 Communications and Network, 2013, 5, 601-605 http://dx.doi.org/10.4236/cn.2013.53B2108 Published Online September 2013 (http://www.scirp.org/journal/cn) Copyright © 2013 SciRes. CN Threshold Selection Study on Fisher Discriminant Analysis Used in Exon Prediction for Unbalanced Data Sets Yutao Ma1, Yanbing Fang2, Ping Liu1, Jianfu Teng3 1School of Physics and Electrical Information Engineering, Ningxia University, Yinchuan, China 2School of Mathematics and Computer Science, Ningxia University, Yinchuan, China 3School of Electronic Information Engineering, Tianjin University, Tianjin, China Email: yutao_ma@163.com, nxfangzi@nxu.edu.cn, liuping@nxu.edu.cn Received May 2013 ABSTRACT In gene prediction, the Fisher discriminant analysis (FDA) is used to separate protein coding region (exon) from non- coding regions (intron). Usually, the positive data set and the negative data set are of the same size if the number of the data is big enough. But for some situations the data are not sufficient or not equal, the threshold used in FDA may have important influence on prediction results. This paper presents a study on the selection of the threshold. The eigen value of each exon/intron sequence is computed using the Z-curve method with 69 variables. The experiments results suggest that the size and the standard deviation of the data sets and the threshold are the three key elements to be taken into consideration to improve the prediction results. Keywords: Fisher Discriminant Analysis; Threshold Selection; Gene Prediction; Z-Curve; Size of Data Set 1. Introduction Protein coding region and non-coding region of a DNA sequence are also called exon and intron respectively. Gene prediction of eukaryotes is still one of the most important research domains of bioinformatics for the prediction accuracy is needed to be improved [1]. Fisher Discriminant analysis/algorithm (FDA) is widely used in solving binary classification problems like fault classifi- cation [2], gene expression data classification [3], image categorization [4], integrating heterogeneous data sets [5] and DNA sequence classification [6,7], and it has at- tracted more and more attention. Kernel FDA (KFDA) may present out performance in both simulation time consume and classification precision than support vector machine (S VM) for i t does not need t o solve any quadratic problem [8,9]. For maximizing the uniformity of class- pair separabilities and class separability in kernel space simultaneously, a novel kernel FDA kernel parameters optimization criterion is presented [10]. A novel dimen- sionality reduction algorithm based on FDA is proposed for ranking applications such as visual search re-ranking [11]. Usually, the size of the training and test data sets are selected equal and the threshold is determined by making the false negative rate and the false positive rate equal [6]; but for some real situations, the threshold could not be obtained by the method because the sizes of the positive and negative data sets are unbalanced. In the latter situa- tion, there are five possible thresholds for making the coding/non-coding decision. Using the exon and intron data sets downloaded from the website: http://www.fruitfly.org/seq_tools/datasets/Human/, the FDA based experiments show that the size and the standard deviation of the data sets are the two key elements to be taken into consideration to improve the prediction re- sults. 2. Data Sets and Methods 2.1. The Data Sets The Data sets used in this paper were downloaded from the above net address. The Data were separated into 7 parts in the website, and each part consists of an exon sub-set and an intron sub-set. There are 301 DNA frag- ments at least an d 448 at most in each sub-set as detailed in Table 1. The difference values between the numbers of exons and introns in each part are all 66 from the Table 1. Table 1. The details of each part of the data sets. Part0 Part1 Part2 Part3 Part4 Part5 Part6 Exon 367 448 447 389 399 402 391 Intron 301 382 381 323 333 336 325 Total 668 830 828 712 732 738 716  Y.-T. MA ET AL. Copyright © 2013 SciRes. CN 2.2. The Z Curve Methods The Z curve method is a powerful tool to visualize and analyze DNA sequences. It is also one of the most widely used mapping methods which maps a DNA sequence into three digital sequences. According to [6], the predic- tion results of the Z curve method with 69 variables are almost the same with that of 169 variables ones. So, the Z curve method with 69 variables is applied in this paper. The 69 variables are composed with 9 Z curve parame- ters for frequencies of phase-specific mononucleotides, 12 Z curve parameters for frequencies of phase indepen- dent di-nucleotides and 48 Z curve parameters for fre- quencies of phase independent tri-nucleotides. These Z curve parameters are defined below. Let A, T, C and G represent base adenine, thymine, cytosine and guanine respectively. The bases A, T, C, G are occurr ing in a DNA fragment at positions 1, 4, 7, …; 2, 5, 8, …; and 3, 6, 9, …, with frequencies , , , ; , , , ; , , , respectively. Then by using the Z-transform defined by Equation (1), a fragment of DNA sequence is transformed into the 9 Z curve parameters for frequencies of phase-specific mo- nonucleotides. ()( ) ()( ) ( )() ,,[1, 1 ],1,2,3 ii iii ii iii iiii i iii x agct y acgt z atgc xyz i = +−+ =+−+ = +− + ∈− = . (1) Let be the frequency of the di-nucleotides ( ), the 12 variables for frequen- cies of phase independent di-nucleotides are given by [ ()()][ ()()] [ ()()][ ()()] [ ()()][ ()()] ,,, X X X xpXApXGp XCP XT ypXApXCp XGP XT zpXAp XTp XGP XC XACGT =+−+ =+− + =+−+ = . (2) The last 48 Z curve parameters for frequencies of phase independent tri-nucleotides can be obtained using the similar notations by [ ()()][ ()()] [ ()()][ ()()] [ ()()][()()] ,,,, ,,, XY XY XY xp XYAp XYGp XYCPXYT yp XYApXYCp XYGP XYT zp XYAp XYTp XYGP XYC XACGT YAC GT =+−+ =+− + =+−+ = = . (3) Next, the 6 9 variabl e s of each exon/intron are 999 12 1212 484848 129 1212 1248 ,,,,,,,,, ,,uuuuuuuuu . They are defined by Equations (4) to (8). These 99 9 112233 99 9 4 15263 99 9 7 18293 ,,, ,,, ,, uxuxux uyuyuy uzuzuz = == = == = == . (4) 121212 12 1 234 1212 12 12 5 678 12 12 1212 910 11 12 , , ,, ,,,, ,,, AC GT AC GT ACG T uxuxuxux uyuyuyuy uzuzuzuz === = == = = = = == . (5) 48 4848 48 1234 4848 48 48 5678 48 484848 910 11 12 48 484848 13 141516 ,,,, ,, ,, ,,,, ,,,, AAACAG AT CACCCG CT GA GCGG GT TA TCTGTT uxuxuxux uxuxuxux uxuxuxux uxuxuxux = == = == = = = === = === . (6) 48 4848 48 17 1819 20 4848 4848 2122 2324 48 484848 25 262728 48 484848 29 303132 ,,,, ,, ,, ,,,, , ,,, AAACAG AT CACC CG CT GA GC GGGT TATCTG TT uyuyuyuy uyuyuyuy uyuyuyuy uyuyuyuy = === = = = = = === = === . (7) 48 484848 33 343536 48484848 37383940 48 484848 41 424344 48 484848 45 464748 ,,,, ,,,, ,,,, ,,,. AAACAG AT CACC CG CT GA GC GGGT TATC TG TT uzuzuzuz uzuzuzuz uzuzuzuz uzuzuzuz = == = == == = === = === . (8) 2.3. The Fisher Discriminant Analysis/Algorithm Let be the data matrix, where N and M are the number of data/samples and the dimension of each data respectively. The M is also the number of the best FDA coefficients, which are . In this paper the dimension of the data is 69, that is . Each exon/Intron is described by a point or row vector in a 69-dimensional (69-D) spa c e spanned by 999 121212 484848 129 1212 1248 1 269 (,,,,,,,,, ,, ) (, ,,) T T uuuu uuu uu xx x = = x , (9) There are two groups of samples in the training data set used in FDA. One contains positive samples which belong to sample space , and another contains the negative samples which belong to sample space . The positive samples are DNA fragments from real exons and the negative samples are from the real introns. All the sequences longer than 2000 bases were cut down to 2000 bases. The numbers of positive and negative samples are and respectively, and . The mean vector of the positive/negative samples in input space is defined by n 11 , ,, MM pn pn pn RR NN ∈∈ ==∈∈ ∑∑ p xw xw ux uxuu . (10) The best FDA coefficients consists a column vector . Using the divergence matrix W S, the vector is defined as the best projecting direc tion (BP D ) a n d i s given by ( ) ( ) ( ) 1 12 ,,, , TM pn MW uu uR − == −∈uSuuu , (11) where “T” indicates the transpose of a matrix, and the  Y.-T. MA ET AL. Copyright © 2013 SciRes. CN divergence matrix is defined as ( )( ) 2 , j T W ijij jpni N= ∈ = −− ∑∑ Sxu xu . (12) Once the and the threshold are obtained, the dis- crimination of exon/intron for each DNA fragment in the test set is carried out by . 2.4. The Threshold Selection of FDA For situations where numbers of positive and negative samples are not equal, in other words , there are five different thresholds ( , , ) to be selected, which are represented by Equations (13) to (17). , (13) , (14) , (15) ( ) 4 Tpp nn pn c σσ σσ + =+ uuu , (16) where and are the average variances of the positive and negative samples respectively. ( ) 5 Tnp pn pn c σσ σσ + =+ uu u . (17 ) For situation where , the threshold is uniquely determined by letting the false negative rate (FNR) be equal to the false positive rate (FPR) [6,7]. The details will be given in the next section of this paper. 2.5. The Z Curve Mark of th e DNA Sequence The best projecting direction can be interpreted as the weight vector corresponds to the 69 Z curve values of each test sample. The Z curve score (ZCS) of each DNA sequence is defined as , where the “×” is the multiplication cross symbol, “ ”and “ ” are the best project direction and row vector of a DNA sequence. The ZCS is too small to be display directly, so a transform named remainder and multiple (RM) is carried out. The RM transform is defined as ()(1000, ) ()(1000 /) R ZCSMODZCSM MZCSfloorZCSM = × = × , (18) where “MOD” and “floor” are two MATLAB function names. Function “mod(x, y)” returns the modulus of “x” after it is devided by “y”, and function “floor(x)” rounds the elements of “x” to the nearest integers. In this work, the “M” in the RM transform is set to 3. Thus, the ZCS of a DNA sequence can be displayed in a two dimen- sional surface with the rounds ( ) and the mod- ulus ( ) are the two coordinate axis. 3. Results and Discussion 3.1. The Prediction Accuracy Measures To measure the prediction accuracy, the sensitivity and the specificity are applied. They are defined by Equations (19) and (20) respectively. (19) (20) where is the number of true exons/introns which were predicted as exons/introns, and the FP/FN is the number of true introns/exons which were predicted as exons/introns [12]. The prediction average accuracy is defined as . The FPR and the FNR are de- fined by Eq ua tions (20) and (21) respectively. (21) (22) Let , we have . For the situation equals to , the expressions and are satisfied. Then we have , and the threshold can be expressed as () ( ) 0 / /2 TT ppnnp n c NNN=+=+uuuuu u . (23) The Statistics of the ZCS of Data Sets. To clear the relationship between the data sets and the prediction results, the statistics nature of the 7 data parts are presented in Tables 2 and 3. These statistics nature include mean and standard deviation of the Z curve value. Figure 1 gives the ZCS scatter diagram of the Part0 to Part6 with the RM transform is applied to the ZCS of each sequence. Table 2. The ZCS statistics nature of the exon sequences. Data sets Part0 Part1 Part2 Part3 Part4 Part5 Part6 mean 0.0053 0.0039 0.0043 0.0035 0.0043 0.0053 0.0043 SDa 0.0053 0.0043 0.0039 0.0043 0.0044 0.0045 0.0041 Table 3. The ZCS statistics nature of the intron sequences. Data sets Part0 Part1 Part2 Part3 Part4 Part5 Part6 mean −0.0103 −0.0086 −0.0055 −0.0078 −0.0075 −0.0065 −0.0064 SDa 0.0041 0.0034 0.0028 0.0036 0.0034 0.0033 0.0035 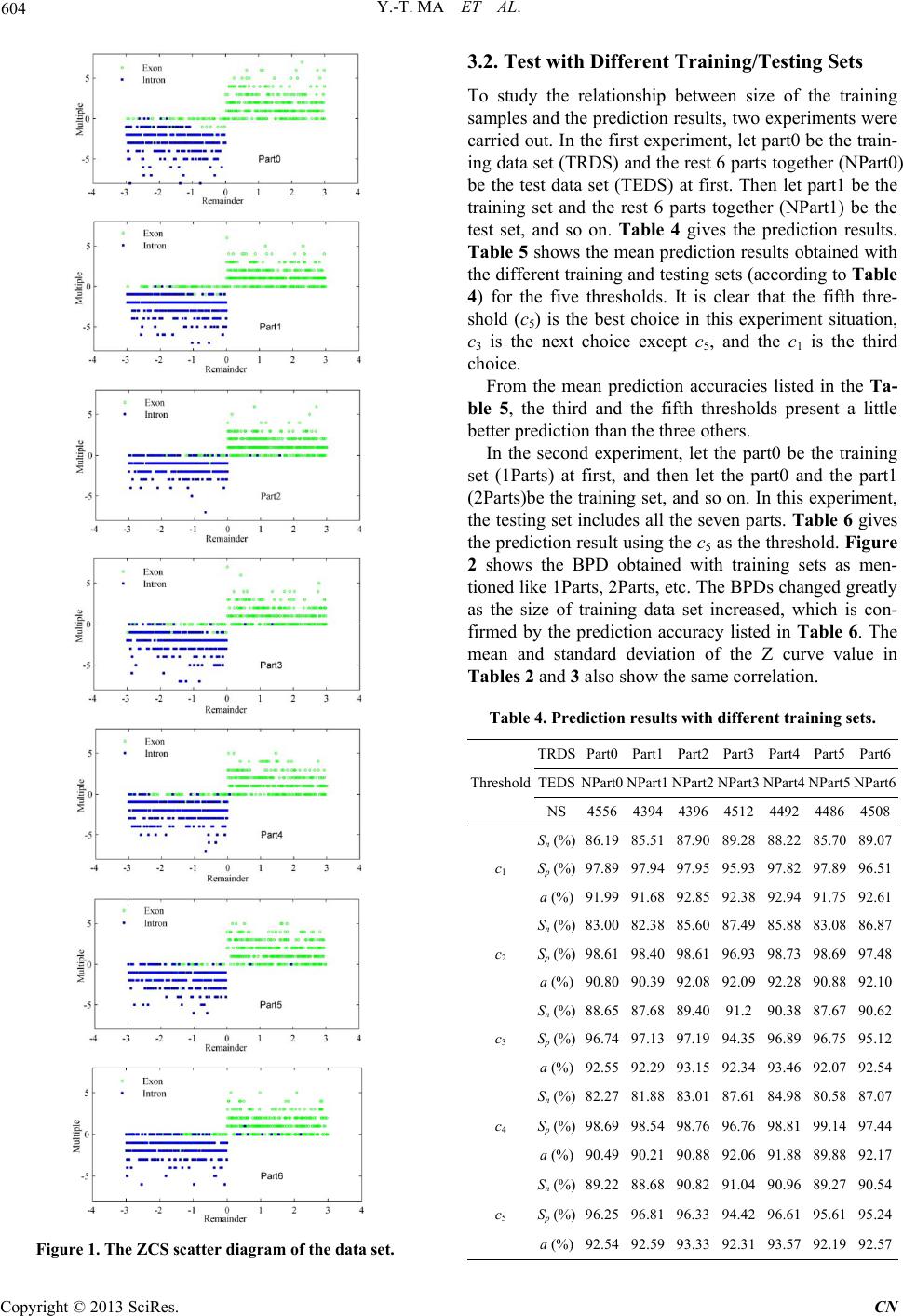 Y.-T. MA ET AL. Copyright © 2013 SciRes. CN Figure 1. The ZCS scatter diagram of the data set. 3.2. Test with Different Training/Testing Sets To study the relationship between size of the training samples and the prediction results, two experiments wer e carried out. In the first experiment, let part0 be the train- ing data set (TRDS) and th e rest 6 parts together (NPart0) be the test data set (TEDS) at first. Then let part1 be the training set and the rest 6 parts together (NPart1) be the test set, and so on. Table 4 gives the prediction results. Table 5 shows the mean prediction resu lts obtained with the different training and testing sets (according to Table 4) for the five thresholds. It is clear that the fifth thre- shold (c5) is the best choice in this experiment situation, c3 is the next choice except c5, and the c1 is the third choice. From the mean prediction accuracies listed in the Ta- ble 5, the third and the fifth thresholds present a little better prediction than the three others. In the second experiment, let the part0 be the training set (1Parts) at first, and then let the part0 and the part1 (2Parts)be the training set, and so on. In this experiment, the testing set includes all the seven parts. Table 6 gives the prediction r esult using the c5 as the threshold. Figure 2 shows the BPD obtained with training sets as men- tioned like 1Parts, 2Parts, etc. The BPDs changed greatly as the size of training data set increased, which is con- firmed by the prediction accuracy listed in Table 6. The mean and standard deviation of the Z curve value in Tables 2 and 3 also show the same correlation. Table 4. Prediction results with different training sets. Threshold TRDS Part0 Part1 Part2 Part3 Part4 Part5 Part6 TEDS NPart0 NPart1 NPart2 NPart3 NPart4 NPart5 NPart6 NS 4556 4394 4396 4512 4492 4486 4508 c1 Sn (%) 86.19 85.51 87.90 89.28 88.22 85.70 89.07 Sp (%) 97.89 97.94 97.95 95.93 97.82 97.89 96.51 a (%) 91.99 91.68 92.85 92.38 92.94 91.75 92.61 c2 Sn (%) 83.00 82.38 85.60 87.49 85.88 83.08 86.87 Sp (%) 98.61 98.40 98.61 96.93 98.73 98.69 97.48 a (%) 90.80 90.39 92.08 92.09 92.28 90.88 92.10 c3 Sn (%) 88.65 87.68 89.40 91.2 90.38 87.67 90.62 Sp (%) 96.74 97.13 97.19 94.35 96.89 96.75 95.12 a (%) 92.55 92.29 93.15 92.34 93.46 92.07 92.54 c4 Sn (%) 82.27 81.88 83.01 87.61 84.98 80.58 87.07 Sp (%) 98.69 98.54 98.76 96.76 98.81 99.14 97.44 a (%) 90.49 90.21 90.88 92.06 91.88 89.88 92.17 c5 Sn (%) 89.22 88.68 90.82 91.04 90.96 89.27 90.54 Sp (%) 96.25 96.81 96.33 94.42 96.61 95.61 95.24 a (%) 92.54 92.59 93.33 92.31 93.57 92.19 92.57 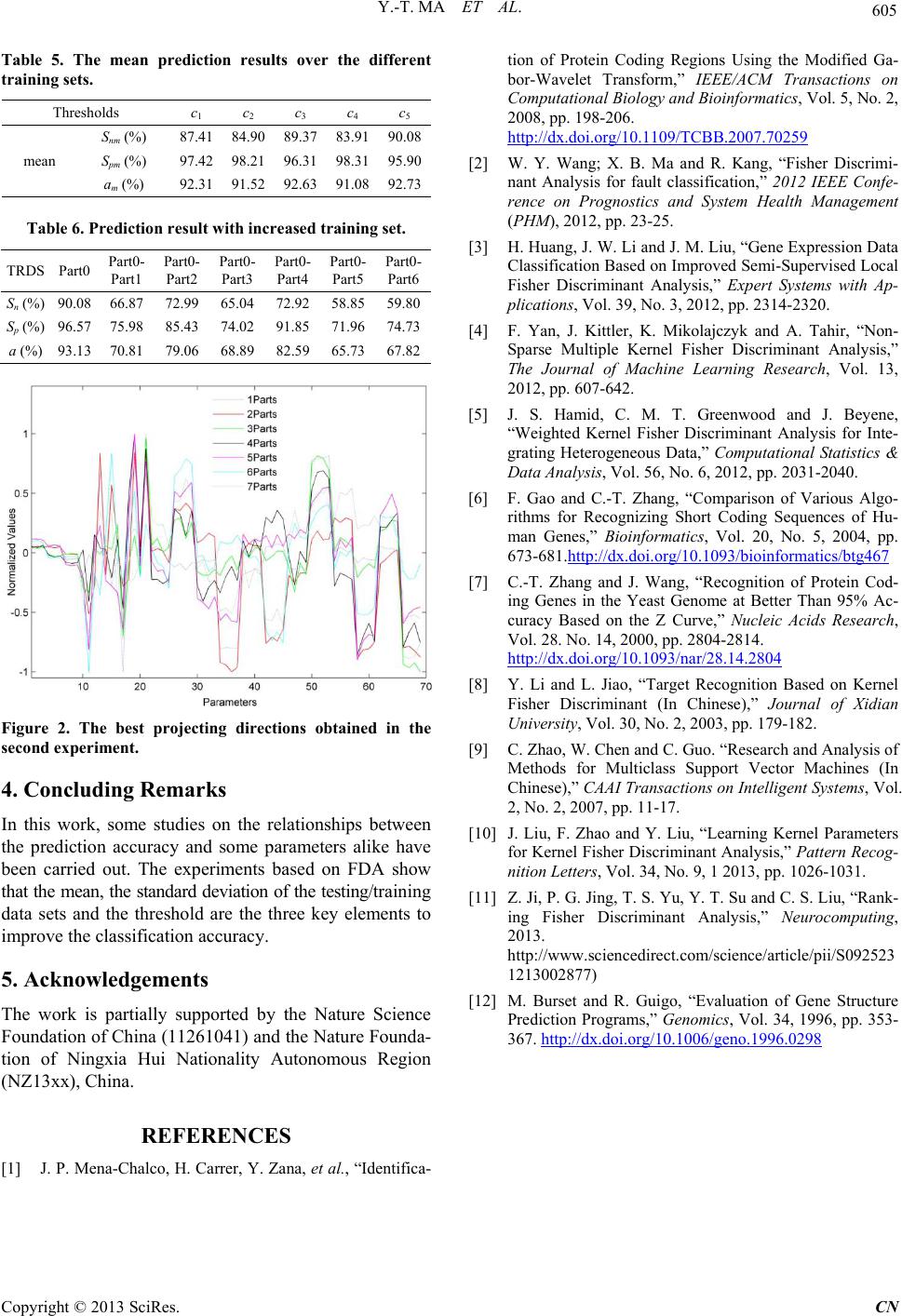 Y.-T. MA ET AL. Copyright © 2013 SciRes. CN Table 5. The mean prediction results over the different training sets. Thresholds c1 c2 c3 c4 c5 mean Snm (%) 87.41 84.90 89.37 83.91 90.08 Spm (%) 97.42 98.21 96.31 98.31 95.90 am (%) 92.31 91.52 92.63 91.08 92.73 Table 6. Prediction result with increased training set. TRDS Part0 Part0- Part1 Part0- Part2 Part0- Part3 Part0- Part4 Part0- Part5 Part0- Part6 Sn (%) 90.08 66.87 72.99 65.04 72.92 58.85 59.80 Sp (%) 96.57 75.98 85.43 74.02 91.85 71.96 74.73 a (%) 93.13 70.81 79.06 68.89 82.59 65.73 67.82 Figure 2. The best projecting directions obtained in the second experiment. 4. Concluding Remarks In this work, some studies on the relationships between the prediction accuracy and some parameters alike have been carried out. The experiments based on FDA show that the mean, the standard deviation of the testing/training data sets and the threshold are the three key elements to improve the classification accuracy. 5. Acknowledgements The work is partially supported by the Nature Science Foundation of China (11261041) and the Nature Founda- tion of Ningxia Hui Nationality Autonomous Region (NZ13xx), China. REFERENCES [1] J. P. Mena-Chalco, H. Carrer, Y. Zana, et al., “Identifica- tion of Protein Coding Regions Using the Modified Ga- bor-Wavelet Transform,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, Vol. 5, No. 2, 2008, pp. 198-206. http://dx.doi.org/10.1109/TCBB.2007.70259 [2] W. Y. Wang; X. B. Ma and R. Kang, “Fisher Discrimi- nant Analysis for fault classification,” 2012 IEEE Confe- rence on Prognostics and System Health Management (PHM), 2012, pp. 23-25. [3] H. Huang, J. W. Li and J. M. Liu, “Gene Expression Data Classification Based on Improved Semi-Supervised Local Fisher Discriminant Analysis,” Expert Systems with Ap- plications, Vol. 39, No. 3, 2012, pp. 2314-2320. [4] F. Yan, J. Kittler, K. Mikolajczyk and A. Tahir, “Non- Sparse Multiple Kernel Fisher Discriminant Analysis,” The Journal of Machine Learning Research, Vol. 13, 2012, pp. 607-642. [5] J. S. Hamid, C. M. T. Greenwood and J. Beyene, “Weighted Kernel Fisher Discriminant Analysis for Inte- grating Heterogeneous Data,” Computational Statistics & Data Analysis, Vol. 56, No. 6, 2012, pp. 2031-2040. [6] F. Gao and C.-T. Zhang, “Comparison of Various Algo- rithms for Recognizing Short Coding Sequences of Hu - man Genes,” Bioinformatics, Vol. 20, No. 5, 2004, pp. 673-681.http://dx.doi.org/10.1093/bioinformatics/btg467 [7] C.-T. Zhang and J. Wang , “Recognition of Protein Cod- ing Genes in the Yeast Genome at Better Than 95% Ac- curacy Based on the Z Curve,” Nucleic Acids Research, Vol. 28. No. 14, 2000, pp. 2804-2814. http://dx.doi.org/10.1093/nar/28.14.2804 [8] Y. Li and L. Jiao, “Target Recognition Based on Kernel Fisher Discriminant (In Chinese),” Journal of Xidian University, Vol . 30, No. 2, 2003, pp. 179-182. [9] C. Zhao, W. Chen and C. Guo. “Research and Analysis of Methods for Multiclass Support Vector Machines (In Chinese),” CAAI Transactions on Intelligent Systems, Vol. 2, No. 2, 2007, pp. 11-17. [10] J. Liu, F. Zhao and Y. Liu, “Learning Kernel Parameters for Kernel Fisher Discriminant Analysis,” Pat tern Recog- nition Letters, Vol. 34, No. 9, 1 2013, pp. 1026-1031. [11] Z. Ji, P. G. Ji ng, T. S. Yu, Y. T. Su and C. S. Liu, “Rank- ing Fisher Discriminant Analysis,” Neurocomputing, 2013. http://www.sciencedirect.com/sci ence/article/pii/S092523 1213002877) [12] M. Burset and R. Guigo, “Evaluation of Gene Structure Prediction Programs,” Genomics, Vol. 34, 1996, pp. 353- 367. http://dx.doi.org/10.1006/geno.1996.0298
|