 Journal of Biomaterials and Nanobiotechnology, 2011, 2, 1-7 doi:10.4236 /jbnb.2011.21001 Published Online January 2011 (http://www.SciRP.org/journal/jbnb) Copyright © 2011 SciRes. JB NB Self-Assembly of Colloidosome Shells on Drug-Containing Hydrogels Rachel T Rosenberg, Nily Dan Department of Chemical and Biological Engineering, D r exel U niversity, Philadelphia, Pennsylvania 19104 Email: dan@coe.dr exel.edu Received August 30th, 2010; revised September 22nd, 2010; accepted Sept ember 28th, 2010. ABSTRACT Colloidosomes are composed of an aqueous or hydrogel corethat is coated by a semi-permeable colloidal shell. The properties of the shell can be varied to control the rate of release of encapsulated components such as drugs. Specifi- cally, the pores formed between the colloidal particles suppress transport of large components, while allowing diffusion of smaller on es. Self-assembly of colloidal particles on hydrogel films is a convenient method forcolloidosome synthesis, but to d ate little is kno wn rega rding th e effect (if an y) of the encapsulated drug on the shell packing density. In this pa- per we examined self-assembly of colloidal shells on alginate films containing four model drugs: aspirin, caffeine, theophylline and theobromine. We find that the packing density in the colloidal shells is low for all drugs, and ranges between 0.16 and 0.3. There is no clear correlation between drug properties (in particular, water solubility) and the packing density of the self-assembled colloidal shell. Keywords: Drug delivery, Diffusion, Colloids, Colloidosomes 1. Introduction Selective control of drug transport is of interest for pharmaceutical, cosmetic, and food applications [1-6]. Colloidosomes [7] are aqueous microcapsule volumes coated by a shell of colloidal particles (see Figure 1) [7-16]: The packed colloidal particles form a barrier for transport, whose pore size may be controlled through choice of the colloidal particle size and the packing den- sity in the shell [7-16]. As a result, colloidosomes allow size exclusion, namely, inhibition of transport of com- ponents whose size is larger than a critical value, while allowing transport of smaller molecules. Several methodologies have been developed to form the colloidal shells of colloidosomes. Typically, they utilize the tendency of the particles to assemble at the interface between hydrophilic and hydrophobic fluids [7,13,14,15]. Although effective, the conditions involved are often not compatible with those needed for biological agents. As a result, they cannot be used for drug encap- sulation. Kim, et al [11] utilized a different method for colloi- dosome for mation: The co lloid al particles in t he shell are driven to self-assemble on the surface of a hydrogel core by electrostatic interactions. The assembly is conducted in one step, and under benign environmental condition (i.e. aqueous solutions- includ ing, potentially, serum, at room or body temperature). We recentl y studied t he relea se of model drug s from hydrogels coated by self-assembled colloidal mono- layers [17,18]. Quite surprisingly, we found that the release profiles for each drug were insensitive to the size of the particles in the shell, for p[articles ranging from 20nm to 3.3μm [17]. To understand these results, we adapted a two-film diffusion model[18], where the hydrogel is taken to be one film, and the shell coating the second film. The shell is modeled as a composite of an impenetrable phase (the colloidal particles) and a pe- netrable one that is s imilar in pr op erties to the aqueo us or hydrogel core (see Figure 1). The mod el fi nds t hat in th e case of a loosely packed monolayer such as that of the self assembled shell, the release profile is indeed inde- pendent of the colloidal particle size, a function of three parameters only: the diffusion coefficient of the drug in the hydrogel core, the dimension (thickness) of the hy- drogel core, and the packing density of particles in the shell. Thus, controlling the release rate of a drug from a colloidosome may be achieved through control of the shell packing density (since the diffusion coefficient is set by the hydrogel properties, and core size is t ypically set by the specific system needs).  Self-Assembly of Colloidosome Shells on Drug-Containing Hydrogels Copyright © 2011 SciRes. JBNB Figure 1. A schematic of the system examined. Left: A hy- drogel containing a diffusant, or drug, is coated by a self-assembled colloidal monolayer, formed through ad- sorption of colloidal particles onto the oppositely charged hydrogel. Due to the self-assembly process, the shell is loosely packed, typically w ith a colloi dal volu me fra ction of order ¼. The coll oidal s hell may be take n to be a co mposite of an impenetrable phase (the particles) and a penetrable one (hydrogel or aqueous solution). Right: The two-film diffusion mod el used t akes t he hydr ogel to be one fi lm, an d the colloidal shell the other. The drug diffuses through the hydrogel until reaching the interface with the colloi dal shell. It cannot penetrate through the particles, and must go through t he pores i n betw een t he coll oids , so th at the rat e of release is reduced when compared to the uncoated hydro- gel. Fitting our expe rimental re sults to the theoretic al mod- el [18] yielded that the packing density in the self-assembled colloidal shells is independent of the col- loidal particle size. Moreover, it was similar for caffeine and aspirin, despite the differences in their chemical struct ure. This observation is so mewhat surprising, since it has been previously found that the properties of a drug can significantly affect the properties of the encapsulat- ing hydrogel, and thus the release profile: For example, studies of drug release from biodegradable polymeric films have clearly demonstrated that the drug properties can significantly affect the degradation rate, and thus release profile [19,20]. It may be expected that the pres- ence of a drug in the hydrogel would affect the adsorp- tion, and thus packing, of the colloidal particles. To determine whether drug properties affect the pack- ing density of a colloidal shell, and thus the release pro- file of the drug, we compared systems containing four model drugs: caffeine, theophylline, theobromine and aspirin. The first three are similar in structure, and even cause similar physiological effects (stimulation of the central nervous system and gastric acid secretion) [21]. Yet, they differ significantly in their solubility in water. The solubility of caffeine is 21.7 mg/mL [22], theophyl- line is 8 mg/mL [22], and theobromine is 0.5 mg/mL [23]. All drugs, including aspirin, have similar MWs (180 for aspirin, theophylline and theobromine, and 194 for caffe ine), a ltho ugh asp irin d iffer s fro m the thr ee dr ugs in its chemical structure. The solubility of aspirin in water has an intermediate value similar to that of theophylline, of order 3-10 mg/ml [22]. It is somewhat dif ficult to predict, a-prio ri, what effect would drug properties have on the self-assembled col- loidal shell. Self-assembly in these systems is driven by electrostatic interactions between charged groups on the hydrogel surface, and oppositely charged ones on the colloidal particles. It is possible that a hydrophobic drug (as determined by low solubility in water) may cause the hydrogel to collapse, thereby reducing the availability of the hydrogel surface groups to bonding with the colloidal particles. On the other hand, highly hydrophilic drugs may interact with the charged surface groups, thereby reducing particle adsorption. Regardless of the mechan- ism, it is expected that if the encapsulated drug has an effect on shell formation (and thus on the rate of drug release), the packing density of the colloidal particles in the she ll would scale in so me manner with dr ug solubil i- ty in aqueous solutions. We find tha t the two-film model fit the release rate of all drugs well, for both uncoated and colloidal-shell coated hydrogels. However, the effect of the encapsu- lated drugs on the packing density of the shells is found to be weak: In all systems, the packing density of the colloids in the shell (namely, the volume fraction of par- ticles) was calculated to be in a narrow range of 0.16-0.3. The packing density did not vary systemat ically with the drug properties, and in particular did correlate with the drug’s water solubility. Indeed, the packing density for theobromine, which had the lowest solubility of all the drugs tested, was the lowest (~0.16). However, the high- est packing density was found for theophylline (~0.3), which has intermediate water solubility. 2. Materials and Methods 2.1. Materials Sodium alginate, calcium chloride, aspirin, caffeine, theobromine and theophylline were purchased from Sigma-Aldrich (St. Louis, MO and Milwaukee, WI, USA). 3.3 µm amidine (C(NH2)=NH+) functionalized polystyrene particles were purchased from Invitrogen Molecular Probes. All reagents were used as purchased with no further purification. 2.2. Synthe sis of Hydrogel Fi lms To a llow consistent meas urements o f transport thro ugh a colloidal shell, the system consisted of an alginate hy- drogel film (rather than spherical drops) impregnated with a given concentration of the chosen drug. The film was then coated by a monolayer of colloidal particles, and transport determined by measurement of the drug concentration in the medium surrounding the film  Self-Assembly of Colloidosome Shells on Drug-Containing Hydrogels Copyright © 2011 SciRes. JBNB [17,18]. Unlike transport from microgel colloidosomes, this geometry allows exact control over the system pa- rameters (e.g. drug content in the hydrogel). The sur- rounding medium was kept, for all four drugs, at a con- centration that is well below the relevant solubility limit even at 100% release. A 2% (weight/volume) sodium alginate in water solu- tion was shaken and sonicated to remove any excess air. 1 mg of a model molecule was dissolved in 400 µL of the sodium alginate solution and placed in a 30 mL (25 mm x 95 mm) glass vial. The vials were placed in a convec- tion oven at 40ºC overnight to evaporate the excess sol- vent. 500 µL of a 10% wt. solution of calcium chloride in water was added to the alginate and allowed to crosslink for 5 minutes. Excess calcium chloride was removed and polystyrene particles were added to the hydrogel film and incubated overnight. The films were subsequently washed with deionized water. 2.3. Ultraviolet Spectroscopy Concentrations of molecules released were calculated using a linear calibration curve. 8 samples were made with pure water as the lowest concentration. The absorp- tion spectra showed peaks for the following drugs: caffeine at 273 nm, aspirin at 275 nm, theophylline at 271 nm and theobromine at 272 nm. All further mea- surements were taken at respective peak wavelengths. Concentrations were in the linear regime to minimize error. 2.4. Diffusion Model The hydrogel-colloidal shell system is modeled as a two-fil m syst em [ 18] , wher e t he hydr o gel is o ne film a nd the colloidal shell t he other (see Figure 1 ). The transport of drug in such two-film systems is defined by three pa- rameters: The hydrogel film thickness, a, the diffusion coefficient of the drug in the hydrogel, D, and a dimen- sionless parameter L that accounts for the shell permea- bility: L is infinity when there is no shell (or when the shell does not offer resistance to transport), and zero when the shell is completely impenetrable to the drug [18]. The fraction of drug released at time t is given in this approach by () ( ) 22 22 222 2 11 1 1 n D t/a nn nn nn e f(t) LL LL β ββ ββ − ∞∞ = = = −+− +− ∑∑ (1) where βn are the roots of the expression [18] (2) Modeling the colloidal shell as a composite of an im- penetrable phase (the colloidal particles) and a penetrable one yielded, in the case of a colloidal monolayer, the following relationship for L [18]: ( ) ( ) 21 33 1 216L πϕ ϕπϕ ≈− + (3) where φ is the volume fraction of colloids in the shell. Equation (3) is appropriate only for a loosely packed colloida l shell (where the p acking de nsity of the pa rticles in the shell is less than ~0 .5) and whe re t he h ydro gel core thickness a is much larger than the co lloid al par ticle shell thickness. The thickness of the monolayer colloidal shell is equal to the dia meter of the co lloid al pa rticles which i n this case, is much smaller than the thickness of the hy- drogel film. From (1-3) we see that the transport of a drug through a colloid al shell coating a hydrogel is expe cted to depend onl y on t he h ydro gel cor e th ic kness a, the dif fusio n co ef- ficient throug h the hydrogel D, and the c olloidal packing density (volume fraction) in the shell φ. It should not vary with the particle diameter, as indeed verified by our previous experiments [17,18]. 3. Results and Discussion Our earlier work [17,18] suggests that the release profile of drugs from hydrogels coated by a (loosely packed) self-assembled colloidal shell depends only on three parameters: The hydrogel film thickness, the diffusion coefficient of the drug in the hydrogel, and the packing density of the particles in the shell. Therefore, to deter- mine t he di ffusi on co effi cie nt o f the d rug s in our algi nate gels, we measured the release of the four drugs from un- coated hydrogels, as shown in F igure 2. Literature values were published for the diffusion coefficient of some of the drugs in water [25,26]. How- ever, those values cannot be directly applied to transport of these drugs through other media - such as our hydro- gel. Moreover, altho ugh it might have b een a ssume d that the relative ratios of the diffusion coefficients would be independent of the type of media (namely, if diffusion coefficient of one drug in water is higher than that of a second one, the same could be said of their rates in other aque ous-based media), studies show that this is not the case: For example, in water, caffeine and theophylline have similar diffusion coefficients [25,26]. Additionally, their release rate from alginate gels [24] and through Buccal tissue [27] has been found to be similar as well. Yet, in other media- transdermal tissue [27] and pig ear skin [28]- the transport rate of caffeine was found to be two orders of magnitude faster than that of theophylline. Besides the medium, the specific formulation also af- fects the absolute and relative rates of the transport of different drugs [27,28]. In our case, the formulation of the hydrogel used for 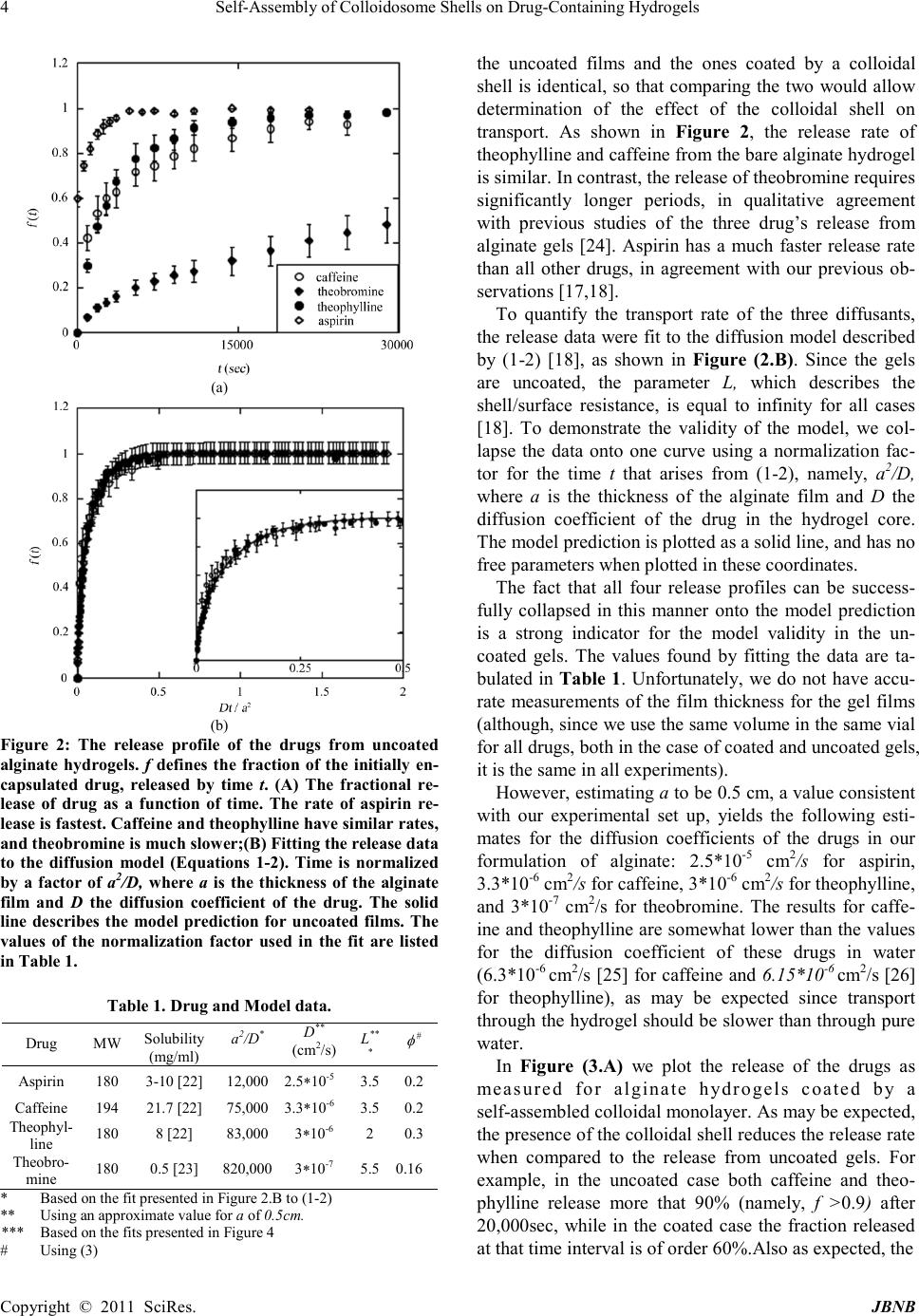 Self-Assembly of Colloidosome Shells on Drug-Containing Hydrogels Copyright © 2011 SciRes. JBNB (a) (b) Figure 2: The release profile of the drugs from uncoated alginate hydrogels. f defines the fraction of the initially en- capsulated drug, released by time t. (A) The fractional re- lease of drug as a function of time. The rate of aspirin re- lease is fast est. Caffei ne a nd theop hyll ine hav e si milar rates , and theobromine is much slower;(B) Fi tting the release data to the diffusion model (Equations 1-2). Time is normalized by a factor of a2/D, where a is the thickness of the alginate film and D the diffusion coefficient of the drug. The solid line describes the model prediction for uncoated films. The values of the normalization factor used in the fit are listed in Table 1. Table 1. Drug and Mod el data. Drug MW Solubility (mg/ml) a2/D* (cm2/s) L** * φ # Aspirin 180 3-10 [22] 12,000 2.5*10-5 3.5 0.2 Caffein e 194 21. 7 [22] 75,000 3.3*10-6 3.5 0.2 line 180 8 [22] 83,000 3*10-6 2 0.3 180 0.5 [23] 820,000 3*10-7 5.5 0.16 * Based on the fit presented in Figure 2.B to (1-2) ** Usin g an approxim ate value for a of 0.5cm. *** Based on the fits presented in Figure 4 # Using (3) the uncoated films and the ones coated by a colloidal shell is identical, so that comparing the two would allo w determination of the effect of the colloidal shell on transport. As shown in Figure 2, the release rate of theophylline and caffeine from the bare alginate hydrogel is similar. In contrast, the release of theobromine requires significantly longer periods, in qualitative agreement with previous studies of the three drug’s release from alginate gels [24]. Aspirin has a much faster release rate than all other drugs, in agreement with our previous ob- servations [17,18]. To quantify the transport rate of the three diffusants, the release data were fit to the diffusion model described by (1-2) [18], as shown in Figure (2.B). Since the gels are uncoated, the parameter L, which describes the shell/surface resistance, is equal to infinity for all cases [18]. To demonstrate the validity of the model, we col- lapse the data onto one curve using a normalization fac- tor for the time t that arises from (1-2), namely, a2/D, where a is the thickness of the alginate film and D the diffusion coefficient of the drug in the hydrogel core. The model prediction is plotted as a solid line, and has no free parameters when plotted in these coordinates. The fact that all four release profiles can be success- fully collapsed in this manner onto the model prediction is a strong indicator for the model validity in the un- coated gels. The values found by fitting the data are ta- bulated in Table 1. Unfortunately, we do not have accu- rate measurements of the film thickness for the gel films (although, since we use the same volume in the same vial for all drugs, both in the case of coated and uncoated gels, it is the same in all experiments) . Ho wever, esti mati ng a to be 0.5 cm, a value consistent with our experimental set up, yields the following esti- mates for the diffusion coefficients of the drugs in our formulation of alginate: 2.5*10-5 cm2/s for aspirin, 3.3*10-6 cm2/s for caffeine, 3*1 0-6 cm2/s for theophylline, and 3*10-7 cm2/s for theobromine. The results for caffe- ine and theophylline are somewhat lower than the values for the diffusion coefficient of these drugs in water (6.3*10-6 cm2/s [25] for caffeine and 6.15*10-6 cm2/s [26] for theophylline), as may be expected since transport thro ugh the hydro gel s hould b e slo wer t han thr ough pure water. In Figure (3.A) we plot the release of the drugs as measured for alginate hydrogels coated by a self-assembled colloidal monolayer. As may be expected, the presence of the colloidal shell reduces the release rate when compared to the release from uncoated gels. For example, in the uncoated case both caffeine and theo- phylline release more that 90% (namely, f >0.9) after 20,000sec, while in the coated case the fraction released at that time interval is of order 60%.Also as expected, the  Self-Assembly of Colloidosome Shells on Drug-Containing Hydrogels Copyright © 2011 SciRes. JBNB (a) (b) Figure 3. The release of caffeine, theophylline and theo- bromine from alginate hydrogels coated by a monolayer of 3.3mm colloidal particles. f defines the fraction of the in- itially encapsulated diffusant, released by time t. The sym- bols are as defi ned in Fig ure 2. (A) The fracti onal rele ase as a function of time; (B) The fractional release as a function of the normalized time Dt/a2, where the value of D/a2is as calculated from the fit to the uncoated hydrogel (Figure 2.B). The solid line describes the model prediction for the uncoate d gels. relative rates remain the same as in the uncoated case: Aspirin is released most rapidly, theophylline and caffe- ine have similar rates, and theobromine requires the longest time. However, the raw data cannot distinguish between the effects of the diffusion coefficient in the hydrogel, and the potential effect of the self assembled colloida l mono layer. To determine the effect of the colloidal shell, we again normalize time the value of a2/D as calculated from the uncoated gels (Figure 3.B). If the effect of the shell is simil ar fo r a ll d r ugs, t hen t hey sho uld again co ll ap se o nto the same cur ve, although this one would differ from that of the uncoated system, as given in (1-3). We see, how- ever, that the normalized release profiles do not overlap: Although all are slower than the predicted rate for the uncoated gels (as depicted by the solid line), caffeine and theobromine seem to have a faster rate than that of aspi- rin, while theophylline is clearly slower than all other drugs. The two film model suggests that these differences in the release rates may be due to the packing density of the colloidal particles in the shell; since the shell is self as- sembl ed, the densi ty ma y var y bet ween the d iffer ent s ys- tems. Therefore, we use (1-2) to fit the release p rofiles of the four drugs to evaluate the value of L, the shell trans- port retardation parameter, and (3) to correlate this para- meter to the colloidal packing density. The fits are pre- sented in Figure 4, a nd the values l iste d in Table 1. We find that the values of the shell permeability para- meter L fall, for all drugs, in the range of 2-5, which translated to a range for the packing volume fraction of particles in the shell of 0.3-0.16. For both caffeine and aspirin, the best fit is obtained with a value of L=3.5, which translates to a colloidal packing density in the shell using (3 ) of ≈0.2. For theophylline, the best fit is for L=2 , namely, ≈0.3 , while for theobromine the best fit is for L=5.5, corresponding to ≈0.16 . While there are clear differences between these values, they are all within the same range for the packing density. Moreover, there is no clear tr end li nking the dr ug pro per ties and the colloidal packing density in the shell: Caffeine and aspi- rin have similar packing densities, despite the differences in str uctur e, M W and sol ubili ty. The op hylli ne, which ha s a similar solubility to that of aspirin- which is interme- diate between caffeine and theobromine- has the highest packing density, while theobromine has the lowest. 4. Conclusions Previous studies have shown that the properties of an encapsulated drug can significantly affect the properties of the encapsulating hydrogel. In this paper we investi- gate the effect of drugs, encapsulated in hydrogel films, on the self-assembly of a colloidal shell. Fourmodel drugs are investigated: aspirin, caffeine, theophylline and theobromine. These drugs are similar in MW, and three of them (caffeine, theophylline and theobromine) have a similar structure and even similar physiological effects. However, they differ significantly in their solubility in water. We find that the release profiles in all systems (both uncoated hydrogels and ones coated by the self assem- bled colloidal shells) can be fit well with a t wo-film dif- fusion model [18]. The packing density in the self-assembled colloidal layers is found to vary, between the drugs, in the range of 0.16-0.3. The lowest packing  Current Distortion Evaluation in Traction 4Q Constant Switching Frequency Converters Copyright © 2011 SciRes. JB NB Figure 4. Fitting the release rate from coated hydrogel to the two-film model (equations 1-2). The dotted line is for a shell where L=5, and the dashed line where L=2. a ) Aspirin. The solid line is the model with a value of L=3.5; b) Caffeine. The solid line is the model with a value of L=3.5; c) Theophylline; d) Theobromine. The so lid line i n the inset is for L=5.5 . density is found for theobromine, which has the lowest solub ility i n water . Ho wever, the hig hest pa ckin g densit y is fou nd for theophylline, which hasanintermediate water solubility (see Table 1). Caffeine and aspirin have iden- tical packi ng densities, d espite the fact that t he solubility of caffeine in water is three times as high as that of aspi- rin. We conclude that there is some effect of the drug properties on the packing density in the self assembled colloidal shells. However, this effect is weak, and cannot be directly linked to the drug properties, in particular solubility in water. 5. Acknowledgements Thanks to NSF/IDBR award number 0649897 for finan- cial support. RTR acknowledges the NSF-IGERT DGE-0654313 fellowship. REFERENCES [1] T. Kaasgaard and T. L. Andresen, “Liposomal Cancer Therapy: Exploiting Tumor Characteristics”, Expert Opinion on Drug Delivery, Vol. 7, No. 2 , 2010, pp. 225-243. doi:10.1517/17425240903427940 [2] W. N. Hait, “Targeted Cancer Therapeutics”, Cancer Research , Vol. 69, No. 4, 2009, pp. 1263-1267 . doi:10.1158/0008-5472.C AN-08-3836 [3] L. Sagalowicz and M. E. Leser, “Delivery Systems for Liquid Food Products”, Current Opinion in Colloid and Interf ace Science, Vol. 15, No. 1-2, 2010, pp. 61 -72. doi:10.1016/j.cocis.2009.12.003 [4] X. Wu and R. H. Guy, “Applications of Nanoparticles in Topical Drug Delivery and in Cosmetics”, Journal of Drug Delivery Science and Technology, Vol. 19, No. 6, 2009, pp . 371-384. [5] J. Pardeike, A. Hommoss and R. H. Muller, “Lipid Nanoparticles (SLN, NLC) in Cosmetic and Pharmaceutical Dermal Products”, International Journal of Pharmaceutics, Vol. 366, No. 1-2, 2009, pp 170-184. doi:10.1016/j.ijpharm.2008.10.003 [6] D. S. Pisal, M. P. Kosloski and S. V. Balu-I yer, “Delivery of Therapeutic Proteins” Journal of Pharmaceutical Scien ces, Vol. 99, No. 6, 2010, pp. 2557-2575. doi:10.1002/jps.22054 [7] A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marqu ez, A.R. Bau sch and D. A. Weitz, “Colloidosomes: Selectively Permeable Capsules Composed of Colloidal Particles” Scien ce, Vol. 298, No. 5595, 2002, pp. 1006-1009. doi:10.1126/science.1074868 [8] D. Lee and D. A Weitz, “Double Emul- sion-Templatednanoparticlecolloidosomes with Selective Permeability”, Advanced Materials, Vol. 20, No. 18,  Self-Assembly of Colloidosome Shells on Drug-Containing Hydrogels Copyright © 2011 SciRes. JBNB 2002, pp . 3498-3503. doi:10.1002/adma.200800918 [9] D. Lee and D.A Weitz, “Nonspherical Colloidosomes with Multiple Compartments from Double Emulsions”, Small, Vol. 5, No. 17 , 2009, pp . 1932 -1935. doi:10.1002/smll.200900357 [10] S. Shilpi, A. Jain, Y. Gupta and S. K. Jain “Colloido- somes: an Emergin g V esicu lar Syste m in Dru g D eli ver y”, Critical Reviews in Therapeutic Drug Carrier Systems, Vol. 24, N o. 4, 20 07, pp. 361-391. [11] J. W. Kim, A. Fernandez-Nieves A, N. Dan N, A. S. Utada AS, M. Marquez and D. A. Weitz, “Colloidal Assembly Route for Responsive Colloidosomes with Tunable Permeability” Nano Letters, Vol. 7, No. 9, 2007, pp. 2876-2880. doi:10.1021/nl0715948 [12] A. S. Miguel, J. Scrimgeour, J. E. Curtis and S. H. Behrens, “Smart Colloidosomes with a Dissolution Trigger”, Soft Matter, Vol. 6, No. 14, 2010, pp. 316 3-3166. doi:10.1039/c002930k [13] R. McGor ty, J. Fung, D. Kaz and V. N. Manoharan , “Col- loidal Self-Assembly at an Interface”, Materials Today, Vol. 13, No. 6, 20 10 , pp. 34-42. doi:10.1016/S1369-7021(10)70107-3 [14] M. H. Lee, V. Prasad and D. Lee, “Microfluidic Fabrica- tion of Stable Nanoparticle-Shelled Bubbles”, Langmuir, Vol. 26, N o. 4, 20 10, pp. 22 27-2230. doi:10.1021/la904425v [15] J. Texter, “Templating Hydrogels”, Colloid and Polymer Scien ce, Vol. 28 7, No. 3, 2009, pp. 313-321. doi:10.1007/s00396-008-1990-z [16] X. C. Yang and Z. H. Mo “Microcapsules from the Self-Assembly of Nanoparticles at Interfaces”, Progress in Chemist ry, Vol. 22, No. 9, 20 1 0, pp. 1735-1740. [17] R. T. Rosenberg and N. R. Dan, “Controlling Surface Porosity and Release from Hydrogels Using a Colloidal Particle Coating”, Journal of Colloid and Interface Scien ce, Vol. 349, No. 2, 20 10 , p p. 498 -504. doi:10.1016/j.jcis.2010.05.095 [18] R. T. Rosenberg and N. R. Dan, “Diffusion through Colloidosome Shells”, Journal of Colloid and Interface Scien ce, 2 010. In press. [19] S. J. Si egel, J. B. Kah n, K. Metzger K , K. I. Winey KI, K. Werner K and N. Dan, “Effect of Drug Type on the De- gradation Rate of PLGA Matrices”, European Journal of Pharmaceutics and Biopharmaceutics, Vol. 64, No. 3, 2006, pp . 287-293. doi:10.1016/j.ejpb.2006.06.009 [20] R. Rosenberg, W. Devenney, S. Siegel and N. Dan, “Anomalous Release of Hydrophilic Drugs from Poly(Epsilon-Caprolactone) Matrices”, Molecular Phar- maceut i cs, Vol. 4, No. 6. 20 07 , pp. 943-948. doi:10.1021/mp700097x [21] A. Meyer, T. Ngiruwonsanga and G. Henze, “Determination of Adenine, Caffeine, Theophylline and Theobromine by HPLC with Amperometric Detection”, Journal of Analytical Chemistry, Vol. 356, 1996, pp. 284-287. [22] I. P. o. C. S. (IPCS), http://www.inchem.org/documents/pims/pharm/aspirin.ht m#SectionTitle:3.2%20Chemical%20structure [23] J. B. Tho mas, J. H. Yen, M . M. Sch antz, B. J. P orter, and K.E. Sharpless, “Determination of Caffeine, Theobromine, and Theophylline in Standard Reference Material 2384, Baking Chocolate, Using Reversed-Phase Liquid Chromatography”, Journal of Agricultural and Food Chemistry, Vol. 52, No. 11, 2004, pp. 3259-3263. doi:10.1021/jf030817m [24] P. Sriamornsak and R. A. Kennedy, “Effect of Drug So- lubility on Release Behavior of Calcium Polysaccharide Gel-coated Pellets”, European Journal of Pharmaceutical Scien ces, Vol. 32, No. 3, 2007, pp. 231-239. doi:10.1016/j.ejps.2007.08.001 [25] D. W. Green and R. H. Perry, “Perry's Chemical Engineers' Handbook”, 8th ed., McGraw-Hill, Columbus, 2008. [26] M. Grassi, I. Colombo and R. Lapasin, “Experimental Determination of the Theophylline Diffusion Coefficient in Swollen Sodium-Alginate Membranes”, Journal of Controlled Release, V ol. 76, No. 1-2, 2001, pp. 93 -105. doi:10.1016/S0168-3659(01)00424-2 [27] R. A. Thakur and B. B. Michniak, “Transdermal and Buccal Delivery of Methylxanthines Through Human Tissue In Vitro”, Drug Development and Industrial Pharmacy, V ol. 33, No. 5 , 2007, pp. 513-521. doi:10.1080/03639040600901994
|