Paper Menu >>
Journal Menu >>
 J. Mod. Phys., 2010, 1, 226-235 doi:10.4236/jmp.2010.14034 Published Online October 2010 (http://www.SciRP.org/journal/jmp) Copyright © 2010 SciRes. JMP Study of the Electric Explosion of Titanium Foils in Uranium Salts Leonid I. Urutskoev1,2, Dmitry V. Filippov1 1RECOM, National Research Center “Kurchatov Institute”, Moscow, Russia 2Moscow State University of Printing Arts, Moscow, Russia E-mail: filippov-atom@ya.ru, urleon@ya.ru Received June 9, 2010; revised July 27, 2010; accepted August 2, 2010 Abstract The results of experiments on electroexplosion titanic foil in water solutions of salts of uranium are pre- sented in this paper. It is shown, that as a result of electroexplosion occurs appreciable (to 20%) distortion of an initial isotope parity of uranium. In the most solution parts, observable isotope distortion occurs in favour of enrichment by 235U. At the moment of electroexplosion it was not observed an appreciable stream of the neutrons. By means of Cs label and by methods by α, β, γ-spectrometry and mass-spectrometry it have been shown, that isotope distortion occurs at the expense of non-uniform “disappearance” of both isotopes from a solution. The isotope distortion leads to infringement of the 234Th secular equilibrium in the uranyl solution. The equilibrium infringement between the 234Th and 234mPa, i.e. within the proper thorium decay chain, was observed also. The assumption about that the effects are caused of low-energy nuclear reactions at the mo- ment of electroexplosion is suggested. Keywords: Electric Explosion, Nuclear Decay 1. Introduction Currently, it is beyond doubt that strong external elec- tromagnetic fields can considerably change the probabil- ity of nuclear decay and even change the conditions of nuclear stability [1-7]. For example, complete ionization of 187Re increases the probability of -decay (due to de- cay to a bound electron state) [2], while complete ioniza- tion of stable isotopes 163Dy, 193Ir, 205Tl makes them –-active (the half-life time of completely ionized 163Dy was 47 5 days [3]). Now it is clear that strong external fields affect to one or another extent the probabilities of all nuclear processes, the major effect being mediated by deformation of the atomic electron states (both occupied by electrons and free) [4-7]. This influence depends ap- preciably on the decay type and energy, on the degree of prohibition, and, in the general case, on the spins and evenness of the initial and final states of the decaying nucleus. In some cases, the dependence of the change in the decay probability on the intensity of external action can be non-monotonic. Usually, the effect is stronger for hindered (forbidden) decay channels but it is noteworthy that a change in the decay probability induced by exter- nal fields depends on many parameters and every nu- cleus is in a certain sense unique. For the nuclei that de- cay along several channels having different parameters and degrees of prohibition, the effect of external fields changes the ratio of decay intensities along different channels. When the fields act on a substance containing a mixture of isotopes, the final ratio of the radioactive iso- topes should change as the effects may be different for different isotopes. It is known that high-current electric explosion of metal wires in a liquid induces strong magnetic fields (H ~ 1 MG) and high pulse pressures (Р ~ 105 atm) [8,9]. To study the effect of external action on the decay of ura- nium series isotopes, we carried out experiments on the electric explosion of titanium foil in a solution of uranyl sulfate in doubly distilled water. The very idea to inves- tigate impacts of strong electromagnetic fields on the uranium isotopic decay dawned on the authors of this articles resulting from multiple and repeated attempts to grasp the physical mechanism of nuclear reactor runaway which actually took place in the Chernobyl NPP in 1986 [10]. The results of preliminary experiments of this series were published previously [11].  L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 227 2. Description of the Experimental Setup The experiments outlined below were carried out using one of the capacitor banks of the setup described in detail previously [12]. The energy store of the setup was W ~ 22 kJ, the charge voltage was U = 4.8 kV, the current amplitude was I ~ 120 kA, and the pulse duration was 130 s. Unlike the explosion chamber used previously [12], the explosion chamber presented in Figure 1 was sealed and allowed mounting of only one electrode. The internal part of explosion chamber 1 was made of poly- ethylene and was arranged in a lower part of a tough stainless-steel case 2, which ensured air tightness. The voltage was supplied through the upper part of a metal container, which served simultaneously as a gas-col- lecting chamber 3 with the volume V ~ 3 ℓ (prechamber). The gas-collecting chamber is required due to the fact that all attempts to make the gas-water mixture formed during current passage stay in the explosion chamber failed, resulting only in the collapse of the explosion chamber. In a number of experimental series, a pre- chamber made of organic glass was used to provide the possibility of high-speed optical recording. Uranium solution 4 with the volume υ ~ 20 cm3 was poured into the internal part of explosion chamber 1. Two types of solutions were used, namely, a solution of uranyl sulfate enriched in 235U and a solution of uranyl nitrate with the natural isotope distribution of uranium. The concentration of the uranium solutions used in ex- periments varied from 10–2 to 10–1 g.-mol/liter. A bulk central titanium electrode 5 bearing foil 6 welded to it 1 2 3 4 5 6 7 8 Figure 1. Schematic drawing of experimental setup: 1–interior of the explosion chamber; 2–stainless steel frame; 3–gas-collecting chamber; 4–liquid; 5–titanium electrode; 6–titanium foil; 7–tightening; 8–lid. was immersed in the solution. Thus, the welded foil acted as a cable armoring short-circuited to the cable central core. The amount of the titanium foil (load) var- ied in different experiments from one to four 1 cm-wide bands, each with the thickness ∆ = 50 μm and the length L = 4.0 cm. The weight of each band was m = (90 5) 10–3 g. The discharge parameters were monitored using coax- ial shunts and a voltage-ratio box to record current and voltage pulses. The signals were recorded by means of an AD converter with a sampling rate of 1 GHz. Typical current and voltage oscillograms were presented in our earlier publications [12,13]. It is well known that electric explosion of a conductor in water produces an intense electromagnetic radiation [8,9], which requires taking special measures to provide the interference immunity of the signals. Note that if an uranium solution is used as the liquid in electric explo- sion, the level of electromagnetic radiation increases by a large factor, especially in the second half of the pulse duration. During current passage, -radiation was recorded. This was done by scintillator-based detectors (NaI, CsI) and photomultipliers, which were placed 1.5 to 2 meters away from the location of electric explosion. No notice- able excess over the background -radiation during the current pulse was detected. The neutron yield was also monitored. For sufficiently efficient recording of fission neutrons, a special neutron detector was assembled from nine slow neutron counters (СНМ-17). The whole assembly was surrounded by a 8 cm-thick polyethylene moderator. The electromagnetic interference immunity was provided by placing the whole detector block together with preamplifiers inside a copper screen. For elimination of false pulses (com- mon-mode interferences), the slow neutron counters were split into two groups. 3. The Procedure of Measurements For determining the efficiency of neutron detection, a neutron calibration source 252Cf was placed in the explo- sion chamber. The measured detection efficiency of fis- sion neutrons was 0.04%. Every detected pulse was iden- tified as a neutron if it coincided with the pulse from the calibration source in the shape and amplitude. It follows from the measurements that the neutron flux after elec- tric explosion did not exceed I 103 neutrons per pulse. Note that in the experiments using organic glass pre- chamber 3, the neutron detector also recorded some pulses other than neutron pulses. Figure 2(а) shows a neutron signal from the calibration source and a typical “false” signal. It follows from Figure 2 that duration of 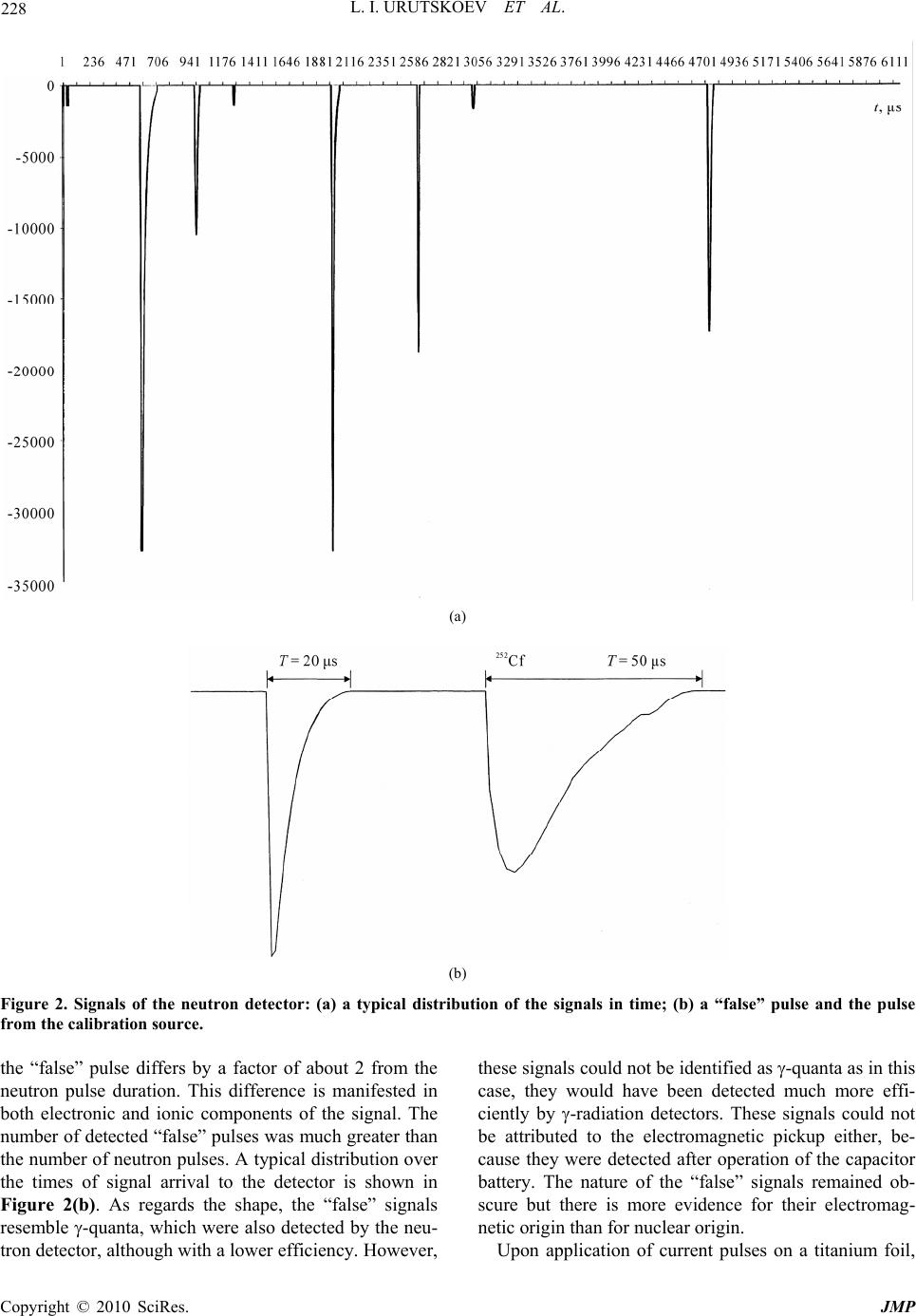 L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 228 (a) (b) Figure 2. Signals of the neutron detector: (a) a typical distribution of the signals in time; (b) a “false” pulse and the pulse from the calibration source. the “false” pulse differs by a factor of about 2 from the neutron pulse duration. This difference is manifested in both electronic and ionic components of the signal. The number of detected “false” pulses was much greater than the number of neutron pulses. A typical distribution over the times of signal arrival to the detector is shown in Figure 2(b). As regards the shape, the “false” signals resemble -quanta, which were also detected by the neu- tron detector, although with a lower efficiency. However, these signals could not be identified as -quanta as in this case, they would have been detected much more effi- ciently by -radiation detectors. These signals could not be attributed to the electromagnetic pickup either, be- cause they were detected after operation of the capacitor battery. The nature of the “false” signals remained ob- scure but there is more evidence for their electromag- netic origin than for nuclear origin. Upon application of current pulses on a titanium foil,  L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 229 the foil explodes and the pressure in the explosion chamber sharply increases. For this reason, most of the uranium solution breaks to prechamber 3 through seal 7. This gives two different solution volumes, and, accord- ing to preliminary results [11], they differed substantially in the isotope composition of uranium. For this reason, sampling of the uranium solution after the “shot” was performed from both upper (hereinafter “up”) and lower (hereinafter “lw”) volume. For convenience of collecting the solution, special groove was envisaged in the poly- ethylene lid 8. The samples were taken by a disposable syringe, placed in standard 2-mℓ or 5-mℓ plastic tubes and imme- diately sealed by PARAFILM “M”. The dosage accuracy was relatively low being equal to ~5%. Therefore, the measured volumetric activity values were used only for rough estimates and the results of , , and spectrome- try measurements were represented as ratios of the val- ues obtained in one and the same measurement. This presentation of measurement results ensured minimiza- tion of possible errors caused by dosage errors. In order to avoid procedural errors and to enable com- parison of the results of measurements of the same val- ues obtained by different procedures, two samples with identical volumes were measured for each procedure. One sample (reference sample, “rs”) contained the initial solution of uranium for this experiment and the other sample (experimental sample, “ex”) was the uranium solution taken from the explosion chamber after the elec- tric explosion. This made it possible to compare the re- sults of, for example, - and mass spectrometry for ex- periments carried out with natural and isotope-enriched solutions of uranium by plotting the results together as the ratio R = (235U/238U)ex/(235U/238U)rs. Laser mass spectrometry was chosen as the procedure for direct determination of the elemental and isotope compositions. Having rather high accuracy (10–4-10–5 at%), this procedure gave an error of determination of trace impurities of about 10-15%. The accuracy of de- termination of the isotope composition of elements was 1.5-2% for minor isotopes and less than 1% for principal isotopes, which is consistent with published data [14]. During our study, the samples taken from all “shots” were measured by mass spectrometry but the most thor- ough isotope analysis was carried out for the following chemical elements: Fe, Ti and U. The results of meas- urements for initial components and samples from one experiment with a solution of enriched uranyl sulfate are summarized in Table 1. The errors of measurements of percentages of all chemical elements and isotopes pre- sented in Table 1 fall into the corridor of errors dis- cussed above. The independent measurements of the uranium isotope Table 1. Elemental and isotope compositions of the sam- ples, %. Control Contr. foil Exp Na 0.135 0.0000 4.380 Mg 0.001 0.0056 0.170 Al 0.060 0.0032 0.550 Si 0.117 0.0067 0.500 P 0.037 0.0025 0.015 S 49.250 0.0038 3.750 Cl 0.321 0.0044 0.093 K 0.047 0.0050 3.500 Ca 0.034 0.0163 1.970 Ti 0.434 99.8606 80.080 V 0.002 0.0018 0.002 Cr 0.002 0.0121 0.039 Mn 0.005 0.0020 0.023 Fe 0.077 0.0694 0.690 Ni 0.009 0.0060 0.013 Co 0.002 0.0002 0.009 Cu 0.005 0.0002 0.021 Zn 0.011 0.0000 0.045 Zr 0.000 0.0003 0.000 Ag 0.000 0.0000 0.510 U 49.453 0.0000 3.610 235U 21 24.56 238U 79 75.44 46Ti 8.0 8.00 47Ti 7.3 8.07 48Ti 73.8 71.48 49Ti 5.5 6.16 50Ti 5.4 6.27 ratio and concentrations of some of its decay products were accomplished using liquid scintillation , -spec- trometry and solid-state -spectrometry procedures. The counting sample for liquid scintillation spec- trometry was prepared in the following way. A 10-ℓ  L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 230 aliquot portion of the test solution was thoroughly mixed with the scintillating solution ULTIMA GOLD AB. The counting sample obtained in this way was measured by a TRI-CARB 2550 TR/AB counter (Canberra-Packard) equipped with a time discriminator of alpha and beta pulses and a multichannel analyzer. The identification and activity calculation of isotopes were performed using a special program Spectradec for spectrum processing, which makes use of the library of instrumental spectra of separate isotopes. The result was found by comparing the studied convoluted instrumental spectrum with this spec- trum obtained by simulation. This procedure has sub- stantial errors in determination of most isotopes and, hence, this analysis was semiquantitative. The contents of alpha-emitting isotopes were deter- mined on a standard Canberra -spectrometric complex, which comprised four -spectrometers of design 7401 with PIPS detectors. The active area of each detector was 600 mm2. The counting sample was prepared by electro- chemical deposition of the emitters from a solution of sodium ammonium sulfate at pH ~ 2.2-2.5 onto stainless- steel polished discs. To decrease the effect of conversion electrons, the measurements were carried out at a 14 mm distance from the detector surface. This effect was manifested as asymmetry of the right “shoulder” of the peaks of the instrumental -spectrum, giving rise to an additional error in their approximation. The average time of meas- urement was ~1.5 105 s. The quality of the prepared counting sources was estimated based on 234U peak resolution at the 4774.6 keV line (yield 72.4%). The en- ergy resolution of the measurements was at least 23.3 keV, while the contribution of the 234U radiation peak base to the low-energy 235U and 238U isotopes was less than 0.1% and could be properly taken into account. The isotope ratios were calculated from the peak areas. Since 235U gives rise to seven intense lines which overlap with all -decaying nuclei present in the mixture (234U, 236U, 238U), this was calculated using the algorithm im- plemented in RadSpectraDee software. This implies ap- proximation of peaks by an intricate function combining an asymmetric Gaussian, an exponent and a hyperbola. In the -spectrometric procedure, a 40 cm3 germanium detector and a standard Canberra spectrometer were used. The energy resolution of the gamma-spectrometer de- termined from the 137Cs line (662 keV) was 1.8 keV. The effect of geometric factor on the results was eliminated by mounting samples right on the detector end face using a specially manufactured holder. Since the fine titanium powder suspended in an uranium solution precipitated over 24 hours and Th was found to interact with titanium oxides, then in order to avoid additional errors in the measurements, the uranium solution container was mounted in a strictly horizontal position relative to the ground surface. In order to eliminate the possible errors related to the time drift of -spectrometer parameters, the experimental and reference samples of uranium solution were measured in turn. The spectra were processed in the energy regions of 92 keV (92.38 keV and 92.8 keV are -lines of 234Th, which is a daughter product of 238U), 1 MeV (1001 keV is the -line of 234mPa, a daughter product of 234Th) and 186 keV (185.7 keV is the -line of 231Th, a daughter product of 235U). Single peaks were first approximated by a Gaussian and then the areas under them were calcu- lated. The region of -doublet at 92.5 keV shows an overlap of the 234Th and 231Pa -lines (93.063 keV) with the K X-ray lines of actinides, resulting in a complex background. The uranium K1 and K2 X-ray lines ap- pear due to uranium self-fluorescence, the K2 peak con- tributing directly to the 92.5 keV doublet. The contribu- tion of thorium K1 and K2 lines related to 235U decay was negligibly small. The resolution of the spectrum at 82-102 keV into components was done by a computer program. 4. Experimental Results As shown by preliminary studies [11], an electric explo- sion of titanium foil in a solution of uranyl sulfate in- duces a distortion of the initial U isotope ratio and dis- turbance of the 234Th secular equilibrium. Our study con- firmed the conclusions of the preliminary experiments and, resorting to additional procedures, provided a more detailed picture of the phenomenon. The use of both natural and enriched U in the experiments increased the accuracy of measurements and provided a more reliable knowledge of the process. For instance, experiments with enriched U allowed us to attain a satisfactory accuracy in determining the uranium isotope ratio by mass spec- trometry, and the use of natural uranium markedly in- creased the accuracy of measurement of the uranium isotope ratio from the ratio of 186/1001 -lines. When using different procedures we often faced the so-called “first measurement problem”. In liquid - spectrometry and solid-state -spectrometry, this was a distortion of the studied spectrum. In -spectrometry, the effect was manifested as an increase in the fraction of conversion electrons, and in -spectrometry, this was appearance of an additional low-energy peak. In the measurements by -spectrometric procedure, the “first measurement effect” was in the fact that the measure- ment that was performed first differed most often from the subsequent measurements by a value markedly ex- ceeding three standard deviations. After several hours, even in the second measurement, the effect usually dis- 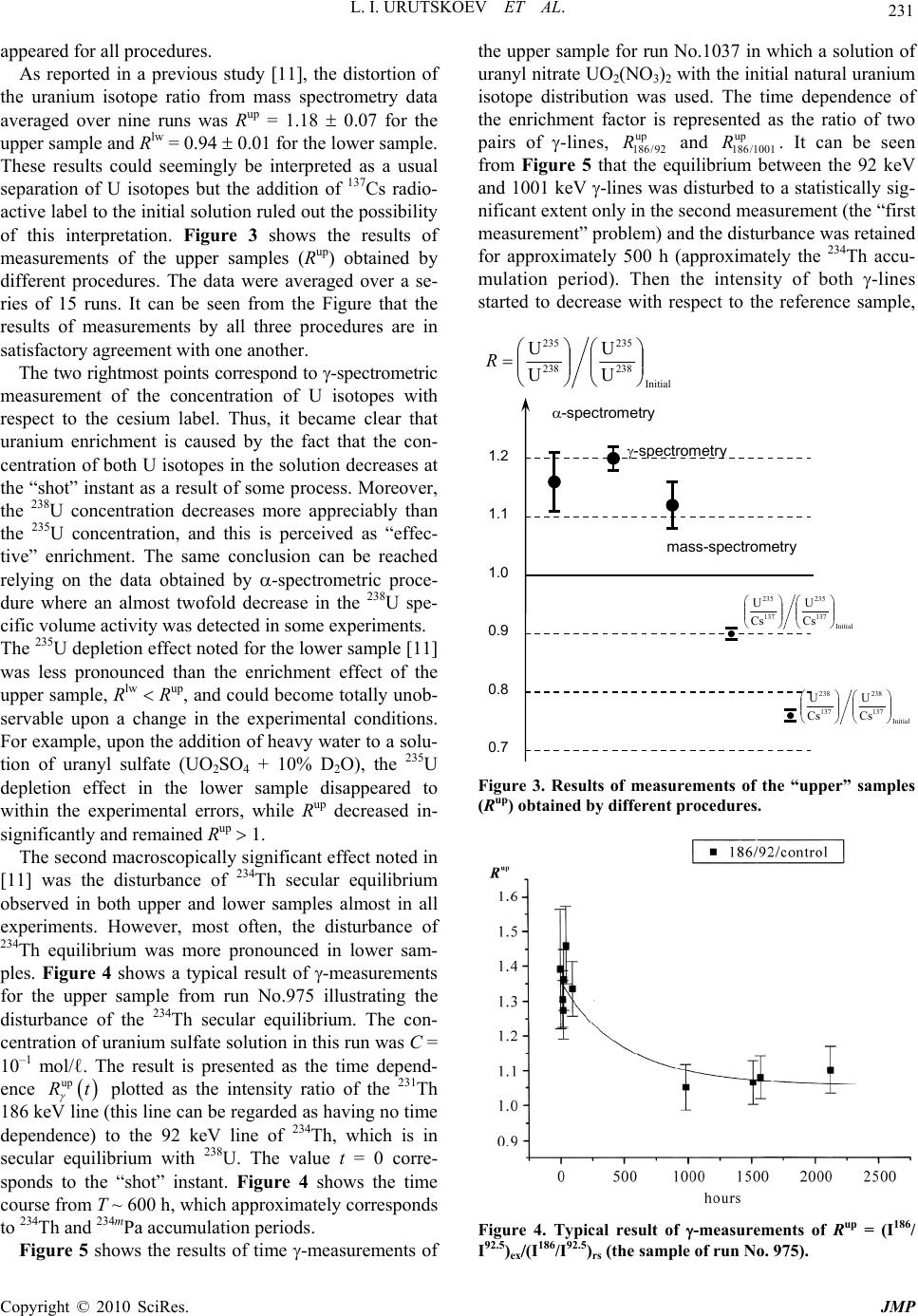 L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 231 appeared for all procedures. As reported in a previous study [11], the distortion of the uranium isotope ratio from mass spectrometry data averaged over nine runs was Rup = 1.18 0.07 for the upper sample and Rlw = 0.94 0.01 for the lower sample. These results could seemingly be interpreted as a usual separation of U isotopes but the addition of 137Cs radio- active label to the initial solution ruled out the possibility of this interpretation. Figure 3 shows the results of measurements of the upper samples (Rup) obtained by different procedures. The data were averaged over a se- ries of 15 runs. It can be seen from the Figure that the results of measurements by all three procedures are in satisfactory agreement with one another. The two rightmost points correspond to -spectrometric measurement of the concentration of U isotopes with respect to the cesium label. Thus, it became clear that uranium enrichment is caused by the fact that the con- centration of both U isotopes in the solution decreases at the “shot” instant as a result of some process. Moreover, the 238U concentration decreases more appreciably than the 235U concentration, and this is perceived as “effec- tive” enrichment. The same conclusion can be reached relying on the data obtained by -spectrometric proce- dure where an almost twofold decrease in the 238U spe- cific volume activity was detected in some experiments. The 235U depletion effect noted for the lower sample [11] was less pronounced than the enrichment effect of the upper sample, Rlw Rup, and could become totally unob- servable upon a change in the experimental conditions. For example, upon the addition of heavy water to a solu- tion of uranyl sulfate (UO2SO4 + 10% D2O), the 235U depletion effect in the lower sample disappeared to within the experimental errors, while Rup decreased in- significantly and remained Rup 1. The second macroscopically significant effect noted in [11] was the disturbance of 234Th secular equilibrium observed in both upper and lower samples almost in all experiments. However, most often, the disturbance of 234Th equilibrium was more pronounced in lower sam- ples. Figure 4 shows a typical result of -measurements for the upper sample from run No.975 illustrating the disturbance of the 234Th secular equilibrium. The con- centration of uranium sulfate solution in this run was С = 10–1 mol/ℓ. The result is presented as the time depend- ence up Rt plotted as the intensity ratio of the 231Th 186 keV line (this line can be regarded as having no time dependence) to the 92 keV line of 234Th, which is in secular equilibrium with 238U. The value t = 0 corre- sponds to the “shot” instant. Figure 4 shows the time course from T ~ 600 h, which approximately corresponds to 234Th and 234mPa accumulation periods. Figure 5 shows the results of time -measurements of the upper sample for run No.1037 in which a solution of uranyl nitrate UO2(NO3)2 with the initial natural uranium isotope distribution was used. The time dependence of the enrichment factor is represented as the ratio of two pairs of -lines, up 186 /92 R and up 186 /1001 R. It can be seen from Figure 5 that the equilibrium between the 92 keV and 1001 keV -lines was disturbed to a statistically sig- nificant extent only in the second measurement (the “first measurement” problem) and the disturbance was retained for approximately 500 h (approximately the 234Th accu- mulation period). Then the intensity of both -lines started to decrease with respect to the reference sample, Initial 238 235 238 235 U U U U R 0.7 0.8 0.9 1.0 1.1 1.2 Initial 137 235 137 235 Cs U Cs U Initial 137 238 137 238 Cs U Cs U mass-spectrometry -spectrometry -spectrometry Figure 3. Results of measurements of the “upper” samples (Rup) obtained by different procedures. Figure 4. Typical result of -measurements of Rup = (I186/ I92.5)ex(I186/I92.5)rs (the sample of run No. 975). 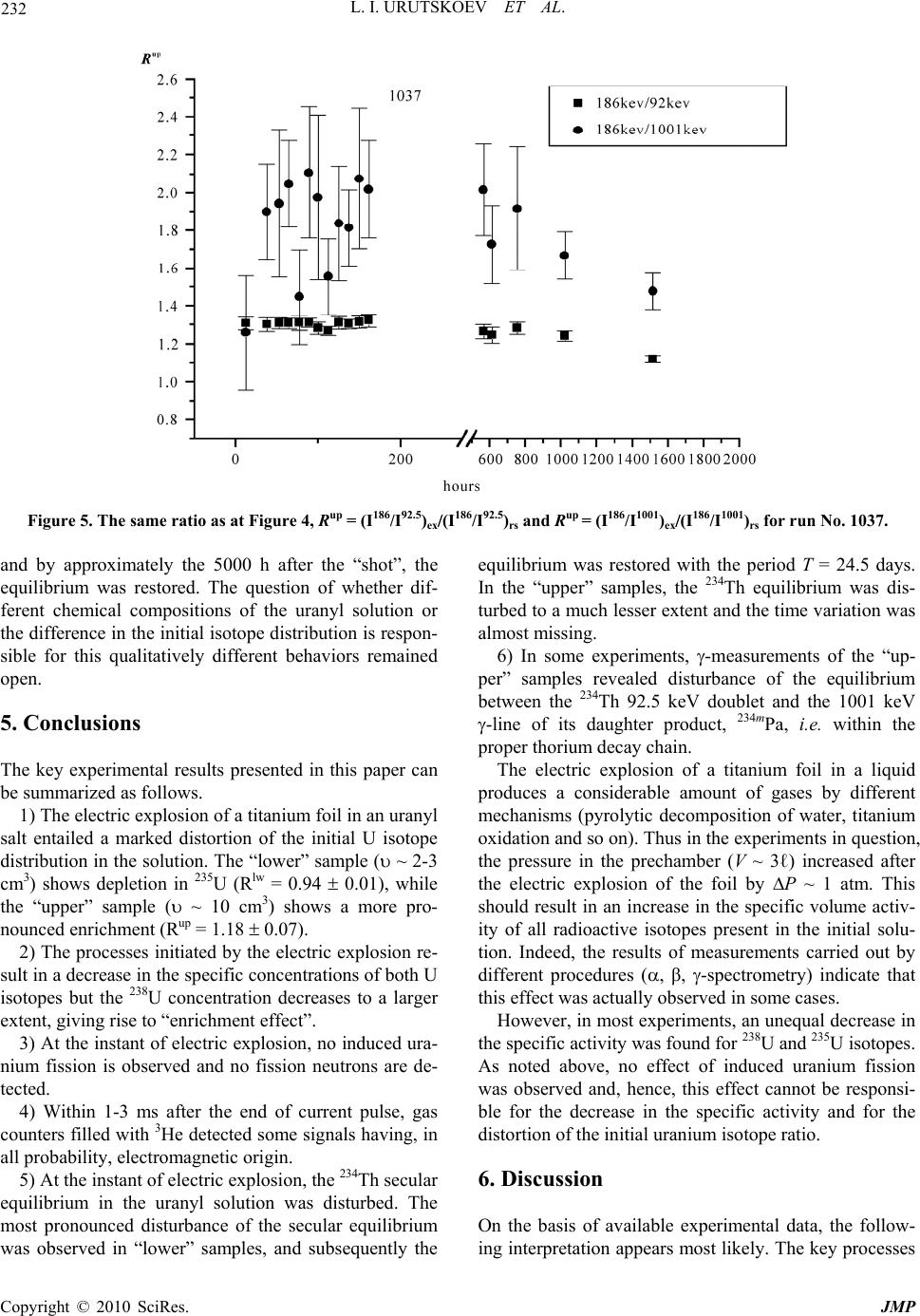 L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 232 Figure 5. The same ratio as at Figure 4, Rup = (I186/I92.5)ex(I186/I92.5)rs and Rup = (I186/I1001)ex(I186/I1001)rs for run No. 1037. and by approximately the 5000 h after the “shot”, the equilibrium was restored. The question of whether dif- ferent chemical compositions of the uranyl solution or the difference in the initial isotope distribution is respon- sible for this qualitatively different behaviors remained open. 5. Conclusions The key experimental results presented in this paper can be summarized as follows. 1) The electric explosion of a titanium foil in an uranyl salt entailed a marked distortion of the initial U isotope distribution in the solution. The “lower” sample ( ~ 2-3 cm3) shows depletion in 235U (Rlw = 0.94 0.01), while the “upper” sample ( ~ 10 cm3) shows a more pro- nounced enrichment (Rup = 1.18 0.07). 2) The processes initiated by the electric explosion re- sult in a decrease in the specific concentrations of both U isotopes but the 238U concentration decreases to a larger extent, giving rise to “enrichment effect”. 3) At the instant of electric explosion, no induced ura- nium fission is observed and no fission neutrons are de- tected. 4) Within 1-3 ms after the end of current pulse, gas counters filled with 3He detected some signals having, in all probability, electromagnetic origin. 5) At the instant of electric explosion, the 234Th secular equilibrium in the uranyl solution was disturbed. The most pronounced disturbance of the secular equilibrium was observed in “lower” samples, and subsequently the equilibrium was restored with the period T = 24.5 days. In the “upper” samples, the 234Th equilibrium was dis- turbed to a much lesser extent and the time variation was almost missing. 6) In some experiments, -measurements of the “up- per” samples revealed disturbance of the equilibrium between the 234Th 92.5 keV doublet and the 1001 keV -line of its daughter product, 234mPa, i.e. within the proper thorium decay chain. The electric explosion of a titanium foil in a liquid produces a considerable amount of gases by different mechanisms (pyrolytic decomposition of water, titanium oxidation and so on). Thus in the experiments in question, the pressure in the prechamber (V ~ 3ℓ) increased after the electric explosion of the foil by ∆P ~ 1 atm. This should result in an increase in the specific volume activ- ity of all radioactive isotopes present in the initial solu- tion. Indeed, the results of measurements carried out by different procedures (, β, -spectrometry) indicate that this effect was actually observed in some cases. However, in most experiments, an unequal decrease in the specific activity was found for 238U and 235U isotopes. As noted above, no effect of induced uranium fission was observed and, hence, this effect cannot be responsi- ble for the decrease in the specific activity and for the distortion of the initial uranium isotope ratio. 6. Discussion On the basis of available experimental data, the follow- ing interpretation appears most likely. The key processes  L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 233 resulting in the “effective” uranium enrichment take place in the area of current passage and in the area di- rectly adjoining the plasma channel, since the major en- ergy contribution is made in this area and strong mag- netic fields and high pressures develop at the pulse in- stant. As regards the space marked by a dashed line in Figure 1, only a pressure pulse is transmitted there, which is apparently insufficient to give rise to the “en- richment” effect. The amount of the solution remaining on the bottom of the explosion chamber after the electric explosion was in line with this interpretation. This fact could possibly be explained by assuming that the decrease in the specific radioactivity of uranium and other radioactive isotopes is a consequence of low-energy nuclear reactions. Below we call this hypothetical phe- nomenon the low energy transformation of nuclei (here- inafter LET [15]). Indeed, since the electric explosion of a titanium foil in both distilled water [12,16,17] and ura- nium salt resulted in depletion of the natural titanium isotope mixture in 48Ti isotope, it is reasonable to assume that this phenomenon is caused by the same physical mechanism in both cases. The experimental research of the regular features of LET that we have carried out for many years showed that even-even isotopes of chemical elements with atomic numbers divisible by 16 (А = 16 is oxygen) are more prone to be transformed than their neighbors. Yet another hypothetical mechanism responsible for the uranium isotope distortion could be a change in -decay periods caused by either ionization or action of intense magnetic fields. The atomic electrons increase the -decay probability with respect to that for a com- pletely ionized atom. First, the field of atomic electrons decreases the barrier for an -particle and, second, the nuclear charge decreases by 2 units upon -decay, which changes the electron shell energy. Taking account of the effect of atomic electrons results in the necessity of re- placing energy of an -particle in the calculation of the -decay constant by an “effective” energy, which is greater than the real energy 5 433 73 65EE ZZ eV [18]. Due to the exponential dependence of the -decay probability on the -particle energy 1 ln constpE , the effect of atomic electrons may be pronounced. As was to be expected, the atomic electrons affect low-en- ergy processes to a larger extent. For example, for 147Sm (-particle energy ~ 2.31 MeV; T½ = 7 1011 years), the presence of the electron shell increases the probability of -decay 2.6-fold compared with the nucleus of a fully ionized atom [18]. The influence of a superstrong magnetic field on the -decay probability can be qualitatively described in the following way. An external superstrong magnetic field changes the energy of the atomic electron shell [19] and, hence, changes also the energy of any nuclear decay, as the decay energy is equal to the difference between the total energies of the initial and final systems with allow- ance for ionization energies of atoms or ions [1]. For -decay, the presence of an external superstrong mag- netic field results in an increase in the decay energy and, hence, in an increase in the -decay probability. Since the 238U -decay energy is lower than the energies of the principal channels of 235U -decay, the relative increase in the 238U -decay probability caused by ionization and by the effect of an external superstrong magnetic field on the atomic electron shell would be more pronounced than that the relative increase in the 235U -decay probability. On the other hand, an external magnetic field changes the geometry of the problem: the spherical symmetry is replaced by the preferential direction along the magnetic field. This effect is ambiguous and would be considera- bly different for the even-even 238U nucleus having a zero magnetic moment and the 235U nucleus where the -decay occurs between states of nuclei with nonzero spins. It can be seen without difficulty that since the above- discussed effects have different signs with respect to the change in the specific volume activity of isotopes, then depending on the experimental conditions, either an in- crease or a decrease in the resulting specific activity with respect to different U isotopes is possible. This is actu- ally observed in experiments. One more reason supporting LET is the disturbance of 234Th secular equilibrium observed in experiments. The disturbance of the equilibrium was due to a decrease in the 234Th specific concentration in an uranyl solution. This conclusion follows from data of and -spec- trometry. Indeed, if only the 238U specific concentration decreased at the instant of electric explosion, then the specific -activity of 234Th would decrease with the pe- riod T = 24.5 days. However, the sharp drop of the spe- cific -activity was detected during the first 24 hours after the electric explosion. The same conclusion can be drawn from the -intensity ratio of E = 92.5 keV lines of the sample and the initial solution, which was 92.5 92.5 ex rs1II . Thus, the sign of this effect and the fact of the subsequent 234Th accumulation with time preclude interpreting the disturbance of secular equilibrium as being due to the decrease in the 238U concentration alone. As has already been noted in the Introduction, the macroscopic nature of the observed distortions of the initial uranium isotope ratio implies that the phenomenon responsible for this effect occurs in a solution volume considerably exceeding the plasma channel volume. This is evidence supporting the hypothesis that some radiation arises during the electric explosion [12]. The detection of pulses of obviously electromagnetic origin by gas detec-  L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 234 tors is indirect evidence in favor of the electromagnetic nature of the arising radiation. It is noteworthy that very similar signals but in much lower quantities were also detected in those experiments with the organic-glass prechamber in which H2O, D2O or aqueous glycerol were used instead of the uranyl solution. Several recent publications reported the observation of neutrons in high-voltage electrolysis in D2O or during cavitation (e.g., [20]). In those cases where the neutron spectrum was not directly measured and neutron detec- tors were used in the counting mode, an error could arise in the interpretation of the results of measurements due to detection of “false” pulses. The disturbance of the equilibrium between 234Th and its daughter 234mPa nucleus detected by -spectrometry can be a result of the change in the –-decay probabilities along different channels. This item was discussed in more detail previously [21]. Note that the specific features arising during the spec- trometric measurements and observed during the first 24 hour after electric explosion (the “first measurement” problem) coincide, as regards the time scale of the effect, with the period for disappearance of the deformation of 57Fe Mössbauer spectrum noted previously [13]. Coinci- dence of the time scales of these effects can suggest a common origin. Thus, summarizing the results of these studies pro- vides the following conclusions. Additional evidence supporting the emergence of lepton type “strange” radia- tion at the instant of electric explosion of a conductor in a liquid was obtained [12]. The electric explosion of tita- nium foil in an uranium salt was shown to initiate a number of phenomena with still obscure physical mechanisms. It can be seen from the presented experi- mental results that all the observed effects are closely related to one another but the available experimental data do not allow their final separation into constituents. 7. Acknowledgements The authors wish to express their gratitude to staff members of the Russian Scientific Center Kurchatov Institute V. L. Kuznetsov and S. V. Zhukov for perform- ing numerous measurements. We are grateful to RE- KOM staff members and list the persons without whose active participation it would be just impossible to per- form the experiments: A. G. Volkovich, S. V. Smirnov, A. A. Gulyaev, A. P. Govorun, P. F. Strashko, V. L. Shevchenko, A. B. Gaverdovsky, and V. N. Bayushkin. We thank A. A. Rukhadze for useful discussion and for the support of these works. The experiments were carried out at the RECOM (an affiliate company of the I. V. Kurchatov Institute of Atomic Energy) at the Kurchatov Institute territory since 2002-2004. 8. References [1] L. I. Urutskoev and D. V. Filippov, “Beta-Stability Con- dition for the Nuclei of Neutral Atoms,” Physics-Uspekhi, Vol. 47, No. 12, 2004, pp. 1257-1260. [2] F. Bosch, T. Faestermann, J. Friese, F. Heine, P. Kienle, E. Wefers, K. Zeitelhack, K. Beckert, B. Franzke, O. Klepper, C. Kozhuharov, G. Menzel, R. Moshammer, F. Nolden, H. Reich, B. Schlitt, M. Steck, T. Stöhlker, T. Winkler and K. Takahashi, “Observation of Bound-State beta-Decay of Fully Ionized 187Re: 187Re-187Os Cosmo- chronometry,” Physical Review Letters, Vol. 77, No. 26, 1996, pp. 5190-5193. [3] M. Jung, F. Bosch, K. Beckert, H. Eickhoff, H. Folger, B. Franzke, A. Gruber, P. Kienle, O. Klepper, W. Koenig, C. Kozhuharov, R. Mann, R. Moshammer, F. Nolden, U. Schaaf, G. Soff, P. Spädtke, M. Steck, T. Stöhlker and K. Sümmerer, “First Observation of Bound-State Beta-De- cay,” Physical Review Letters, Vol. 69, No. 15, 1992, pp. 2164-2167. [4] D. V. Filippov, “Increase in the Probability of Allowed Electron Beta Decays in a Superstrong Magnetic Field,” Physics of Atomic Nuclei, Vol. 70, No. 2, 2007, pp. 258- 264. [5] D. V. Filippov, “Decrease in the Probability of Tritium Decay in an External Electric Field,” Physics of Atomic Nuclei, Vol. 70, No. 11, 2007, pp. 1840-1845. [6] D. V. Filippov, “Increase in the Probability of Forbidden Electron Beta Decays in a Superstrong Magnetic Field,” Physics of Atomic Nuclei, Vol. 70, No. 12, 2007, pp. 2016 -2024. [7] K. A. Kouzakov and A. I. Studenikin, “Bound-state Beta- Decay of a Neutron in a Strong Magnetic Field,” Physical Review C, Vol. 72, No. 1, 2005, p. 15502. [8] W. G. Chace and H. K. Moore, “Exploding wires,” Ple- num Press, New York, 1962. [9] Yu. L. Bakshaev, P. I. Blinov, V. V. Vikhrev, E. M. Gordeev, S. A. Dan’ko, V. D. Korolev, S. F. Medovsh- chikov, S. L. Nedoseev, E. A. Smirnova, V. I. Tumanov, A. S. Chernenko and A. Yu. Shashkov, “Study of the Plasma in a Preformed Z-pinch Constriction,” Plasma Physics Reports, Vol. 27, No. 12, 2001, pp. 1039-1047. [10] D. V. Filippov, L. I. Urutskoev, G. Lochak and A. A. Rukhadze, “On the Possible Magnetic Mechanism of Shortening the Runaway of RBMK-1000 Reactor at Chernobyl Nuclear Power Plant,” in Condensed Matter Nuclear Science, J. P. Biberian, Ed., World Scientific Publishing Co., Singapore, 2006, pp. 838-853. [11] A. G. Volkovish, A. P. Govorum, A. A. Gulyaev, S. V. Zhukov, V. L. Kuznetsov, A. A. Rukhadze, A. V. Ste- blevskii and L. L. Urutskoev, “Experimental Observation of the Distortion of The Uranium Isotopic Relationship and Violation of the Thorium-234 Secular Equilibrium Upon Electric Explosion,” Annales de la Fondation Louis  L. I. URUTSKOEV ET AL. Copyright © 2010 SciRes. JMP 235 de Broglie, Vol. 30, No. 1, 2005, pp. 63-70. [12] L. I. Urutskoev, V. I. Liksonov and V. G. Tsinoev, “Ob- servation of Transformation of Chemical Elements during Electric Discharge,” Annales de la Fondation Louis de Broglie, Vol. 27, No. 4, 2002, pp. 701-726. [13] N. G. Ivoilov and L. I. Urutskoev, “The Influence of “Strange” Radiation on Mössbauer Spectrum of Fe57 in Metallic Foils,” Annales de la Fondation Louis de Broglie, Vol. 29, No. Hors ser. 3, 2004, pp. 1177-1186. [14] G. I. Ramendik, “Elemental Mass-Spectrometric Analysis of Solids,” Khimiya, Moscow, 1993 [in Russian]. [15] D. V. Filippov and L. I. Urutskoev, “On the Possibility of Nuclear Transformation in Low-Temperature Plasma from the Viewpoint of Conservation Laws,” Annales de la Fondation Louis de Broglie, Vol. 29, No. Hors ser. 3, 2004, pp. 1187 -1206. [16] V. D. Kuznetsov, G. V. Mishinsky, F. M. Penkov, V. I. Arbuzov and V. I. Zhemenik, “Low Energy Transmuta- tion of Atomic Nuclei of Chemical Elements,” Annales de la Fondation Louis de Broglie, Vol. 28, No. 2, 2003, pp. 173- 214. [17] D. Priem, G. Racineux, G. Lochak, C. Daviau, D. Fargue, M. Karatchentzeff and H. Lehn, “Explosion électrique d’un fil de titane dans de l’eau en milieu confiné,” An- nales de la Fondation Louis de Broglie, Vol. 33, No. 1-2, 2008, pp. 129-138. [18] V. A. Erma, “Electron Effects on Barrier Penetration,” Physical Review, Vol. 105, No. 6, 1957, pp. 1784-1787. [19] B. B. Kadomtsev and V. S. Kudryavtsev, “Atoms in a Superstrong Magnetic Field,” JETP Letters, Vol. 13, No. 1, 1971, pp. 42-44. [20] R. P. Taleyarkhan, C. D. West, J. S. Cho, R. T. Lahey Jr., R. I. Nigmatulin and R. C. Block, “Evidence for Nuclear Emissions During Acoustic Cavitation,” Science, Vol. 295, No. 5561, 2002, pp. 1868-1873. [21] F. Cardone, R. Mignani and A. Petrucci, “Piezonuclear Decay of Thorium,” Physics Letters A, Vol. 373, No. 22, 2009, pp. 1956-1958. |