP N28 BD*( *)N27-C30 14.22 BD () N28-C30 BD*(*)C21-C25 28.46 LP N31 BD*(*)N28-C30 64.29 BD ( ) N31-H33 BD*( *)N27-C30 6.28 BD* () N19-C20 BD*(*)C21-C25 29.24 CR O26 RY* C24 5.11 BD* () N28-C30 BD*(*)C21-C25 72.65 LP N22 BD*(*)N19-C20 48.98 Intermolecular Interactions LP N19 BD*( *)N1-C4 35.26  M. O. ALMEIDA, S. H. D. M. FARIA 130 the stabilization energy of the electronic transition at the acceptor antibonding orbitals σ* of the carbons of the triangular cycle C4-N1-C5. The data indicate that the electronic transition at the antibonding orbital σ* N1-C4 stabilizes the system more, making that this carbon (C4) be more susceptible to a nucleophilic attack. Looking at the data regarding state 3, at Table 1, an increase of around 20% can be noticed at the stabilization energy E2 of the acceptor antibonding σ* of the train- gular cycle carbons C4-N1-C5. It is also observed the ap- pearance of two new intramolecular transitions of the aziridinium ion in which the electronic donor is the bond orbital σ N 1-C4, another indication that this bond is the most probable to be cleaved with the guanine nucleo- philic attack. The electronic transitions obtained for the guanine are the typical resonance transitions of the aro- matic rings and of the carbonyl formed by the atoms C24 and O26. The most important transitions that occur at the guanine were with 72.65 kcal·mol−1 and with 64.29 kcal·mol−1. * N28-C30 C21-C25 ππ * π N31N28-C30 Another relevant electronic transition is the intermo- lecular transition of E2 = 35.26 kcal·mol−1 indicating that a nucleophilic attack of the N19 (the guanine N7 mentioned at the references [1,2,9]) pos- sibly contributes to the N1-C4 bond break. LP * N19 N1-C4 LP The algorithm QST3 that determined the geometry of theaziridinium ion of mechlorethamine transition state (state 4) shows that before the alkylation, there is a break at the triangular cycle C4-N1-C5. This new geometry is obtained with the bond break of σ N 1-C4, what was al- ready predicted by the NBO analysis of Table 1 and by the new intramolecular transition with 10.75 kcal·mol−1 from Table 2. Among the guanine intra- molecular transitions at this reaction state, two Lone Pair transitions of the C21, e can be highlighted, with stabilization energies of 244.46 and 97.92 kcal·mol−1 respectively. * N1-C4 N1-C5 * C21 N22-C25 LP π * * C21 C24-O26 LP π LP It is interesting to notice that at this reaction step, where there is the transition state, the intermolecular transition N19N1-C4 presents a second order en- ergy of E2 = 63.59 kcal·mol−1. This value corresponds to an increase of 80.35% in the E2 energy of this transi- tion when compared to the value obtained at the previous state (3). This result demonstrates that there is a transi- tion state (4) of the aziridinium ion of mechlorethamine that favors the nucleophilic attack of the guanine N7 at the ion C4. It is thought that this fact happens due to the proximity of the C4 to N7 at the transition state (4), because the distance between these two atoms at the state (4) is 2.11 Å, while at state (3) this same distance is 3.34 Å (Fig- ure 2). 3.3. Application of the QTAIM Theory to Obtain the Electron Density and the Laplacian of the Electron Density of Molecules at the Four Reaction Sites The QTAIM theory (Quantum Theory: Atoms in Mole- cules) was used to calculate the electron density and the Laplacian of the electron density for the critical points (3,−1) and (3,−3) and for all the structures at each alkyla- tion reaction state of the mechlorethamine with the gua- nine (Figure 3). It can be noticed by Table 3, that in all changes of state, the critical points of electron density variation (State 3) 3.34 Å (State 4) 2.11 Å Figure 2. Distance between the N19 (guanine N7) and themechlorethamine C4 at the state (3) and (4) of the alkylation reaction. Open Access OJPC  M. O. ALMEIDA, S. H. D. M. FARIA 131 Table 2. Electronic Transitions obtained by NBO Analysis of state (4) with the wavefunction B3LYP/6-31G (d, p). Donor NBO Acceptor NBO E2 (kcal/mol) Donor NBO Acceptor NBO E2 (kcal/mol) Intramolecular Interactions of Aziridinium Ion (TS) BD ( ) N1-C4 BD*( *)N1-C5 10.75 Intramolecular Interactions of Guanine (Cluster) BD ( ) N19-C20 LP C21 26.91 LP N19 BD*( *)C20-N22 6.81 BD ( ) C20-N22 BD*( *)C25-N28 5.40 LP C21 BD*(*)N19-C20 60.94 BD ( ) N19-C21 BD*( *) C21-C24 5.28 LP C21 BD*(*)N22-C25 244.46 BD () N22-C25 LP C21 12.34 LP C21 BD*(*)C24-O26 97.92 BD () N22-C25 BD*(*) N19-C20 29.35 LP O26 RY* C24 14.07 BD () C24-O26 LP C21 6.89 LP O26 BD*( *)C21-C24 16.87 BD ( ) C25-N28 BD*( *)C30-N31 5.33 LP O26 BD*( *)C24-N27 29.32 BD ( ) N28-C30 BD*( *)N22-C25 6.84 LP N27 BD*(*)C24-O26 49.44 BD () N28-C30 BD*(*)N22-C25 40.59 LP N27 BD*(*)N28-C30 58.95 BD ( ) N31-H33 BD*( *)N27-C30 6.19 LP N28 BD*( *)C21-C25 10.84 CR O26 24 * C RY 5.40 LP N28 BD*( *)N27-C30 14.64 Intermolecular Interactions LP N19 BD*( *)N1-C4 35.26 (State 1) (State 3) (State 2) (State 4) Figure 3. Structures of the 4 reaction states with the critical points obtained by the QTAIM theory. The red critical points are the Bond Critical Points (BCP), and the yellow critical points are the Ring Critical Points (RCP). Open Access OJPC 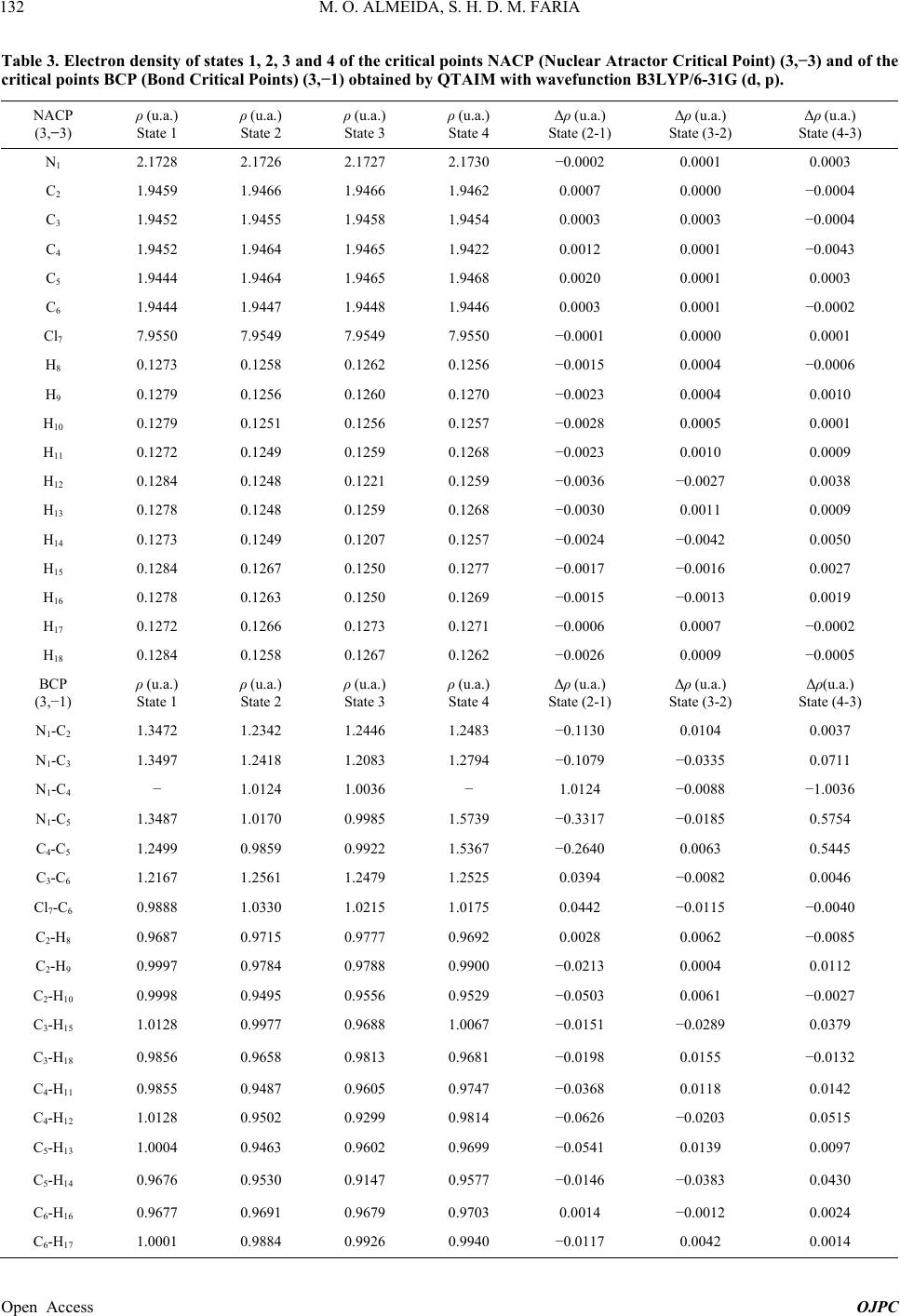 M. O. ALMEIDA, S. H. D. M. FARIA 132 Table 3. Electron density of states 1, 2, 3 and 4 of the critical points NACP (Nuclear Atractor Critical Point) (3,−3) and of the critical points BCP (Bond Critical Points) (3,−1) obtained by QTAIM with wavefunction B3LYP/6-31G (d, p). NACP (3,−3) ρ (u.a.) State 1 ρ (u.a.) State 2 ρ (u.a.) State 3 ρ (u.a.) State 4 Δρ (u.a.) State (2-1) Δρ (u.a.) State (3-2) Δρ (u.a.) State (4-3) N1 2.1728 2.1726 2.1727 2.1730 −0.0002 0.0001 0.0003 C2 1.9459 1.9466 1.9466 1.9462 0.0007 0.0000 −0.0004 C3 1.9452 1.9455 1.9458 1.9454 0.0003 0.0003 −0.0004 C4 1.9452 1.9464 1.9465 1.9422 0.0012 0.0001 −0.0043 C5 1.9444 1.9464 1.9465 1.9468 0.0020 0.0001 0.0003 C6 1.9444 1.9447 1.9448 1.9446 0.0003 0.0001 −0.0002 Cl7 7.9550 7.9549 7.9549 7.9550 −0.0001 0.0000 0.0001 H8 0.1273 0.1258 0.1262 0.1256 −0.0015 0.0004 −0.0006 H9 0.1279 0.1256 0.1260 0.1270 −0.0023 0.0004 0.0010 H10 0.1279 0.1251 0.1256 0.1257 −0.0028 0.0005 0.0001 H11 0.1272 0.1249 0.1259 0.1268 −0.0023 0.0010 0.0009 H12 0.1284 0.1248 0.1221 0.1259 −0.0036 −0.0027 0.0038 H13 0.1278 0.1248 0.1259 0.1268 −0.0030 0.0011 0.0009 H14 0.1273 0.1249 0.1207 0.1257 −0.0024 −0.0042 0.0050 H15 0.1284 0.1267 0.1250 0.1277 −0.0017 −0.0016 0.0027 H16 0.1278 0.1263 0.1250 0.1269 −0.0015 −0.0013 0.0019 H17 0.1272 0.1266 0.1273 0.1271 −0.0006 0.0007 −0.0002 H18 0.1284 0.1258 0.1267 0.1262 −0.0026 0.0009 −0.0005 BCP (3,−1) ρ (u.a.) State 1 ρ (u.a.) State 2 ρ (u.a.) State 3 ρ (u.a.) State 4 Δρ (u.a.) State (2-1) Δρ (u.a.) State (3-2) Δρ(u.a.) State (4-3) N1-C2 1.3472 1.2342 1.2446 1.2483 −0.1130 0.0104 0.0037 N1-C3 1.3497 1.2418 1.2083 1.2794 −0.1079 −0.0335 0.0711 N1-C4 − 1.0124 1.0036 − 1.0124 −0.0088 −1.0036 N1-C5 1.3487 1.0170 0.9985 1.5739 −0.3317 −0.0185 0.5754 C4-C5 1.2499 0.9859 0.9922 1.5367 −0.2640 0.0063 0.5445 C3-C6 1.2167 1.2561 1.2479 1.2525 0.0394 −0.0082 0.0046 Cl7-C6 0.9888 1.0330 1.0215 1.0175 0.0442 −0.0115 −0.0040 C2-H8 0.9687 0.9715 0.9777 0.9692 0.0028 0.0062 −0.0085 C2-H9 0.9997 0.9784 0.9788 0.9900 −0.0213 0.0004 0.0112 C2-H10 0.9998 0.9495 0.9556 0.9529 −0.0503 0.0061 −0.0027 C3-H15 1.0128 0.9977 0.9688 1.0067 −0.0151 −0.0289 0.0379 C3-H18 0.9856 0.9658 0.9813 0.9681 −0.0198 0.0155 −0.0132 C4-H11 0.9855 0.9487 0.9605 0.9747 −0.0368 0.0118 0.0142 C4-H12 1.0128 0.9502 0.9299 0.9814 −0.0626 −0.0203 0.0515 C5-H13 1.0004 0.9463 0.9602 0.9699 −0.0541 0.0139 0.0097 C5-H14 0.9676 0.9530 0.9147 0.9577 −0.0146 −0.0383 0.0430 C6-H16 0.9677 0.9691 0.9679 0.9703 0.0014 −0.0012 0.0024 C6-H17 1.0001 0.9884 0.9926 0.9940 −0.0117 0.0042 0.0014 Open Access OJPC 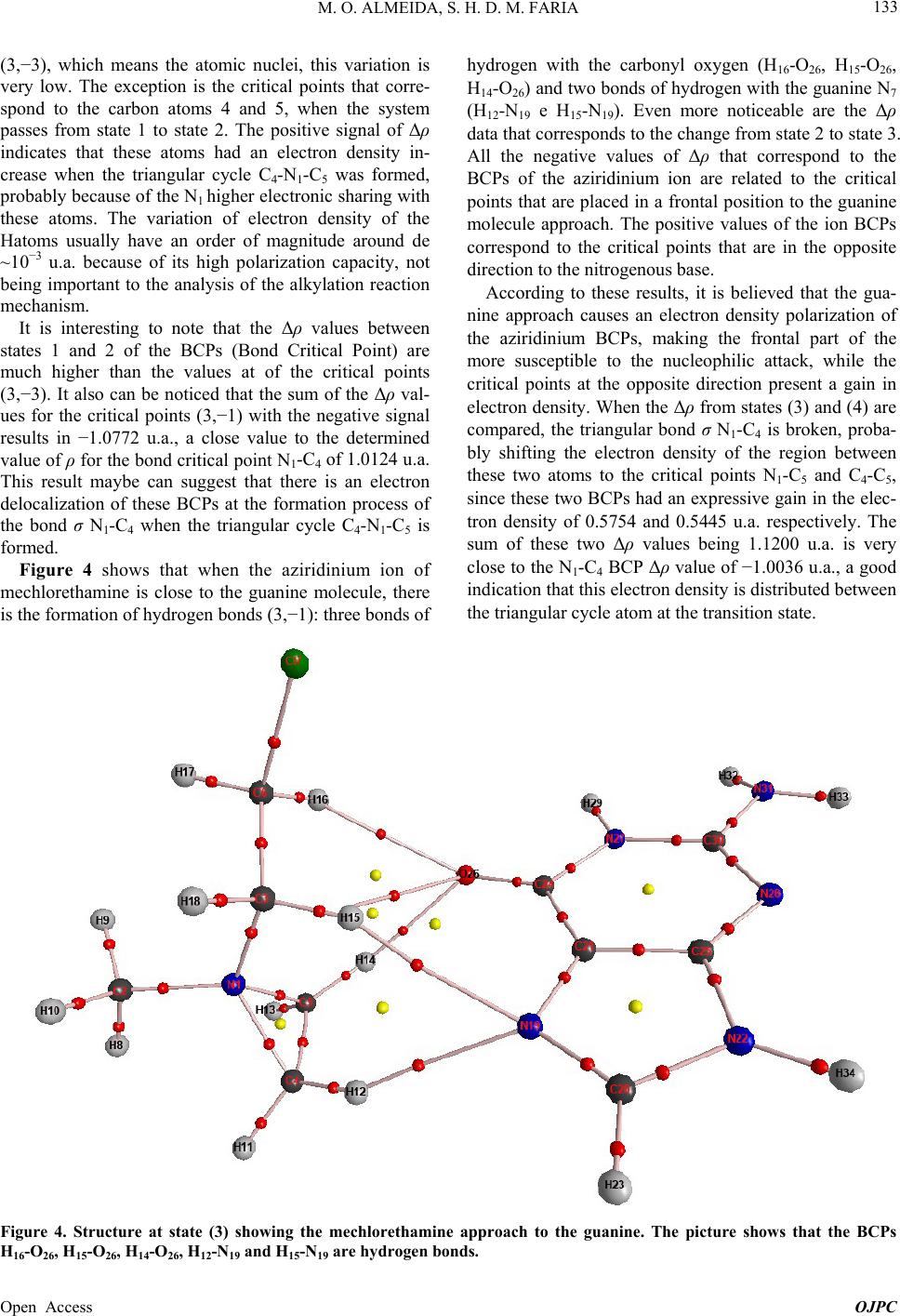 M. O. ALMEIDA, S. H. D. M. FARIA Open Access OJPC 133 (3,−3), which means the atomic nuclei, this variation is very low. The exception is the critical points that corre- spond to the carbon atoms 4 and 5, when the system passes from state 1 to state 2. The positive signal of Δρ indicates that these atoms had an electron density in- crease when the triangular cycle C4-N1-C5 was formed, probably because of the N1 higher electronic sharing with these atoms. The variation of electron density of the Hatoms usually have an order of magnitude around de ~10−3 u.a. because of its high polarization capacity, not being important to the analysis of the alkylation reaction mechanism. hydrogen with the carbonyl oxygen (H16-O 26, H15-O26, H14-O26) and two bonds of hydrogen with the guanine N7 (H12-N19 e H15-N19). Even more noticeable are the Δρ data that corresponds to the change from state 2 to state 3. All the negative values of Δρ that correspond to the BCPs of the aziridinium ion are related to the critical points that are placed in a frontal position to the guanine molecule approach. The positive values of the ion BCPs correspond to the critical points that are in the opposite direction to the nitrogenous base. According to these results, it is believed that the gua- nine approach causes an electron density polarization of the aziridinium BCPs, making the frontal part of the more susceptible to the nucleophilic attack, while the critical points at the opposite direction present a gain in electron density. When the Δρ from states (3) and (4) are compared, the triangular bond σ N1-C4 is broken, proba- bly shifting the electron density of the region between these two atoms to the critical points N1-C5 and C4-C5, since these two BCPs had an expressive gain in the elec- tron density of 0.5754 and 0.5445 u.a. respectively. The sum of these two Δρ values being 1.1200 u.a. is very close to the N1-C4 BCP Δρ value of −1.0036 u.a., a good indication that this electron density is distributed between the triangular cycle atom at the transition state. It is interesting to note that the Δρ values between states 1 and 2 of the BCPs (Bond Critical Point) are much higher than the values at of the critical points (3,−3). It also can be noticed that the sum of the Δρ val- ues for the critical points (3,−1) with the negative signal results in −1.0772 u.a., a close value to the determined value of ρ for the bond critical point N1-C4 of 1.0124 u.a. This result maybe can suggest that there is an electron delocalization of these BCPs at the formation process of the bond σ N1-C4 when the triangular cycle C4-N1-C5 is formed. Figure 4 shows that when the aziridinium ion of mechlorethamine is close to the guanine molecule, there is the formation of hydrogen bonds (3,−1): three bonds of Figure 4. Structure at state (3) showing the mechlorethamine approach to the guanine. The picture shows that the BCPs 16-O26, H15-O26, H14-O26, H12-N 19 and H15-N19 are hydrogen bonds. H  M. O. ALMEIDA, S. H. D. M. FARIA 134 In the Table 4 that refers to the Laplacian of the elec- tron density, it is noticed that at the aziridinium ion of mechlorethamine (state 2), there is a low asymmetry be- tween the Δ2ρ values of the carbon 4 and 5 (3.3838 and Table 4. Laplacian of the Electron density of states 1, 2, 3 and 4 of the critical points NACP (Nuclear Attractor Critical Point) (3,−3) and the critical points BCP (Bond Critical Points) (3,−1) obtained by QTAIM with wave function B3LYP/6-31G (d, p). NACP (3,−3) Δ2ρ (u.a.) State 1 Δ2ρ (u.a.) State 2 Δ2ρ (u.a.) State 3 Δ2ρ (u.a.) State 4 2 (u.a.) State (2-1) 2 ) (u.a.) State (3-2) 2 (u.a.) State (4-3) N1 2.4213 2.4235 2.4248 2.4250 0.0022 0.0013 0.0002 C2 3.3851 3.3858 3.3833 3.3857 0.0007 −0.0025 0.0024 C3 3.3832 3.3832 3.3853 3.3859 0.0000 0.0021 0.0006 C4 3.3839 3.3838 3.3830 3.3627 −0.0001 −0.0008 −0.0203 C5 3.3824 3.3836 3.3829 3.3864 0.0012 −0.0007 0.0035 C6 3.3828 3.3878 3.3882 3.3854 0.0050 0.0004 −0.0028 Cl7 25.1423 25.1530 25.1589 25.1635 0.0107 0.0059 0.0046 H8 0.2475 0.2448 0.2458 0.2442 −0.0027 0.0010 −0.0016 H9 0.2499 0.2440 0.2450 0.2475 −0.0059 0.0010 0.0025 H10 0.2498 0.2429 0.2443 0.2452 −0.0069 0.0014 0.0009 H11 0.2489 0.2421 0.2447 0.2462 −0.0068 0.0026 0.0015 H12 0.2511 0.2420 0.2344 0.2429 −0.0091 −0.0076 0.0085 H13 0.2489 0.2420 0.2447 0.2472 −0.0069 0.0027 0.0025 H14 0.2475 0.2421 0.2306 0.2440 −0.0054 −0.0115 0.0134 H15 0.2510 0.2466 0.2417 0.2493 −0.0044 −0.0049 0.0076 H16 0.2489 0.2457 0.2428 0.2469 −0.0032 −0.0029 0.0041 H17 0.2489 0.2464 0.2480 0.2474 −0.0025 0.0016 −0.0006 H18 0.2510 0.2446 0.2468 0.2461 −0.0064 0.0022 −0.0007 BCP (3,−1) Δ2ρ (u.a.) State 1 Δ2ρ (u.a.) State 2 Δ2ρ (u.a.) State 3 Δ2ρ (u.a.) State 4 2 (u.a.) State (2-1) 2 (u.a.) State (3-2) 2 (u.a.) State (4-3) N1-C2 −0.7180 −0.7079 −0.7014 −0.6660 0.0101 0.0065 0.0354 N1-C3 −0.7388 −0.7514 −0.7232 −0.7027 −0.0126 0.0282 0.0205 N1-C4 - −0.5191 −0.5182 - −0.5191 0.0009 0.5182 N1-C5 −0.7373 −0.5172 −0.5129 −0.9286 0.2201 0.0043 −0.4157 C4-C5 −0.5391 −0.2799 −0.2855 −0.6744 0.2592 −0.0056 −0.3889 C3-C6 −0.5080 −0.5379 −0.5305 −0.5367 −0.0299 0.0074 −0.0062 Cl7-C6 −0.5648 −0.5726 −0.5808 −0.5702 −0.0078 −0.0082 0.0106 C2-H8 −0.1888 −0.1724 −0.1778 −0.1714 0.0164 −0.0054 0.0064 C2-H9 −0.2020 −0.1749 −0.1758 −0.1876 0.0271 −0.0009 −0.0118 C2-H10 −0.2020 −0.1499 −0.1557 −0.1632 0.0521 −0.0058 −0.0075 C3-H15 −0.2245 −0.2033 −0.1746 −0.2156 0.0212 0.0287 −0.0410 C3-H18 −0.2068 −0.1694 −0.1854 −0.1828 0.0374 −0.0160 0.0026 C4-H11 −0.2069 −0.1389 −0.1464 −0.1509 0.0680 −0.0075 −0.0045 C4-H12 −0.2245 −0.1401 −0.1292 −0.1634 0.0844 0.0109 −0.0342 C5-H13 −0.1970 −0.1360 −0.1462 −0.1674 0.0610 −0.0102 −0.0212 C5-H14 −0.1667 −0.1419 −0.1153 −0.1586 0.0248 0.0266 −0.0433 C6-H16 −0.1668 −0.1701 −0.1682 −0.1715 −0.0033 0.0019 −0.0033 C6-H17 −0.1968 −0.1854 −0.1887 −0.1900 0.0114 −0.0033 −0.0013 Open Access OJPC  M. O. ALMEIDA, S. H. D. M. FARIA 135 3.3836 u.a. respectively), besides there is also an asym- metry to the BCPs N1-C4 and N1-C5 (−0.5191 and −0.5172 u.a.). Can also be observed in the Table 4 that the state (3), with the presence of the guanine, an increase on the Laplacian difference is seen between the BCPs N1-C4 and N1-C5 for states (2) and (3), being now 2 = 0.0053 u.a.. It is also observed at state (3), a reduction in the Laplacian of the electron density of C4 with respect to the state (2), and when states (3) and (4) are compared, this reduction is even greater ( 2u.a. 0.0203. This demonstrates that the guanine approach increases the hole factor at the C4 (Figure 5), and that the nucleophilic attack, based on the lump-hole concept, makes the region of this atom to become the reaction site for this alkyla- tion. C4 1 C5 1 C5 C4 Electrophile or hole (hole factor increased by guanine) Figure 5. Relief maps of the Laplacian of the electron den- sity of the states 2 (up) and 3 (down). It is noticed a greater hole at carbon 4 at the structure on right, emphasizing that the guanine presence increases the electrophilic carater of 4. Conclu this atom. sions NBO analysis clearly showed that e Atoms in Molecules (Q btained for the Laplacian of the electron de 5. Acknowledgements CENAPAD-SP. Michell O. REFERENCES [1] V. L. Almeideina, C. A. Mon- 1 The results of the when the triangular cycle C4-N1-C5 is formed at the cy- clization process, there is a kind of electronic resonance among these atoms. It was also noticed a certain asym- metry between the second order perturbation energy of the C4 and C5 of the triangular cycle, indicating that the C4 is more susceptible to a nucleophilic attack. Finally, the NBO analysis showed that the nucleophilic attack occurs through an electronic transition between the gua- nine N7 lone pair and the C4 of the triangular cycle of the aziridinium ion of mechlorethamine transition state, state found by the algorithm QST3. The data obtained with th TAIM) theory showed that when there is a change in the state of the mechlorethamine molecule, the main variation in electron density is at the critical points (3,−1) and not at (3,−3). It was also noticed that the approach of the guanine molecule possibly generates a polarization of the BCPs electron density of the aziridinium ion of me- chlorethamine, making that the frontal part of the mole- cule (related to the guanine position) becomes more defi- cient in electrons, being more susceptible to a nucleo- philic attack. The values o nsity showed that the region around the C4 of the C4-N1-C5 triangular cycle is the reaction site of the alky- lation reaction of the aziridinium ion of mechlorethamine, due to the presence of a greater hole than the C5. The authors are grateful to Almeida thanks Universidade Paulista (UNIP) for under- graduate student fellowships. a, A. Leitão, L. C. B. R tanari and C. L. Donnici, “Câncer e Agentes Antineo- plásicos, Ciclo-Celular Específicos e Ciclo-Celular não Específicos que Interagem com o DNA: uma Introdução,” Química Nova, Vol. 28, No. 1, 2005, pp. 118-129. http://dx.doi.org/10.1590/S0100-4042200500010002 of [2] P. K. Shukla, P. C. Mishra and S. Suhai, “Reactions DNA Bases with the Anti-Cancer Nitrogen Mustard Mechlorethamine: A Quantum Chemical Study,” Chemi- cal Physics Letters, Vol. 449, No. 4-6, 2007, pp. 323-328. http://dx.doi.org/10.1016/j.cplett.2007.10.072 [3] P. Brookes and P. D. Lawley, “The Reaction of Mono- and Difunctional Alkylating Agents with Nucleic Acids,” Biochemical Journal, Vol. 80, No. 3, 1961, pp. 496-503. [4] S. M. Rink and P. B. Hopkins, “A Mechlorethamine-In- duced DNA Interstrand Cross-Link Bends Duplex DNA,” Biochemistry, Vol. 34, No. 4, 1995, pp. 1439-1445. http://dx.doi.org/10.1021/bi00004a039 Open Access OJPC  M. O. ALMEIDA, S. H. D. M. FARIA 136 [5] J. P. Holley, A. Mather, R. T. Wheelhouse, P. M. Cullis rakash, W. A. Denny, T. A. Gourdie, K. K. , J. A. Hartley, J. P. Bingham and G. M. Cohen, “Targeting of Tumor Cells and DNA by a Chlorambucil-Spermidine Conjugate,” Cancer Research, Vol. 52, 1992, pp. 4190- 4195. [6] A. S. P Valu, P. D. Woodgate and L. P. G. Wakelin, “DNA-Directed Alkylating Ligands as Potential Antitumor Agents: Se- quence Specificity of Alkylation by Intercalating Aniline Mustards,” Biochemistry, Vol. 29, No. 42, 1990, pp. 9799-9807. http://dx.doi.org/10.1021/bi00494a007 [7] I. Carvalho, M. T. Pupo, A. D. L. Borges and L. S. C. Bernardes, “Introdução a Modelagem Molecular de Fár- macos no curso Experimental de Química Farmacêutica,” Quimica Nova, Vol. 26, No. 3, 2003, pp. 428-438. http://dx.doi.org/10.1590/S0100-40422003000300023 [8] H. Broch, A. Hamza and D. Vasilescu, “Quantum Mo- lecular Modeling of the Interaction between Guanine and Alkylating Agents-1-Sulfur Mustard,” Journal of Biomo- lecular Structure Dynamics, Vol. 13, No. 3, 1996, pp. 903-914. http://dx.doi.org/10.1080/07391102.1996.10508905 [9] H. Broch, A. Hamza and D. Vasilescu, “Quantum Mo- lecular between Guanine and Alkylating Agents 2-Nitro- gen-Mustard,” Journal of Biomolecular Structure Dyna- mics, Vol. 13, No. 3, 1996, pp. 915-924. http://dx.doi.org/10.1080/07391102.1996.10508906 [10] H. Broch, R. Viani and D. Vasilescu, “Quantum Molecu- lar Study of the Alkylating Agent Mechlorethamine,” In- ternational Journal of Quantum Chemistry, Vol. 40, No. S18, 1991, pp. 119-130. http://dx.doi.org/10.1002/qua.560400715 [11] D. Vasilescu, M. Adrian-Scotto, A. Fadiel and A. Hamza, “Ab Initio Study of Alkylation of Guanine-Cytosine Base Pair by Sulfur and Nitrogen Mustards,” Journal of Bio- molecular Structure Dynamics, Vol. 27, No. 4, 2010, pp. 465-476. http://dx.doi.org/10.1080/07391102.2010.10507331 [12] S. Kang and J. P. Green, “Correlation between Activity and Electronic State of Hallucinogenic Amphetamines,” Nature, Vol. 226, No. 5246, 1970, pp. 645-645. http://dx.doi.org/10.1038/226645a0 [13] R. T. Lalonde, H. Leo, H. Perakyla, C. W. Dence and R. P. Farrell, “Associations of Halogenated 2 (5H)-Furanones with Their MNDO-PM3 Computed Properties and Mode of Reactivity with Sodium Borohydride,” Chemical Re- search in Toxicology, Vol. 5, No. 3, 1992, pp. 392-400. http://dx.doi.org/10.1021/tx00027a012 [14] O. Kikuchi, “Systematic QSAR Procedures with Quan- tum Chemical Descriptors,” Quantitative Structure-Acti- vity Relationships, Vol. 6, No. 4, 1987, pp. 179-184. http://dx.doi.org/10.1002/qsar.19870060406 [15] B. W. Clare, “Structure-Activity Correlations Psychoto- mimetcis. 1. Phenylalkylamines: Electronic, Volume, and Hidrophobicity Parameters,” Journal of Medicinal Chem- istry, Vol. 33, No. 2, 1990, pp. 687-702. http://dx.doi.org/10.1021/jm00164a036 [16] B. W. Clare, “Charge Transfer Complexes and Frontier Orbital Energies in QSAR: A Congeneric Series of Elec- tron Acceptors,” Journal of Molecular Structure Theo- chem, Vol. 337, No. 2, 1995, pp. 139-150. http://dx.doi.org/10.1016/0166-1280(95)04135-S [17] J. P. Foster and F. Weinhold, “Natural Hybrid Orbitals,” Journal of American Chemical Society, Vol. 102, No. 24, 1980, pp. 7211-7218. http://dx.doi.org/10.1021/ja00544a007 [18] A. E. Reed, L. A. Curtiss and F. Weinhold, “I cular Interactions from a Natural Bon ntermole- d Orbital, Donor- Accpetor Viewpoint,” Chemical Reviews, Vol. 88, No. 6, 1988, pp. 899-926. http://dx.doi.org/10.1021/cr00088a005 [19] L. Pauling, “The Nature Chemical Bond. A Results Obtained from the Quantum Me pplication of chanics and from a Theory of Paramagnetic Susceptibility to the Structure of Molecules,” Journal of American Chemical Society, Vol. 53, No. 4, 1931, pp. 1367-1400. http://dx.doi.org/10.1021/ja01355a027 [20] J. C. Slater, “Directed Valence in Polyatom Physical Review, Vol. 37, No. 5, 1931, p ic Molecules,” p. 481-489. http://dx.doi.org/10.1103/PhysRev.37.481 [21] C. A. Coulson, “Valence,” 2nd Edition, Oxford Univ sity Press, London, 1952. er- rd, 1990. he Laplacian of the [22] R. F. W. Bader, “Atoms in Molecules: A Quantum The- ory,” Claredon Press, Oxfo [23] S. Calvo-Losada and J. J. Q. Sánchez, “Pericyclic versus Pseudopericyclic Reactions. What t Charge Density, ∇2ρ(r), Has to Say about It? The Case of Cycloaddition Reactions,” Journal of Physical Chem- istry A, Vol. 112, No. 35, 2008, pp. 8164-8178. http://dx.doi.org/10.1021/jp711565g [24] M. T. Carroll, J. R. Cheeseman, R. Osman and H stein, “Nucleophilic Addition to Activ . Wein- ated Double Bonds: Predictions of Reactivity from the Laplacian Charge Den- sity,” Journal of Physical Chemistry, Vol. 93, No. 13, 1989, pp. 5120-5123. http://dx.doi.org/10.1021/j100350a019 [25] M. J. Frisch, G. W. Trucks, H. B. Schle M. A. Robb, J. R. Cheeseman, J. A. M gel, G. E. Scuseria, ontgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Po- melli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslo- wski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al- Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, “GAUSSVIEW 3.0, Revision D.02,” Gaussian Inc., Wallington, 2004. [26] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, Open Access OJPC  M. O. ALMEIDA, S. H. D. M. FARIA Open Access OJPC 137 ntgomery Jr., T. les, “AIM2000 Atoms in Mole- emical Reviews, Vol. 98, 1998, M. A. Robb, J. R. Cheeseman, J. A. Mo Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Po- melli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslo- wski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al- Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, “Gaussview 03, Revision D.02,” Gaussian Inc., Wallington, 2004. [27] 2013. http://www.cenapad.unicamp.br/ [28] F. Biegler-König, J. Schönbohm and D. Bay —A Program to Analyze and Visualize cules,” Journal of Computational Chemistry, Vol. 22, No. 5, 2001, pp. 545-559. [29] S. R. Rajski and R. M. Williams, “DNA Cross-Link Agent as Antitumor Drugs,” Ch No. 8, pp. 2723-2795. http://dx.doi.org/10.1021/cr9800199 [30] C. Avendanaño and C. J. Menéndez, try of Anticancer Drugs,” Elsevier, Am “Medicinal Chemis- sterdam, 2008.
|