 International Journal of Analytical Mass Spectrometry and Chromatography, 2013, 1, 61-71 http://dx.doi.org/10.4236/ijamsc.2013.11008 Published Online September 2013 (http://www.scirp.org/journal/ijamsc) Separation Behavior of U(VI) and Th(IV) on a Mixed Ion Exchange Column Using 2,6-Pyridine Dicarboxylic Acid as a Complexing Agent and Determination of Trace Level Thorium in Uranium Matrix Employing High Performance Ion Chromatography Vaibhavi V. Raut1, S. P. Roy2, M. K. Das1, S. Jeyakumar1*, K. L. Ramakumar3 1Radioanalytical Chemistry Division, Bhabha Atomic Research Centre, Mumbai, India 2Product Development Division, Bhabha Atomic Research Centre, Mumbai, India 3Radiochemistry and Isotope Group, Bhabha Atomic Research Centre, Mumbai, India Email: vvraut@barc.gov.in, roy.satyendra@yahoo.com, mkdas@barc.gov.in, *sjkumar@barc.gov.in, klram@barc.gov.in Received July 24, 2013; revised August 25, 2013; accepted September 25, 2013 Academic Editor: Prof. N. Sivaraman, HBNI, India and Head, SCSS, Chemistry Group, Indira Gandhi Centre for Atomic Research, Kalpakkam-603102, INDIA Copyright © 2013 Vaibhavi V. Raut et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Retention behavior of U(VI) and Th(IV) as their 2,6-pyridine dicarboxylic acid (PDCA) complexes on reversed phase and ion exchange (cation, anion and mixed ion exchange) columns was studied and based on the results, a simple ion chromatography method for the determination of trace level thorium in uranium oxide using 0.075 mM 2,6-pyridine dicarboxylic acid (PDCA) and 1 M KNO3 in 1.2 M HNO3 as eluent (flow rate 1 mL/min) was proposed. The advantage of the developed method is that the separation of uranium matrix is not required prior to the ion chromatographic de- termination of trace Th. Separation was carried out on a mixed ion exchange stationary phase and a 10−4 M arsenazo (III) solution was used as post column reagent for detecting the separated metal ions. The separation of Th from uranium using PDCA in the present investigation is attributed through cation exchange mechanism. A calibration plot was con- structed by following the standard addition method over the concentration range of 0.25 to 10 ppm of Th in the presence of uranium matrix, which resulted in a linear regression coefficient of 0.9978. The precision of the method was better than 5% and the LOD for Th was found to be 0.1 ppm (S/N = 3). The method has been validated by comparing the re- sults with the results obtained from ICP-MS analysis where the Th is separated from the uranium matrix. The pro- posed method is simple, rapid, accurate and cost effective compared to techniques like ICP-MS or ICP-AES and is suit- able for the routine kind of analysis. Keywords: Ion Chromatography; Uranium; Thorium; 2,6-Pyridine Dicarboxylic Acid 1. Introduction The development of fast breeder reactors in India is in- evitable as India has limited resources of uranium and the vast thorium resources require breeder cycle for exploita- tion [1,2]. The Proto Type Fast Breeder Reactor (PFBR), a first commercial fast breeder reactor of India, is under construction and it will use a mixed uranium-plutonium oxide (MOX) as fuel. Thorium is a trace impurity associ- ated with all uranium based fuels [3]. Thorium, on irra- diation with a neutron produces 233U, is a useful fissile isotope. Small amounts of 232U isotope are also produced along with 233U. 232U is an undesirable isotope as its ra- dioactive decay chain produces short-lived and gamma emitting radionuclides 212Bi and 208Tl, leading to high radiation exposure [4]. Hence, the uranium based fuels that are used in the fast reactors has stringent specifica- tion for Th and in India, the limit is fixed as 200 ppmw [5]. Hence, determination of trace Th in uranium is in- dispensible in the chemical quality control of fast reactor fuels. *Corresponding author. C opyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 62 Although different analytical techniques are available for the determination of Th at trace levels [6-8], they require quantitative separation of Th from uranium ma- trix which is a prerequisite for analytical determination. An ICP-MS method [9] was earlier reported from our laboratory for the determination of Th in uranium matrix, which employed a two-stage separation sequence in- volving an initial solvent extraction for the removal of uranium matrix followed by an ion exchange purification to get the required quality of sample solutions suitable for ICP-MS analysis. The conventional separations fol- lowed by instrumental analysis employing ICP-MS or AES are time consuming and expensive. To adopt on routine basis, a reliable, cost effective and rapid analyti- cal method is desirable. Ion chromatography (IC) and High performance liquid chromatography (HPLC) are promising techniques for the separation of polyvalent metal cations by adding suitable complexing agents in the mobile phase [10-13]. Many chromatographic studies have been reported on the selective separation of Th and U in variety of samples using reversed phase surfaces with weak organic acids as chelating agents [14,15]. In these separations, the extent of separation depended on the nature of the metal com- plexes formed and their conditional stability constants [16]. Interestingly, it is reported that the elution order of U and Th on a reversed phase surface could be altered by selecting a suitable complexing agent. For example, an elution order of Th followed by U was observed in a RP surface when α-hydroxy isobutyric acid (α-HIBA) was used as a complexing agent [17,18]. However, this elu- tion order got reversed when mandelic acid (MA) was used in place of α-HIBA [19]. The change in elution or- der may be explained on the basis of differences in the complexing abilities of the ligands with respect to indi- vidual metal cation, effective charge, thermodynamic and kinetic stabilities and hydrophobicities of the metal com- plexes formed. Although α-HIBA and MA are widely used as com- plexing agents in the separation of U and Th, 2,6-pyri- dine dicarboxylic acid (PDCA), a non-hydroxy dicar- boxylic acid, has been identified as a promising ligand for the separation of heavy metal ions [20-23]. The re- versed phase surfaces dynamically modified with PDCA [22,24] showed stronger retention for U(VI) than Th(IV) due to the high affinity exhibited by the UO2 2+ ion for PDCA [20]. A study reports [22] that dynamic modifica- tion of a small PRP column with 0.1 mM PDCA in 1 M KNO3 and 0.5 M HNO3 could separate some of the acid hydrolysable heavy metal ions like U, Th, Zr, etc. It also states that the lanthanides and other transition metal ca- tions have no retention on the column and get excluded at the solvent front. Other than RP columns, ion exchange columns were also used for the separation of U and Th. The ion chro- matographic separation of U and Th using pure ion ex- change separation is difficult due to the large differences in their distribution ratios. This is because the UO2 2+ and Th4+ ions have considerable differences in their ion ex- change affinity. Separation of U(VI) as UO2 2+ on a low capacity strong cation exchanger has been reported [25]. Even though few studies reported the separation of U and Th from water and geological samples using a pure ca- tion exchange column with gradient elution of a mobile phase consisted of HCl and Na2SO4 [26,27], use of hy- drochloric acid as eluent was not recommended for rou- tine analysis due to its corrosiveness, which demanded utmost care on the instrument maintenance. Earlier in our laboratory, we have separated uranium and thorium using a short length cation exchange column using a mobile phase consisting of 0.08 mM PDCA in 0.24 M KNO3 and 0.22 M HNO3 (pH ~ 0.6) [23]. This method was found suitable for the rapid separation of both U and Th at trace level concentrations; however, it would not be possible to extend the procedure when one of the metal ions was at higher concentrations. The studies that reported the basis of RP-surface modi- fications and ion exchange separations were capable of providing good separation between U and Th, provided their concentrations were comparable. However, the sepa- ration of U and Th is a difficult task when one of these metal ions presents in large amount. When determining one metal ion at trace level in the presence of large ex- cess of other, the system loses its capacity, resulting in poor resolution between the analytes [21]. The most practicable way is to remove matrix element by means of conventional methods like solvent extraction, column chromatography and ion exchange prior to the IC or HPLC. Such sample preparation procedures are not only time consuming and laborious but also susceptible to cause inconsistency in the recoveries of the analyte of interest. Moreover, methods that follow pre-separation followed by instrumental analysis demand the use of certified reference materials to assess the recoveries. Since the objective of this study is to separate trace thorium from the dissolved uranium oxide samples by direct sample injection, one of the desired conditions to realize the objective is to have an elution order of Th(IV) followed by U(VI) so as to get the elution of Th first and then the matrix U. This elution order can be obtained by separating them on a reversed phase column (modified and unmodified) using α-HIBA as eluent [11]. But, the separation of Th in presence of bulk U could not be real- ized by following the reported procedures. A good sepa- ration of lanthanides, Th and U at relatively large con- centrations was reported [17] where the C-18 RP column was coated with bis-2-ethylhexyl succinamic acid (BE- HSA). However, the same study showed that it could not separate Th from U when the uranium concentration in the sample was more than 5 mg/mL. Mandelic acid, an- Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 63 other complexing agent frequently used in the separation of U and Th, could not be used as it brings reversed order of elution i.e. U followed by Th and both uranium and thorium mandelate complexes interact hydrophobically with the reversed phase surface and exhibit significant retentions. A similar elution order was reported in the case of cation exchange chromatography as uranium forms weaker cationic species with the frequently used eluents like hydrochloric and nitric acids [28]. This elu- tion order may be useful in the present case only when U(VI) has no or least interaction with stationary phase so that it can get eluted at the solvent front. The realization of this condition obliviously depends on the selection of an appropriate stationary phase and a suitable complex- ing agent. Ion exchange separation of metal ions can be affected by adding weak organic acids in the mobile pha- se, which reduces or alters the effective charge of the me- tal ions by forming either cation, neutral or anion com- plexes and the nature and stability of the metal com- plexes would decide the method of ion exchange (anion or cation) separation. Nowadays, stationary phases hav- ing both cation and anion exchange capacities are found to be more useful in separating heavy and transition me- tal ions [29] because the separation is controlled by the concentrations of free metal ions and the various insitu metal complexes formed, which are in an equilibrium with each other [30]. One such mixed ion exchanger namely Ion Pac CS5A has been widely used for the sepa- ration of lanthanides and transition metal cations in sev- eral matrices [31-33]. Hence it is decided to examine Ion Pac CS5A mixed ion exchanger column in the present investigation for the separation of traces of Th from bulk of U as it 1) contains both strong anion and cation exchangers (anion exchange capacity: 40 µeq/column and cation exchange capacity: 20 µeq/column), and 2) displays anion and cation ex- change properties while separating the metal cations by choosing an appropriate complexing agent [34]. In the present investigation, PDCA has been chosen as com- plexing agent as it has high affinity for complex forma- tion with polyvalent metal cations. The main advantages in using PDCA as a complexing agent are: 1) it can be used with low pH or high acid concentration and 2) the concentration of PDCA required in the mobile phase to facilitate the separation is very low (~10−5 M) compared to the concentrations of the hydroxy carboxylic acids (~10−1 M) required. Since the nuclear fuel samples (mg amounts) are often dissolved in mineral acids and subse- quently aliquots are made out of this solution for wet analyses, it is desirable to use PDCA as complexing agent, which would enable the direct injection of the samples into the system. The aim of this investigation was to develop a rapid and reliable method for the separation and determination of trace level thorium in uranium oxide using a mixed ion exchanger column and PDCA as a complexing agent. 2. Experimental 2.1. Instrumentation The IC system used in the present study was DX500 (Dionex CA, USA). It consisted of a solvent delivery pump (GP-50) and an absorption detector (AD20). A pneumatic post column reagent addition facility consist- ing of a 1000 mL reservoir bottle, gas pressure gauge to control the flow rate of the gas, a T-junction and a 75 µL capacity reaction coil was placed between the separator column and the detector. The samples were injected through a 50 µL loop fitted to a Rheodyne six port inject- tor. The separator columns used were 1) Ionpac CS5A (4.6 × 250 mm) with CG5A guard column (4.6 × 50 mm) (Dionex CA, USA), 2) Ionpac AS11 HC (4.6 × 250 mm) with AG11 guard column (4.6 × 50 mm) (Dionex CA, USA) and 3) IC Pak Cation, 4.6 × 50 mm (Waters, USA). The pH measurements were made on a CL-5 pH meter (Toshniwal, India). 2.2. Reagents All the reagents were of analytical grade (AR). Standard stock solution of uranium was prepared by dissolving nuclear grade U3O8 and its concentration was determined by biamperometry method [35]. Standard stock solution of Th (97.8 mg ml−1) was prepared by dissolving nuclear grade Th(NO3)4 (IRE, India) in 3 M HNO3 and the Th concentration was determined by complexometric titra- tion with EDTA using xylenol orange as indicator. 2,6- pyridine dicarboxylicacid (PDCA) (99%, Aldrich Chemi- cal corporation Inc.) was used as such. 0.01 M PDCA solution was prepared in water and appropriate volume of this solution was used in the preparation of mobile phase. Nitric acid was of suprapure grade (Merck, India). All solutions were prepared using high purity deionised water (18.2 MΩ) obtained from a Milli Q water system (Millipore, USA). The eluents were filtered through ny- lon 6,6, membrane filters (0.45 µm) prior to their use. A post column reagent of 4.5 × 10−5M Arsenazo-III in 0.1N HNO3 was added through a PEEK make T-junction with a flow rate of 0.8 ml min−1 for the detection of the sepa- rated metal ions at 655nm. 3. Results and Discussion 3.1. Preliminary Investigation Earlier we reported the separation of U and Th as their PDCA complexes using cation exchange column with a mobile phase of 0.08 mM PDCA in 0.24 M KNO3 and Copyright © 2013 SciRes. IJAMSC 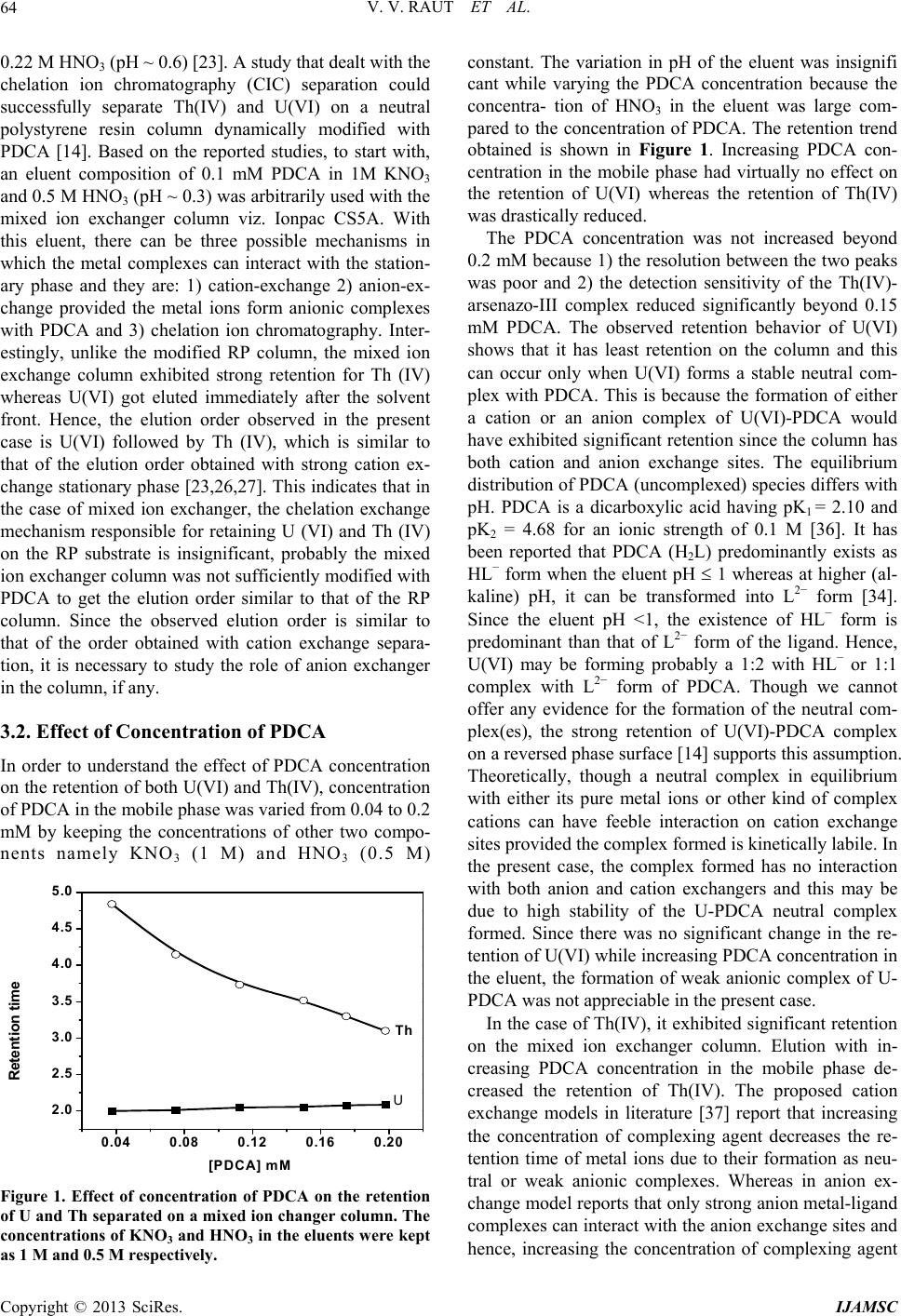 V. V. RAUT ET AL. 64 0.22 M HNO3 (pH ~ 0.6) [23]. A study that dealt with the chelation ion chromatography (CIC) separation could successfully separate Th(IV) and U(VI) on a neutral polystyrene resin column dynamically modified with PDCA [14]. Based on the reported studies, to start with, an eluent composition of 0.1 mM PDCA in 1M KNO3 and 0.5 M HNO3 (pH ~ 0.3) was arbitrarily used with the mixed ion exchanger column viz. Ionpac CS5A. With this eluent, there can be three possible mechanisms in which the metal complexes can interact with the station- ary phase and they are: 1) cation-exchange 2) anion-ex- change provided the metal ions form anionic complexes with PDCA and 3) chelation ion chromatography. Inter- estingly, unlike the modified RP column, the mixed ion exchange column exhibited strong retention for Th (IV) whereas U(VI) got eluted immediately after the solvent front. Hence, the elution order observed in the present case is U(VI) followed by Th (IV), which is similar to that of the elution order obtained with strong cation ex- change stationary phase [23,26,27]. This indicates that in the case of mixed ion exchanger, the chelation exchange mechanism responsible for retaining U (VI) and Th (IV) on the RP substrate is insignificant, probably the mixed ion exchanger column was not sufficiently modified with PDCA to get the elution order similar to that of the RP column. Since the observed elution order is similar to that of the order obtained with cation exchange separa- tion, it is necessary to study the role of anion exchanger in the column, if any. 3.2. Effect of Concentration of PDCA In order to understand the effect of PDCA concentration on the retention of both U(VI) and Th(IV), concentration of PDCA in the mobile phase was varied from 0.04 to 0.2 mM by keeping the concentrations of other two compo- nents namely KNO3 (1 M) and HNO 3 (0.5 M) 0.04 0.08 0.12 0.16 0.20 2.0 2.5 3.0 3.5 4.0 4.5 5.0 U Th Retention tim e [P D C A] m M Figure 1. Effect of concentration of PDCA on the retention of U and Th separated on a mixed ion changer column. The concentrations of KNO3 and HNO3 in the eluents were kept as 1 M and 0.5 M respectively. constant. The variation in pH of the eluent was insignifi cant while varying the PDCA concentration because the concentra- tion of HNO3 in the eluent was large com- pared to the concentration of PDCA. The retention trend obtained is shown in Figure 1. Increasing PDCA con- centration in the mobile phase had virtually no effect on the retention of U(VI) whereas the retention of Th(IV) was drastically reduced. The PDCA concentration was not increased beyond 0.2 mM because 1) the resolution between the two peaks was poor and 2) the detection sensitivity of the Th(IV)- arsenazo-III complex reduced significantly beyond 0.15 mM PDCA. The observed retention behavior of U(VI) shows that it has least retention on the column and this can occur only when U(VI) forms a stable neutral com- plex with PDCA. This is because the formation of either a cation or an anion complex of U(VI)-PDCA would have exhibited significant retention since the column has both cation and anion exchange sites. The equilibrium distribution of PDCA (uncomplexed) species differs with pH. PDCA is a dicarboxylic acid having pK1 = 2.10 and pK2 = 4.68 for an ionic strength of 0.1 M [36]. It has been reported that PDCA (H2L) predominantly exists as HL− form when the eluent pH 1 whereas at higher (al- kaline) pH, it can be transformed into L2− form [34]. Since the eluent pH <1, the existence of HL− form is predominant than that of L2− form of the ligand. Hence, U(VI) may be forming probably a 1:2 with HL− or 1:1 complex with L2− form of PDCA. Though we cannot offer any evidence for the formation of the neutral com- plex(es), the strong retention of U(VI)-PDCA complex on a reversed phase surface [14] supports this assumption. Theoretically, though a neutral complex in equilibrium with either its pure metal ions or other kind of complex cations can have feeble interaction on cation exchange sites provided the complex formed is kinetically labile. In the present case, the complex formed has no interaction with both anion and cation exchangers and this may be due to high stability of the U-PDCA neutral complex formed. Since there was no significant change in the re- tention of U(VI) while increasing PDCA concentration in the eluent, the formation of weak anionic complex of U- PDCA was not appreciable in the present case. In the case of Th(IV), it exhibited significant retention on the mixed ion exchanger column. Elution with in- creasing PDCA concentration in the mobile phase de- creased the retention of Th(IV). The proposed cation exchange models in literature [37] report that increasing the concentration of complexing agent decreases the re- tention time of metal ions due to their formation as neu- tral or weak anionic complexes. Whereas in anion ex- change model reports that only strong anion metal-ligand complexes can interact with the anion exchange sites and hence, increasing the concentration of complexing agent Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 65 in the eluent would increase in the retention time. In or- der to conclude about the probable nature of the metal complexes and their interaction with ion exchange sites, it is necessary to study the retention of U or Th with the components of eluent individually. Initially, separations were carried out by varying the concentration of PDCA between 0.02 mM to 0.4 mM in 0.2 M HNO3 (necessary to avoid hydrolysis of U and Th) without taking KNO3 in the mobile phase. It was ob- served that with 0.02 mM PDCA, U appeared at 4.0 minutes whereas the same appeared at 2.7 minutes when the PDCA concentration was increased to 0.1 mM and subsequent increase of PDCA up to 0.4 mM did not change the retention time significantly. In contrast, Th(IV) peak did not appear at all. A similar observation was made when pure HNO3 (0.5 to 2 M) or pure KNO3 (0.2 to 1.8 M) was used as eluent. Eluents with mixture of HNO3 and KNO3 of varying concentrations were also used and there was no difference in the retention of U(VI) compared to that of the pure HNO3/KNO3. However, interestingly it was observed that Th(IV) appeared when the pH of the mixed eluent was less than 1. Further, it was seen that the elution of thorium was so slow that it resulted in a broad peak. Therefore, it may be concluded that in the absence of PDCA in the eluent, the thorium elution with KNO3+H NO3 may be due to the formation of its nitrato complex and the formation of such nitrato complex is pH dependent. Therefore, it is seen from the experiments carried out with PDCA, KNO3 and HNO3 individually as well as with various combinations that for the separation of Th(IV), for the purpose of analytical determination, a mobile phase with the combination of all three compo- nents is required. The observations showed that at this low concentration of PDCA, the elution of Th(IV) oc- curred only when a KNO3 is added along with minimum 0.1 M HNO3 concentration. It has been observed that eluents with PDCA concentration above 0.08 mM were not found suitable for separating trace Th in the presence of large excess of uranium. Therefore, the PDCA con- centration has been fixed at 0.075 mM. This concentra- tion would reduce the interactions between UO2 2+-PDCA complex with the stationary phase, which is desirable to realize the elution of U peak immediately after or at the excluded peak. 3.3. Effect of Concentration of KNO3 The retention trends of U(VI) and Th(IV) shown in Fig- ure 1 suggest that U(VI) predominantly forms a neutral complex and gets eluted immediately after the excluded peak and Th(IV) forms weak neutral or anionic complex and most probably, the mechanism responsible for the retention of Th(IV) on the column is due to cation ex- change. However, addition of relatively high concentra- tion of KNO3 may suppress the ion exchange process due to the presence of large concentration of K+ ions (acts as competing cation). It was observed that the retention time of U(VI) was unaltered whereas the retention time of Th(IV) decreased when the concentration of KNO3 (0.2 to 1.8 M) was increased in the mobile phase and by keeping the PDCA and HNO3 concentrations as 0.075 mM and 0.2 M, respectively. The retention time of Th(IV) decreases drastically in the concentration range of 0.2 to 1.2 M and subsequently there was a little decrease up to 1.8 M. The drastic re- ducetion in retention time may be attributed due to two reasons: 1) increasing [KNO3] increases the ionic strength of the eluent. While using cation-exchange column with complexing eluent, the affinity of metal ions for ion- exchange resins decreases as ionic strength increases and therefore, the retention time decreases and 2) 3 NO from KNO3 may be competing with PDCA in complex formation. To identify the role of KNO3, separations were carried out with PDCA and HNO3 combinations where the nitrate was supplied in the form of HNO3. For instance, instead of 0.5 M HNO3 + 0.5 M KNO3 + 0.075 mM PDCA (pH 0.3) an eluent of 0.075 mM PDCA in 1 M HNO3 was prepared and pH was adjusted by adding LiOH. It was seen that with the latter mobile phase com- position, Th could not be eluted from the column. This suggests that K+ is acting as competing ion which re- duces the bare metal ions (U and Th) interactions with ion exchange sites. Equation for cation (complex ion) exchange is expre- ssed as loglogloglog log mMA ML DK anCnA where Dm is the distribution coefficient of the metal in presence of complexing agent, KMA is the selectivity co- efficient between the metal and competing cation, aM(L) is complex formation coefficient, C is the capacity of the ion exchanger and [A+] is the concentration of the com- peting cation. The distribution coefficient (Dm) of the metal is a func- tion of the concentration of the competing cation (A+) and that of the complexing agent in the mobile phase. Since in the present case the competing cations in the mobile phase are K+ and H+, the elution mainly depends on the concentration of K+ ions as it has high selectivity over H+. On increasing the KNO3 concentration in the eluent decreased the retention times of only Th(IV) whereas the retention times of U(VI) was unaffected throughout the concentration range (Figure 2(a)). Th(IV) retention times decreased steadily as the KNO3 concen- tration was raised up to 1.2 M and subsequent increment of rise in KNO3 concentration did not reduce the reten- tion times appreciably. As this trend is expected in the case of cation exchange of Th(IV), a plot of log k (reten- Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 66 0.0 0.4 0.81.2 1.6 2.0 0 2 4 6 8 10 12 14 16 18 Th U Reten tion time [KNO3] (a) 0.2 0.4 0.6 0.8 1.0 1.2 1 10 log k' [KNO3] (b) Figure 2. (a) Effect of concentration of KNO3 on the reten- tion of U and Th separated on a mixed ion exchanger col- umn (the concentrations of PDCA and HNO3 in the eluents were 0.075 mM and 0.5 M respectively) and (b) plot of log k (retention factor) of Th against [KNO3]. tion factor) against KNO3 concentration over the range of 0 - 1.2 M brought a straight line with negative slope (Figure 2(b)) indicating the separation is predominantly by cation exchange mechanism. The optimum KNO3 concentration was fixed as 1.0 M as this provided the desired separation between U(VI) and Th(IV). 3.4. Effect of Concentration of HNO3 Addition of HNO3 and its concentration plays an impor- tant role in controlling the effective ligand concentration in the mobile phase, which decides the nature of metal- complex. In addition, HNO3 prevents the hydrolysis of metal cations as these heavy metal ions belong to acid hydrolysable group. Moreover, the H+ ions from the ni- tric acid also act as the competing ion during the elution of metal cations or cation complexes. To investigate the influence of HNO3 concentration on the elution, two sets of experiments were conducted. In the first set, HNO3 concentration was varied over a range between 0.5 and 1.5 M. It was expected that increasing acid concentration would reduce the effective concentration of ligand, whi- ch would affect the complexation of U(VI) leaving more metal cations and in such case, the retention times for both Th(IV) and U(VI) are expected to be increased. However, there was no appreciable change in the reten- tion times observed. Interestingly, there was a steady im- provement in the symmetry of Th(IV) peak was observed. In the second set, the pH of the mobile phase was varied from 0.3 to 4.0. For preparing the eluents, dilute nitric acid of appropriate concentration was used and final pH adjustment was done with dilute LiOH solution. Increas- ing eluent pH caused the retention times of Th(IV) to decrease and of U(VI) to increase. In addition, the or- der of elution reversed from pH 2 onwards (Figure 3). There are two ways in which this can be explained. First, increase of eluent pH leads to increase in the effective ligand (HL−) concentration and therefore, U(VI) tends form anion complex, which interacts with anion ex- changer resulting increasing trend in the retention. On the other hand, it has not resulted in change in the complex forming behavior of Th(IV) significantly and hence, re- tention time decreased. There could also be a possibility of column modification due to the hydrophobic interac- tions between the phenyl group of PDCA and the poly- stryrene-divinyl benzene skeleton of the resin. On in- creasing pH of the eluent, the dissociation of acid groups on the immobilized chelating ligand produces an increase in the conditional stability constants of the surface metal complexes and it causes the retention times of the metal ions to increase [15]. Between Th(IV) and U(VI), it has been reported that the U(VI)-PDCA (1:1) complex has high stability constant (log K1 = 4.72) [38] and hence, it is more retained than Th(VI). The existence of this mechanism would have been confirmed only by carrying out separations at higher pH levels. However, such stud- ies are not affordable in this case as the hydrolysis of heavy metal ions cannot be prevented. From the analytical point of view, considering the de- sired order of elution, separation factor and other chro- matographic parameters, it is desirable to have higher concentration of HNO3 and accordingly the HNO3 con- centration was fixed as 1.2 M in the mobile phase. 3.5. Elution with a Pure Cation Exchange Column Preceding investigations suggest that the elution behavior of Th(IV) was mainly due to the cation exchange mecha- nism. On the other hand U(VI) was eluted as its neutral complex, which gets eluted immediately after the ex- cluded peak indicating that it has least interaction with both cation and anion exchangers. It has been seen that a Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 67 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 10-2 10-1 100 101 Th U log k' (retention factor) Eluent pH Figure 3. Effect of eluent pH on the retention of U and Th on a mixed ion exchanger column. The [PDCA] and [KNO3] were 0.075 mM and 1.0 M respectively. Figure 4. Typical chromatogram obtained for a standard solution of U(VI) and Th(IV) using cation exchanger col- umn. Peaks (1) U(VI) – 3 ppm and (2) Th(IV) 3 ppm. Col- umn: IC Pak Cation (4.6 × 50 mm). Eluent: 0.1 mM PDCA and 0.12 M KNO3 in 0.1 M HNO3; Flow rate: 1 ml/min. Detect io n: P ost col um n addi ti on of 4.5 × 1 0-5 M arsenazo-III and detection at 656 nm. mobile phase of 0.1 mM PDCA and 0.12 M KNO3 in 0.1 M HNO3 brought good separation between U(VI) and Th(IV) and Figure 4 shows a typical chromatogram ob- tained. The order of elution was as same as that of the one obtained with the mixed ion exchanger column. Ef- fects of concentrations of PDCA and KNO3 on the reten- tion of Th(IV) showed similar trend in both mixed ion exchange and cation exchange columns. This suggests that the separation of Th(IV) is mainly due to cation ex- change. In order to understand the separation with mixed ion exchange column and the role of anion exchange, it is necessary to carry out separations with pure anion ex- change column too. 3.6. Separation with Pure Anion Exchange Column When similar investigations were carried out with a pure anion exchange column (IonPac AS11 HC column), the elution order was reversed where Th peak appeared im- mediately after the excluded peak followed by U. while increasing the PDCA concentration (from 0.08 - 0.5 mM) in the mobile phase it was observed that there were no significant changes in the retention time of both U and Th. This indicates that up to 0.5 mM PDCA both U and Th are not forming stable anionic complexes. This ob- servation suggests that Th may be forming weak anionic and neutral complexes with PDCA. In the present case, the appreciable retention of U cannot be explained on the basis of formation of anionic complex because the reten- tion time did not increase while increasing the PDCA concentration. Probably the neutral U-PDCA complex may be interacting on the surface of the column hydro- phobically as it showed in the case of reverse phase sur- face. Therefore, it may be concluded that the cation ex- change sites of the mixed ion exchange column play predominant role in the separation of U and Th. However, the anion exchange sites present in the mixed ion ex- change column may be reducing the hydrophobic inter- action of U-PDCA neutral complex, though we cannot offer any evidence. 3.7. Effect of Other Metal Ions Since the uranium oxide fuel samples are expected to have other metallic impurities like Fe, Cd, Cu, Ca, Mg, Ni, Cr, Mn, Mo, rare earth elements etc. (within the spe- cification limits), separations were carried out in the pre- sence of these metal ions to observe the interference ef- fects, if any, during the separation of Th. For this purpose, two synthetic sample solutions were prepared and used. The first solution was a mixture of thorium with transi- tion metals (Ca and Mg were also added) and the second one consisted thorium and mixture of all lanthanides. The individual metal ion to Th concentration ratio was kept ten in both the solutions. It has been observed that the first solution did not give any peak whereas the second solution containing mixture of lanthanides showed a small triplet peak after the uranium peak with very poor detection sensitivity. Therefore, the formulated eluent composition was found to be suitable for the interference free determination of Th. A typical chromatogram ob- tained for a standard solution containing U and Th is shown Figure 5. 3.8. Effect of Matrix ion on the Separation of Th Under the optimized separation conditions, the uranyl ion eluted very close to the excluded peak and the retention capacity factor (k) for uranium was 0.2. Hence, sepa- Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 68 Figure 5. Chromatogram obtained for a standard mixture solution containing U and Th. Peaks (1) U(VI) 1 ppm and (2) Th(IV) 2.5 ppm. Column: IonPac CS5A (4 × 250 mm). Elu- ent: 0.075 mM PDCA and 1M KNO3 in 1.2 M HNO3. Flow rate: 1 mL/min. Detection: Post column addition with 4.5 × 10-5 M Arsenazo-III in 0.1 N HNO3 at 656 nm; flow rate: 0.8 mL/min. ration of trace Th from bulk uranium may be feasible because most of the uranium may get excluded along with solvent front, which can reduce the possibility of column swamping. Several synthetic samples in dupli- cates having U/Th ratios 1, 2, 20, 200,400 and 1.8 × 104 were prepared and injected for Th separation. The obser- ved recoveries for Th in each case have been listed in Table 1 and the recoveries of Th were found to be more than 90%, which showed that the method has high tolerance for uranium and hence, the direct separation of Th from the dissolved uranium samples is feasible. 3.9. Calibration and Method Validation Since the success of an analytical method is critically dependent upon how accurately the concentrations of the analytes are known and how closely the matrix of the standards match with that of the samples to be analyzed, construction of a calibration plot in presence of matrix is desirable as the instrumental response is read in an al- most identical chemical environment for both samples and calibration standards. This will compensate the ma- trix effect on the measurements. Therefore, high purity UO2 was dissolved in nitric acid and the solution was made up to a known volume. Several aliquots were pre- pared and varying volumes of a standard Th solution were spiked except for one aliquot. The spiked aliquots were made up to a known volume keeping a final con- centration of U in all aliquots at 87 mg/mL. A calibration plot was constructed between the peak areas and their respective Th concentrations in the presence of matrix uranium (87 mg of U/mL of the standard solution), which showed a linear relation with a regression coefficient of 0.9975 and the plot obtained is shown in Figure 6. The Th content of the uranium blank solution was calculated from the intercept of the plot. The limit of detection (LOD) was calculated as 0.1 ppm of Th using S/N = 3 formula. The method was validated by analyzing a U3O8 reference material (ILCE-IV) employing both the present method and ICP-MS (a well established proce- dure [9,39] for the separation of trace Th from the ura- nium matrix prior to the ICP-MS analysis was followed). Accurately weighed quantities (~0.4 g) of this U3O8 ref- erence material were dissolved in Conc.HNO3 and heated to dryness. Further they were re-dissolved in 5 mL of 0.5 M HNO3 and injected into the IC for separation. Another set of dissolved samples were subjected to solvent ex- traction for the removal of matrix U using aliquat-336 [40] and the final separated fractions were taken in 0.5 M HNO3. The values obtained are listed in Table 2 and they are in good agreement with the mean value reported by the ICP-MS method. The chromatograms obtained for a direct injection is shown Figure 7. 3.10. Procedure and Sample Analysis A sample size of 0.4 - 0.5 g was dissolved in Conc.HNO3 Table 1. Effect of concentration of U (matrix) on the sepa- ration of Th. Sample Code [UO22+] µg/mL [Th4+] µg/mL U/Th ratio Observed [Th] µg/mL Recovery Th% S-1 5 5 1 4.9 98.0 S-2 10 5 2 4.78 95.6 S-3 100 5 20 4.81 96.2 S-4 1000 5 200 4.85 97.0 S-5 2000 5 400 4.65 93.0 S-6 88,000 5 1.8 E4 4.73 94.6 0246810 0 200 400 600 800 1000 1200 1400 R^2 = 0.99751 B = 118.87797 Peak A rea Conc. of Th in ppm Figure 6. Calibration plot obtained for Th over a concen- tration range of 0.25 - 10 ppm in the presence of 87 mg of U/ mL of the standard solutions. Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 69 Table 2. Results obtained for a U3O8-refer ence material. Sample Analysis Sample Code [Th4+] by IC (ppmw)b Mean Mean reported by ICP-MS D-1 75.5 D-2 72.7 D-3 74.6 D-4 71.8 Direct Analysisa D-5 75.2 74.0 ± 1.6 72.5 ± 2.2 S-1 72.3 S-2 69.8 71.4 ± 1.3 S-3 72.7 After Solvent Extractionc S-4 70.9 a0.5 g of sample dissolved and made up to ~20 g by 0.1N HNO3 and 50 µL of this solution was injected into IC. beach value quoted is a mean ob- tained from three replicate injections. c0.5 g of sample was dissolved in HCl and U was separated by aliquat-336 [40]in 6 M HCl. Figure 7. Typical chromatograms obtained for a U3O8-Re- ference material sample (A) direct injection of dissolved uranium solution (B) sample injected after removal of ura- nium (matrix) by solvent extraction. and evaporated to dryness. Further, it was re-dissolved in 0.5 M HNO3 to have a final volume of 5 mL and 50 µL of this solution was injected into IC for separation. A mobile phase of 0.075 mM PDCA and 1M KNO3 in 1.2M HNO3 was used at a flow rate of 1 mL/min forseparation. The separated fraction of metals are de- tected and measured by post column addition of 4.5 × 10−4 M arsenazo-III at a flow rate of 0.8 mL/min. The results obtained for typical samples are listed in Table 3. 4. Conclusion The proposed method was successfully used for the de- termination of trace levels of Th in uranium matrix with- out employing prior separation of matrix. The method is simple, rapid, accurate and promising for its adaptation in the routine quality control analysis of uranium based Table 3. Results obtained for uranium samples. Sample Conc. of Th (ppmw)a UO2 – SG 18.3 ± 1.3 U3O8 – NFC-1 15.7 ± 1.4 U3O8 – NFC-2 23.6 ± 1.7 amean of three analyses. fuels. 5. Acknowledgements The authors thank Dr. B. S. Tomar, Head, Radioanalyti- cal Chemistry Division, BARC for his support. They also thank Mr. V. G. Mishra, RACD for his help. REFERENCES [1] H. J. Bhabha and N. B. Prasad, “A Study of the Contribu- tion of Atomic Energy to a Power Programme in India,” Proceedings of the Second United Nations International Conference on the Peaceful Uses of Atomic Energy, Ge- neva, Vol. 1, September 1958, pp. 89-101. [2] M. Dayal, S. R. Paranjape, B. Singh and N. B. Prasad, “Study of Fuel Cycles with Reference to a Power Pro- gramme,” Proceedings of the Second United Nations In- ternational Conference on the Peaceful Uses of Atomic Energy, Geneva, Vol. 13, September 1958, pp. 184-194. [3] T. K. Mukherjee, “Characterization and Quality Control of Nuclear Fuels (CQCNF-2002),” Allied Publishers, New Delhi, 2003. [4] IAEA-TECDOC-1450, “Thorium Fuel Cycle-Potential Benefits and Challenges”, IAEA, Austria, 2005, pp. 68- 69. [5] V. Venugopal, “Chemical Quality Control of Nuclear Materials,” Indian Association for Nuclear Scientists and Allied Chemists (IANCAS) Bulletin, Vol. 4, Mumbai, 2005. [6] S. Joannon, P. Telouk, C. Pin, “Determination of U and Th at Ultra-Trace Levels by Isotope Dilution Inductively Coupled Plasma Mass Spectrometry Using a Geyser- Type Ultrasonic Nebulizer: Application to Geological Samples,” Spectrochimica Acta B, Vol. 52, No. 12, 1997. pp. 1783-1789. http://dx.doi.org/10.1016/S0584-8547(97)00072-4 [7] A. G. Adriaens, J. D. Fassett, W. R. Kelly, D. S. Simons and F. C. Adams, “Determination of Uranium and Tho- rium Concentrations in Soils: Comparison of Isotope Di- lution-Secondary Ion Mass Spectrometry and Isotope Di- lution-Thermal Ionization Mass Spectrometry,” Analy- tical Chemistry, Vol. 64, No. 23, 1992, pp. 2945-2950. http://dx.doi.org/10.1021/ac00047a012 [8] E. H. Borai and A. S. Mady, “Separation and Quantifica- tion of 238U, 232Th and Rare Earths in Monazite Samples by Ion Chromatography Coupled with On-Line Flow Scin- tillation Detector,” Applied Radiation and Isotopes, Vol. 57, No. 4, 2002. pp. 463-469. http://dx.doi.org/10.1016/S0969-8043(02)00089-1 [9] S. B. Deb, M. K. Saxena, B. K. Nagar and K. L. Rama- Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. 70 kumar, “Determination of Trace Amounts of Thorium in a Uranium Matrix by Inductively Coupled Plasma Mass Spectrometry and Validation of the Separation Procedure by Standard Addition and Tracer Techniques,” Atomic Spectroscopy, Vol. 29, No. 2, 2008, pp. 39-44. [10] F. Hao, B. Paull and P. R. Haddad, “Determination of Thorium and Uranyl in Nitrophosphate Solution by On- Line Matrix-Elimination Reversed-Phase Chromatogra- phy,” Chromatographia, Vol. 42, No. 11-12, 1996, pp. 690-696. http://dx.doi.org/10.1007/BF02267703 [11] F. Hao, B. Paull and P. R. Haddad, “Determination of Trace Levels of Thorium(IV) and Uranyl by Reversed- Phase Chromatography with On-Line Preconcentration and Ligand Exchange,” Journal of Chromatography A, Vol. 749, No. 1-2,1996, pp. 103-113. http://dx.doi.org/10.1016/0021-9673(96)00450-5 [12] R. M. Cassidy, “Determination of Rare-Earth Elements in Rocks by Liquid Chromatography,” Chemical Geology, Vol. 67, No. 3-4, 1998. pp. 185-195. http://dx.doi.org/10.1016/0009-2541(88)90127-1 [13] P. E. Jackson, J. Carnevale, H. Fuping and P. R. Haddad, “Determination of Thorium and Uranium in Mineral Sands by Ion Chromatography,” Journal of Chromatography A, Vol. 671, No. 1-2, 1996, pp. 181-191. http://dx.doi.org/10.1016/0021-9673(94)80237-8 [14] R. M. C Sutton, S. J. Hill, P. Jones, A. Sanz-Medel and J. I. Garcia-Alonso, “Comparison of the Retention Behav- iour of Uranium and Thorium on High-Efficiency Resin Substrates Impregnated or Dynamically Coated with Metal Chelating Compounds,” Journal of Chromatogra- phy A, Vol. 816, No. 2, 1998, pp. 286-291. http://dx.doi.org/10.1016/S0021-9673(98)00496-8 [15] R. M. Cassidy and M. Fraser, “Equilibria Effects in the Dynamic Ion-Exchange Separation of Metal Ions,” Chro- matographia, Vol. 18, No. 7, 1984, pp. 369-373. http://dx.doi.org/10.1007/BF02262483 [16] P. Jones and P. N. Nesterenko, “High-Performance Che- lation Ion Chromatography a New Dimension in the Se- paration and Determination of Trace Metals,” Journal of Chromatography A, Vol. 789, No. 1-2, 1997, pp. 413-435. http://dx.doi.org/10.1016/S0021-9673(97)00824-8 [17] Ch. S. K. Raju, M. S. Subramanian, N. Sivaraman, T. G. Srinivasan and P. R. Vasudeva Rao, “Retention Studies on Uranium, Thorium and Lanthanides with Amide Mo- dified Reverse Phase Support and Its Applications,” Jour- nal of Chromatography A, Vol. 1156, No. 1-2, 2007, pp. 340-347. http://dx.doi.org/10.1016/j.chroma.2007.01.010 [18] M. Akhila Maheswari, D. Prabhakaran, M. S. Subrama- nian, N. Sivaraman, T. G. Srinivasan and P. R. Vasudeva Rao, “High Performance Liquid Chromatographic Studies on Lanthanides, Uranium and Thorium on Amide Modi- fied Reversed Phase Supports,” Talanta, Vol. 72, No. 2, 2007, pp. 730-740. http://dx.doi.org/10.1016/j.talanta.2006.12.003 [19] F. Hao, B. Paull and P. R. Haddad, “Retention Behaviour of Thorium(IV) and Uranyl on a Reversed-Phase Column with Glycolate and Mandelate as Eluents,” Journal of Chromatography A, Vol. 739, No. 1-2, 1996, pp. 151-161. http://dx.doi.org/10.1016/0021-9673(96)81462-2 [20] M. J. Shaw, S. J. Hill and P. Jones, “Chelation Ion Chro- matography of Metal Ions Using High Performance Sub- strates Dynamically Modified with Heterocyclic Carbox- ylic Acids,” Analytica Chimica Acta, Vol. 401, No. 1-2, 1999, pp. 65-71. http://dx.doi.org/10.1016/S0003-2670(99)00495-X [21] P. Jones, “Major Sensitivity Improvements in Ion Chro- matography Determinations Involving Post-Column Spe- ctrophotometric Reaction Detectors through Elimination of Pump Noise Using a Dual Wavelength Monitoring Procedure,” Analyst, Vol. 125, No. 5, 2000, pp. 803-806. http://dx.doi.org/10.1039/b001826k [22] M. J. Shaw, S. J. Hill, P. Jones and P. N. Nesterenko, “Determination of Uranium in Environmental Matrices by Chelation Ion Chromatography Using a High Performance Substrate Dynamically Modified with 2,6-Pyridinedi- carboxylic Acid,” Chromatographia, Vol. 51, No. 11-12, 2000, pp. 695-700. http://dx.doi.org/10.1007/BF02505407 [23] S. Jeyakumar, V. G. Mishra, M. K. Das, V. V. Raut, R. M. Sawant and K. L. Ramakumar, “Separation Behavior of U(VI) and Th(IV) on a Cation Exchange Column Using 2,6-Pyridine Dicarboxylic Acid as a Complexing Agent and Its Application for the Rapid Separation and Deter- mination of U and Th by Ion Chromatography,” Journal of Separation Science, Vol. 34, No. 6, 2011, pp. 609-616. http://dx.doi.org/10.1002/jssc.201000907 [24] J. Cowan, M. J. Shaw, E. P. Achterberg, P. Jones and P. N. Nesterenko, “The Ion Chromatographic Separation of High Valence Metal Cations Using a Neutral Polystyrene Resin Dynamically Modified with Dipicolinic Acid,” Ana- lyst, Vol.125, No. 12, 2000, pp. 2157-2159. http://dx.doi.org/10.1039/b008235j [25] “Determiantion of Uranium and Thorium in Complex Matrices Using Chelation Ion Chromatography,” Dionex Application Note, No. 79, 1998, LPN 034670-01. [26] A. W. Al-Shawi and R. Dahl, “Determination of Thorium and Uranium in Nitrophosphate Fertilizer Solution by Ion Chromatography,” Journal of Chromatography A, Vol. 706, No. 1-2, 1995. pp. 175-181. http://dx.doi.org/10.1016/0021-9673(95)00006-9 [27] M. P. Harrold, A. Siriraks and J. Riviello, “High-Perform- ance Ion Chromatographic Separation of Uranium and Thorium in Natural Waters and Geological Materials,” Journal of Chromatography A, Vol. 602, No. 1-2, 1992, pp. 119-125. http://dx.doi.org/10.1016/0021-9673(92)80071-2 [28] F. Nelson, T. Murase and K. A. Kraus, “Ion Exchange Procedures. I. Cation Exchange in Concentration HCl and HClO4 Solutions,” Journal of Chromatography A, Vol. 13, 1964, pp. 503-535. http://dx.doi.org/10.1016/S0021-9673(01)95146-5 [29] H. Hojabri, A. G. Lavin, G. G. Wallace and J. M. Riviello, “Determination of Metal Ions Using Ion Chromatography and Indirect Amperometric Detection,” Analytical Che- mistry, Vol. 59, No. 1, 1987, pp. 54-57. http://dx.doi.org/10.1021/ac00128a011 [30] Y. S. Park, S. H. Han, K. S. Joe, T. Y. Eom and G. Lee, “Retention Behaviour of Transition Metal Ions with Some Copyright © 2013 SciRes. IJAMSC  V. V. RAUT ET AL. Copyright © 2013 SciRes. IJAMSC 71 Complexing Agents on Cation Exchanger,” Bulletin of the Korean Chemical Society, Vol. 14, No. 6, 1993. pp. 692-696. [31] N. Cardellicchio, P. Ragone, S. Cavalli and J. Riviello, “Use of Ion Chromatography for the Determination of Transition Metals in the Control of Sewage-Treatment- Plant and Related Waters,” Journal of Chromatography A, Vol. 770, No. 1-2, 1997, pp. 185-193. http://dx.doi.org/10.1016/S0021-9673(97)00086-1 [32] E. H. Borai, M. A. Eid and H. F. Aly, “Determination of REEs Distribution in Monazite and Xenotime Minerals by Ion Chromatography and ICP-AES,” Analytical and Bioanalytical Chemistry, Vol. 372, No. 4, 2002, pp. 537- 541. http://dx.doi.org/10.1007/s00216-001-1204-8 [33] L. Perna, F. Bocci, L. Aldave de las Heras, J. De Pablob and M. Betti, “Studies on Simultaneous Separation and Determination of Lanthanides and Actinides by Ion Chro- matography Inductively Coupled Plasma Mass Spectro- metry Combined with Isotope Dilution Mass Spectrome- try,” Journal of Analytical Atomic Spectrometry, Vol. 17, 9, 2002, pp. 1166-1171. http://dx.doi.org/10.1039/b202451a [34] P. Janvion, S. Motellier and H. Pitsch, “Ion-Exchange Me- chanisms of Some Transition Metals on A Mixed-Bed Resin with a Complexing Eluent,” Journal of Chroma- tography A, Vol. 715, No. 1, 1995, pp. 105-115. http://dx.doi.org/10.1016/0021-9673(95)00576-9 [35] M. Xavier, P. R. Nair, K. V. Lohithakshan, S. G. Marathe and H. C. Jain, “Determination of Uranium in the Pres- ence of Iron and Plutonium by Ti(III) Reduction and Biamperometric Titration,” Journal of Radioanalytical and Nuclear Chemistry, Vol. 148, No. 2, 1991, pp. 251- 256. http://dx.doi.org/10.1007/BF02060358 [36] L. G. Sillen and A. E. Martell, “Stability Constants of Metal Ion Complexes, spl,” Chemical Society, London, 1964, p. 545. [37] P. R. Haddad and R. C. Foley, “Modelling of Cation Re- tention in Ion Chromatography Using Fixed-Site and Dy- namically Coated Ion-Exchange Columns,” Journal of Chromatography A, Vol. 500, 1990, pp. 301-312. http://dx.doi.org/10.1016/S0021-9673(00)96073-4 [38] A. E. Martell and R. M. Smith “Critical Stability Con- stants,” Vol. I, Amino Acids, Plenum, New York, 1974, p. 377. [39] “DAE-Interlaboratory Comparison Experiment (ILCE) for Trace Metal Assay of Uranium,” Phase 2, Document, BARC, India, 2002. [40] V. R. Bhandiwad, R. Swarup and S. K. Patil, “Extraction of Actinides by Quaternary Amines from Hydrochloric Acid Medium,” Journal of Radioanalytical Chemistry, Vol. 52, No. 1, 1979, pp. 5-14. http://dx.doi.org/10.1007/BF02517694
|