Combination Therapy of Capecitabine with Cyclophosphamide as a Second-Line Treatment after Failure of Paclitaxel
plus Bevacizumab Treatment in a Human Triple Negative Breast Cancer Xenograft Model
1240
colorectal cancer xenograft models [7], PTX has been
reported to increase the level of TP in xenografted tu-
mors [16] and, therefore,, the TP level in tumor would be
increased by PTX + BEV treatment in the 1st-line treat-
ment in our study. However, because antitumor activity
gradually receded during the 1st-line treatment, the amount
of TP induced by PTX might also decrease, if the anti-
tumor activity was attenuated by PTX resistance. To cla-
rify the above hypothesis, the change over time in tumor
TP levels in 1st-line treatment should be examined. In the
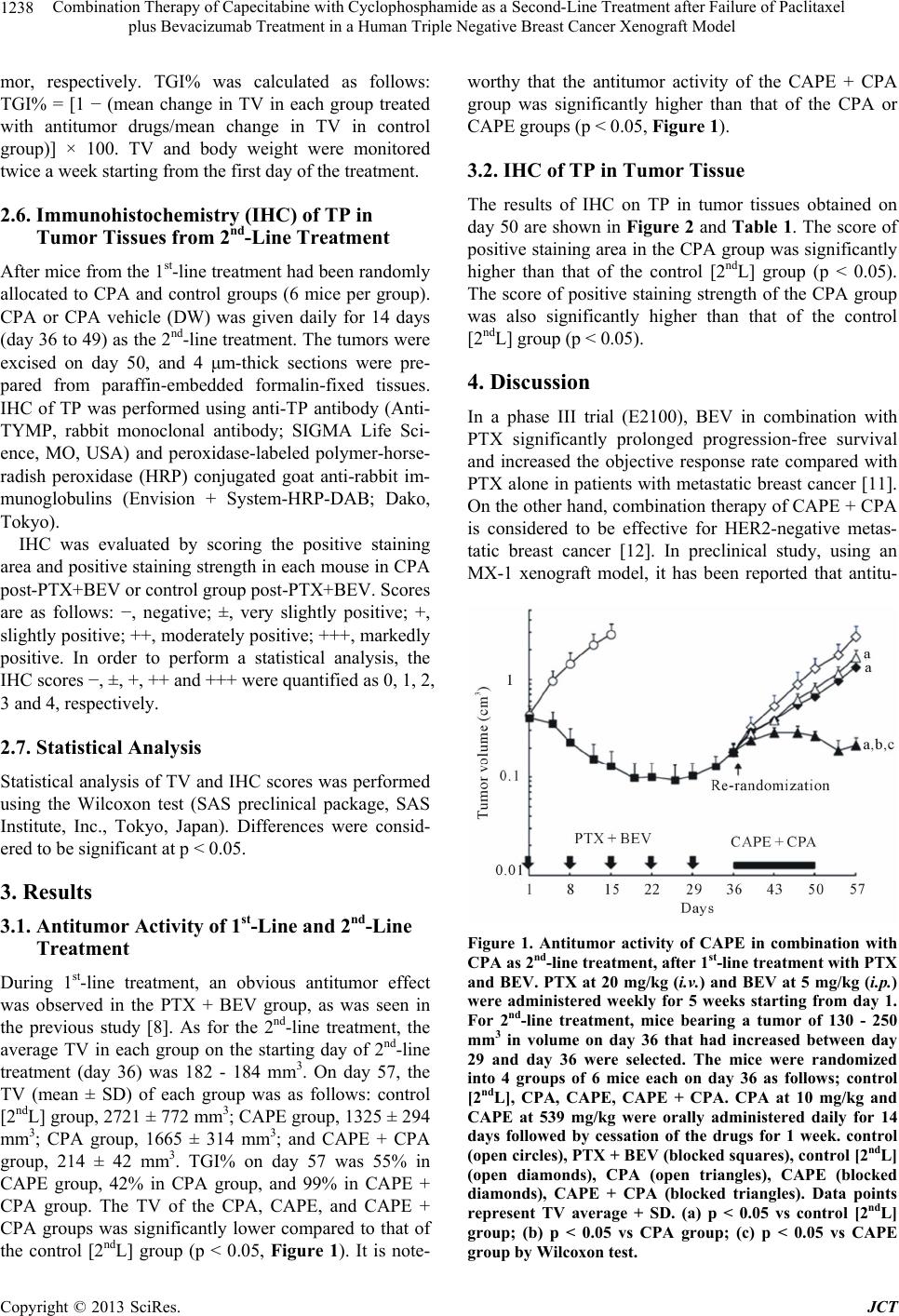
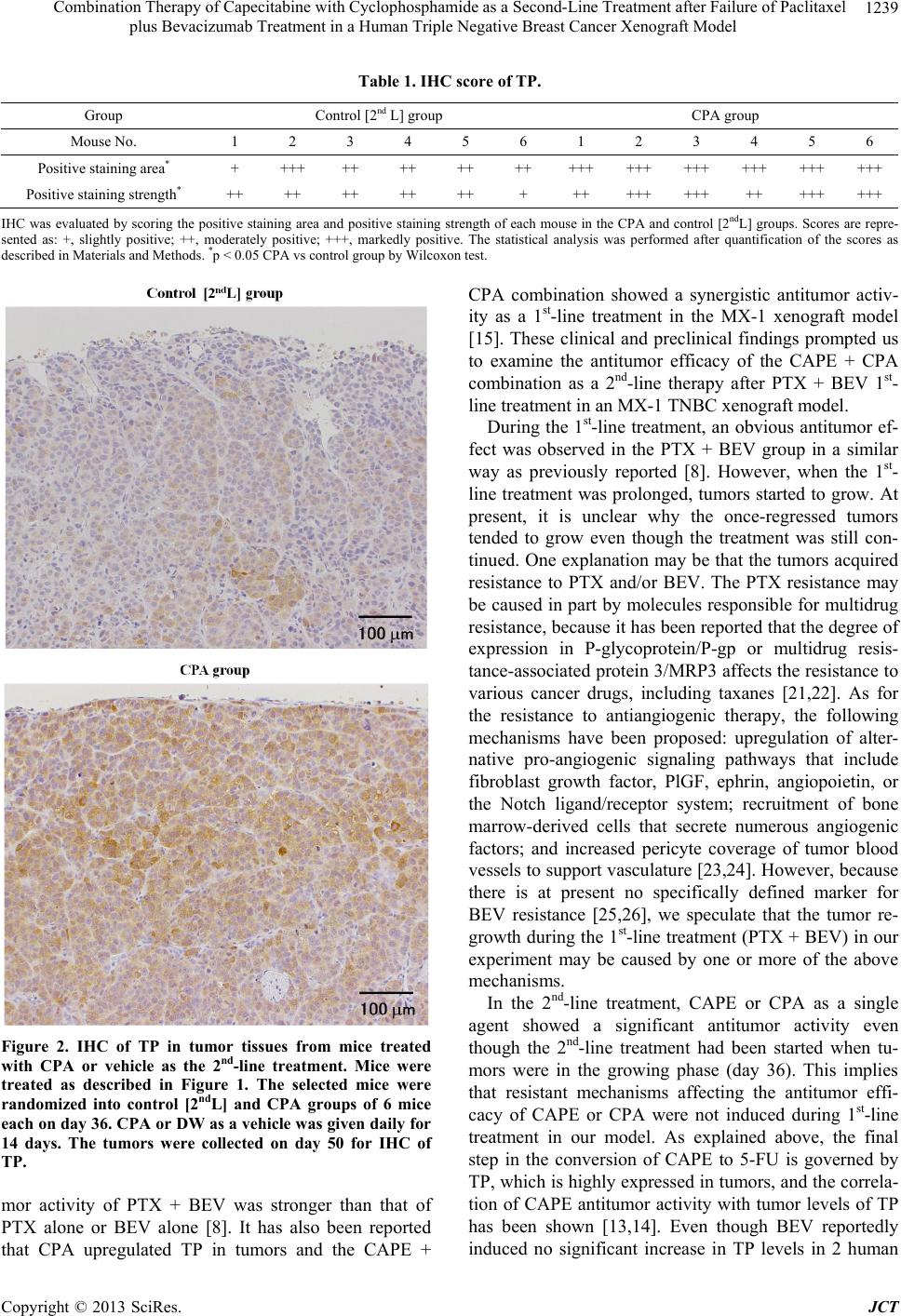
2nd-line treatment, CAPE + CPA combination showed an
extremely high antitumor activity compared to CAPE or
CPA monotherapy. Because the TP level in tumor was
upregulated by CPA treatment, the superior antitumor
effect of CAPE + CPA combination compared to CAPE
monotherapy may be attributed to the increased 5-FU
level in tumor tissue caused by facilitated conversion
from CAPE by TP. These results are similar to that in the
1st-line therapy reported previously [15].
Our preclinical results suggest that the CAPE + CPA
combination therapy may be effective as a 2nd-line ther-
apy for progressive disease after PTX + BEV 1st-line
treatment in TNBC patients.
5. Acknowledgements
We thank Dr. Kazushige Mori for helpful discussion and
comments regarding the manuscript.
REFERENCES
[1] L. G. Presta, H. Chen, S. J. O’Connor, V. Chisholm, Y. G.
Meng, et al., “Humanization of an Anti-Vascular Endo-
thelial Growth Factor Monoclonal Antibody for the Ther-
apy of Solid Tumors and Other Disorders,” Cancer Re-
search, Vol. 57, No. 20, 1997, pp. 4593-4599.
[2] K. J. Kim, B. Li, K. Houck, J. Winer and N. Ferrara, “The
Vascular Endothelial Growth Factor Proteins: Identifica-
tion of Biologically Relevant Regions by Neutralizing
Monoclonal Antibodies,” Growth Factors, Vol. 7, No.1,
1992, pp. 53-64. doi:10.3109/08977199209023937
[3] Y. Wang, D. Fei, M. Vanderlaan and A. Song, “Biologi-
cal Activity of Bevacizumab, a Humanized Anti-Vegf
Antibody in Vitro,” Angiogenesis, Vol. 7, No. 4, 2004, pp.
335-345. doi:10.1007/s10456-004-8272-2
[4] K. J. Kim, B. Li, J. Winer, M. Armanini, N. Gillett, et al.,
“Inhibition of Vascular Endothelial Growth Factor-In-
duced Angiogenesis Suppresses Tumour Growth in Vivo,”
Nature, Vol. 362, No. 6423, 1993, pp. 841-844.
doi:10.1038/362841a0
[5] H. P. Gerber and N. Ferrara, “Pharmacology and Phar-
macodynamics of Bevacizumab as Monotherapy or in
Combination with Cytotoxic Therapy in Preclinical Stud-
ies,” Cancer Research, Vol. 65, No. 3, 2005, pp. 671-680.
[6] P. V. Dickson, J. B. Hamner, T. L. Sims, C. H. Fraga, C.
Y. Ng, et al., “Bevacizumab-Induced Transient Remod-
eling of the Vasculature in Neuroblastoma Xenografts
Results in Improved Delivery and Efficacy of Systemi-
cally Administered Chemotherapy,” Clinical Cancer Re-
search, Vol. 13, No. 13, 2007, pp. 3942-3950.
doi:10.1158/1078-0432.CCR-07-0278
[7] M. Yanagisawa, K. Fujimoto-Ouchi, K. Yorozu, Y. Ya-
mashita and K. Mori, “Antitumor Activity of Bevacizu-
mab in Combination with Capecitabine and Oxaliplatin in
Human Colorectal Cancer Xenograft Models,” Oncology
Reports, Vol. 22, No. 2, 2009, pp. 241-247.
[8] M. Yanagisawa, K. Yorozu, M. Kurasawa, K. Nakano, K.
Furugaki, et al., “Bevacizumab Improves the Delivery
and Efficacy of Paclitaxel,” Anticancer Drugs, Vol. 21,
No. 7, 2010, pp. 687-694.
[9] Y. Yamashita-Kashima, K. Fujimoto-Ouchi, K. Yorozu,
M. Kurasawa, M. Yanagisawa, et al., “Biomarkers for
Antitumor Activity of Bevacizumab in Gastric Cancer
Models,” BMC Cancer, Vol. 12, 2012, p. 37.
doi:10.1186/1471-2407-12-37
[10] S. B. Horwitz, “Mechanism of Action of Taxol,” Trends
in Pharmacological Sciences, Vol. 13, No. 4, 1992, pp.
134-136. doi:10.1016/0165-6147(92)90048-B
[11] K. Miller, M. Wang, J. Gralow, M. Dickler, M. Cobleigh,
et al., “Paclitaxel plus Bevacizumab versus Paclitaxel
Alone for Metastatic Breast Cancer,” New England Jour-
nal of Medicine, Vol. 357, No. 26, 2007, pp. 2666-2676.
doi:10.1056/NEJMoa072113
[12] M. Tanaka, Y. Takamatsu, K. Anan, S. Ohno, R. Nishi-
mura, et al., “Oral Combination Chemotherapy with Ca-
pecitabine and Cyclophosphamide in Patients with Me-
tastatic Breast Cancer: A Phase II Study,” Anti-Cancer
Drugs, Vol. 21, No. 4, 2010, pp. 453-458.
doi:10.1097/CAD.0b013e328336acb1
[13] H. Yasuno, M. Kurasawa, M. Yanagisawa, Y. Sato, N.
Harada, et al., “Predictive Markers of Capecitabine Sen-
sitivity Identified from the Expression Profile of Py-
rimidine Nucleoside-Metabolizing Enzymes,” Oncology
Reports, Vol. 29, No. 2, 2013, pp. 451-458.
[14] T. Ishikawa, F. Sekiguchi, Y. Fukase, N. Sawada and H.
Ishitsuka, “Positive Correlation between the Efficacy of
Capecitabine and Doxifluridine and the Ratio of Thy-
midine Phosphorylase to Dihydropyrimidine Dehydro-
genase Activities in Tumors in Human Cancer Xeno-
grafts,” Cancer Research, Vol. 58, No. 4, 1998, pp. 685-
690.
[15] M. Endo, N. Shinbori, Y. Fukase, N. Sawada, T. Ishikawa,
et al., “Induction of Thymidine Phosphorylase Expression
and Enhancement of Efficacy of Capecitabine or 5’-De-
oxy-5-Fluorouridine by Cyclophosphamide in Mammary
Tumor Models,” International Journal of Cancer, Vol. 83,
No. 1, 1999, pp. 127-134.
doi:10.1002/(SICI)1097-0215(19990924)83:1<127::AID-
IJC22>3.0.CO;2-6
[16] N. Sawada, T. Ishikawa, Y. Fukase, M. Nishida, T. Yo-
shikubo, et al., “Induction of Thymidine Phosphorylase
Activity and Enhancement of Capecitabine Efficacy by
Taxol/Taxotere in Human Cancer Xenografts,” Clinical
Copyright © 2013 SciRes. JCT