Health
Vol.5 No.10A(2013), Article ID:38887,21 pages DOI:10.4236/health.2013.510A1003
Aflatoxins metabolism, effects on epigenetic mechanisms and their role in carcinogenesis
![]()
1Department of Pharmacology and Therapeutics, Makerere University College of Health Sciences, Kampala, Uganda; *Corresponding Author: godfossa@yahoo.com
2Department of Surgery, Mbarara University of Science & Technology Medical School, Mbarara, Uganda
Copyright © 2013 Godfrey S. Bbosa et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 6 September 2013; revised 8 October 2013; accepted 15 October 2013
Keywords: Aflatoxins; Epigenetic Mechanism; CYP450; Metabolism; Hepatocellular Carcinoma
ABSTRACT
Chronic consumption of aflatoxin-contaminated foods is a global problem in both developing and developed countries especially where there is poor regulation of their levels in foods. In the body, aflatoxins (AFBs) mainly AFB1 are biotransformed to various metabolites especially the active AFB1-exo-8,9-epoxide (AFBO). The AFB, AFBO and other metabolites interact with various biomolecules in the body including nucleic acids such as DNA and RNA and the various metabolic pathways such as protein synthesis, glycolytic pathway and electron transport chain involved in ATP production in body cells. The AFB interacts with DNA to form AFBDNA adducts causing DNA breakages. The AFB and its metabolites induce the up regulation of nuclear receptors such as pregnane X receptor (PXR), constitutive androstane receptor (CAR), and aryl hydrocarbon receptor (AhR) through gene expression that regulates the metabolizing enzymes such as CYP450 involved in Phase I and Phase II metabolism of xenobiotics. AFB activates these nuclear receptors to produce the metabolizing enzymes. The AFB1 is metabolized in the body by cytochrome P450 (CYP450) enzyme isoforms such as CYP1A2, CYP1A2, CYP3A4/ CYP3A5, and CYP3A7 in fetus, glutathione Stransferase, aflatoxin B1-aldehyde reductase leading to reactive metabolites, some of which can be used as aflatoxin exposure biomarkers. These enzymes are involved in the Phase I and Phase II metabolic reactions of aflatoxins. The CYP1A2 is the principal metabolizer of aflatoxin at low concentrations while the reverse is true for CYP3A4. The accumulation of AFB and its metabolites in the body especially the AFB1-exo- 8,9-epoxide depletes the glutathione (GSH) due to the formation of high amounts of epoxides and other reactive oxygen species (ROS). The AFB, AFB1-exo-8,9-epoxide and other metabolites also affect the epigenetic mechanisms including the DNA methylation, histone modifications, maturation of miRNAs as well as the daily formation of single nucleotide polymorphism (SNP) where AFB exposure may facilitate the process and induces G:C to T:A transversions at the third base in codon 249 of TP53 causing p53 mutations reported in hepatocellular carcinoma (HCC). The changes in epigenetic mechanisms lead to either epigenetic inactivation or epigenetic derepression and all these affect the gene expression, cellular differentiation and growth. AFB also through epigenetic mechanisms promotes tumorigenesis, angiogenesis, invasion and metastasis in hepatocellular carcinoma. However, the formation of the small amounts of AFB1 from AFB2 is suspected to cause the carcinogenicity of AFB2 in humans and animals. Chronic aflatoxins exposure leads to formation of reactive AFBO metabolites in the body that could activate and de-activates the various epigenetic mechanisms leading to development of various cancers.
1. INTRODUCTION
Aflatoxins are a group of mycotoxins that are derived as fungal metabolites. They are naturally occurring biochemical substances produced from different species of fungi especially Aspergillus species [1]. They commonly contaminate cereals and grains such as rice, maize, sorghum, millet, groundnuts, dried cassava and many others during the storage and poor processing conditions [1,2]. Aflatoxin-food contamination is a global problem especially in the tropical and subtropical regions of the world where warm temperatures and humidity favor the growth of the fungi. They are well known carcinogens especially aflatoxin B1 (AFB1) to humans and animals. There are about 20 aflatoxins related fungal metabolites produced mainly by Aspergillus flavus, A. nominus and A. parasiticus species of fungi [3]. They belong to difuranocoumarins with two important chemical structure series including 1) difurocoumarocyclopentenone series (AFB1, AFB2, AFB2A, AFM1, AFM2, AFM2A and aflatoxicol) and 2) difurocoumarolactone series (AFG1, AFG2, AFG2A, AFGM1, AFGM2, AFGM2A and AFB3) (Table 1) [2,4]. The “B” and “G” refer to the blue and green fluorescent colors produced by aflatoxins under UV light on thin layer chromatography plates, while the subscript numbers 1 and 2 indicate major and minor compounds, respectively. The M designations of M1 and M2 are metabolites of AFB1 and AFB2 respectively that appear in body fluids [2,4].
2. BIOTRANSFORMATION OF AFLATOXINS
In the body, aflatoxins undergo biotransformation mainly in the liver [5]. Biotransformation (metabolism) is the process whereby a chemical substance is changed from one chemical form to another (transformed) by a series of enzymatic or chemical reaction(s) within the body and the eventual excretion of the byproducts or metabolites mainly through renal excretion. Biotransformation in toxicology plays an important defense mechanism in the elimination of the toxic xenobiotics and body wastes in which they are converted into less harmful and polar substances that can easily be excreted [5-7]. The process of elimination (metabolism and excretion) of chemical substances from the body involves two main phases. Phase I metabolism of chemical substances that involves the addition of a small polar group containing both positive and negative charges that are added to the xenobiotics like aflatoxins by the process of oxidation,
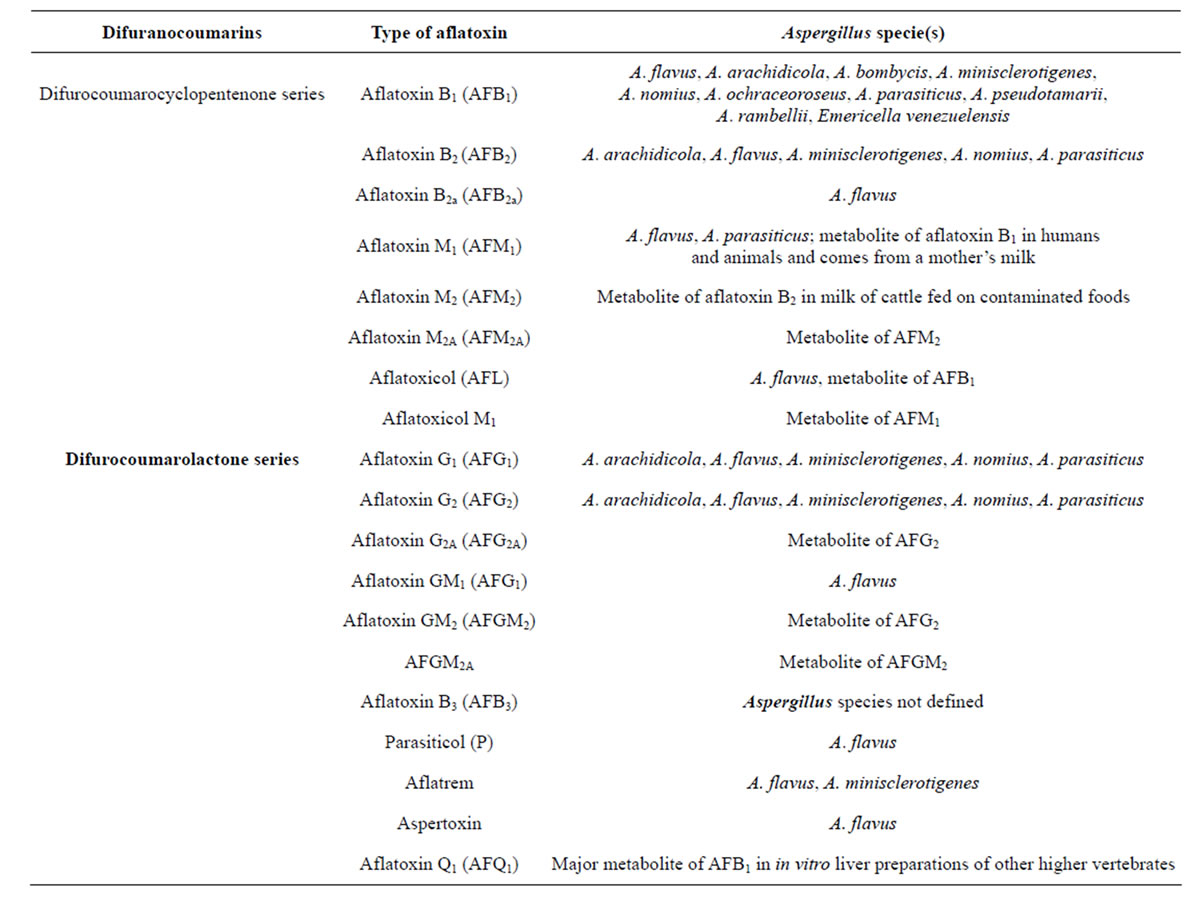
Table 1. Summary of the major aflatoxins produced by the Aspergillus species of Moulds [2,4].
reduction, acetylation and hydrolysis and render it harmless [8]. The process allows the product of Phase I product to “fit’’ into Phase II enzyme system where they become conjugated (joined together) with another substance to produce a polar or water soluble substance that can easily be excreted by the kidney [5,8]. However in some cases, some of the chemical substances may be converted into reactive or harmful products. Phase I is mostly mediated by the Cytochrome P450 (CYP450) enzyme systems. Phase II metabolism involves sulfate, glucuronide, glutathione and amino acid conjugation reactions [5,9].
2.1. Phase I Metabolism of Aflatoxins
Aflatoxins undergo Phase I metabolism by oxidation reactions including epoxidation, hydration, hydroxylation and O-demethylation reactions involving the CYP 450 mainly in the liver to produce AFB1-exo-8,9-epoxide (AFBO), AFB2a, AFM1, AFQ1 and AFP1 that are excreted in bile and urine after conjugation (Figure 1) [5,8-10].
Cytochrome P450s (CYP450s) are a large superfamily of heme-binding enzymes involved in the synthesis and metabolism of endogenous substrates as well as in the biotransformation of xenobiotics like aflatoxins [5,7,11, 12]. The aflatoxin B1 is metabolized in the body by CYP1A2, CYP1A2, CYP3A4/CYP3A5, and CYP3A7 in the fetus [7,11,13,14]. The aflatoxins are metabolized in the liver, mainly by CYP1A2 and CYP3A4 isoenzymes by the initial two electron transfer oxidation reactions [7,13,15]. The CYP3A4 metabolizes AFB1 to its reactive intermediates AFB1-exo-8,9-epoxide and then to aflatoxin Q1 (AFQ1), a less toxic metabolite. Other oxidized metabolites include hydroxylated forms, aflatoxin M1 (AFM1), the O-demethylated form and aflatoxin P1 (AFP) (Figure 2 and Table 1). The AFM, AFP and AFQ are less toxic with reduced carcinogenic potential though AFM retains some carcinogenic activity. The AFB1-exo-8,9- epoxide reacts with the N7 atom of guanine to form a pro-mutagenic DNA adduct (aflatoxin-N7-guanine). The aflatoxin-DNA adduct is unstable and undergoes depurination, leading to its urinary excretion. However, CYP3A4 is reported to have a relatively low affinity for AFB1 epoxidation and is primarily involved in AFB, detoxification through AFQ1 formation [6]. The CYP1A2 metabolises AFB1 to AFM1 and the AFB1-exo-8,9-epoxide. The CYP1A2 is considered as the main effective route of metabolism of aflatoxins [6,16]. CYP1A2 metabolism of AFB leads to formation of some exo-epoxide and a high proportion of endo-epoxide and AFM1 [13,16]. The expression of CYP1A2 and CYP3A4 in the metabolism of aflatoxins depends on affinity and level of expression in the liver. The CYP1A2 has high-affinity for the bioactivation of AFB1 at low substrate concentrations following dietary exposure [13,16]. The AFB1-exo-8,9-epoxide produced by CYP1A2 has a mixture of exoisomer and endoisomer AFBO, with the exoisomer of the epoxide causing genotoxicity. Some of the AFB intermediates undergo further metabolism in Phase II by conjugation with glutathione to produce the polar and less toxic metabolites that are easily excreted in urine and bile. However, the AFBO and AFB1-dihydroxide intermediates causes carcinogenicity while AFB2a causes acute toxicity, liver necrosis and cellular metabolizing enzyme inhibition [8]. The AFB1-8,9-epoxide metabolite form adducts with amino acids and DNA [17]. The DNA adducts are fairly resistant to DNA repair processes and this causes gene mutation and hence the development of cancers especially the hepatocellular carcinomas. CYP3A5 also metabolizes AFB1, mainly to the exo-8,9-epoxide with much less efficient formation of the detoxification product, AFQ1. Aflatoxin cross the placenta and found in cord blood of newborns where it is metabolized to toxic and mutagenic metabolites AFBO by CYP3A7, a major CYP450 in human fetal liver [13,15]. However, some other aflatoxin metabolites other than AFBO, as well as the naturally occurring aflatoxins such as AFG1, AFB2 and AFG2 are poor substrates for epoxidation and there-
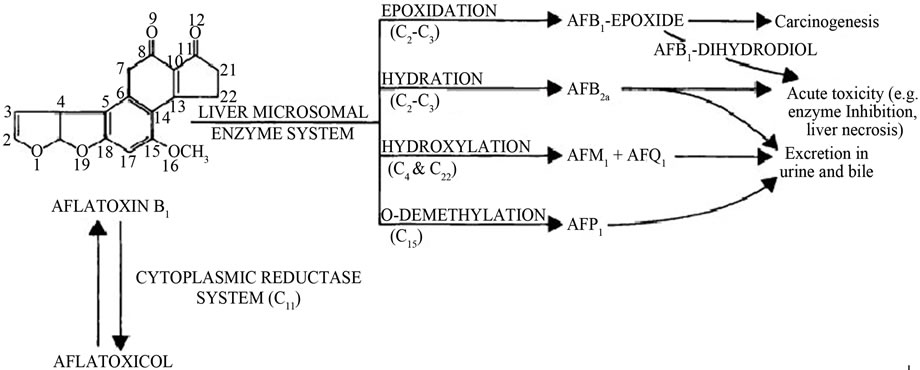
Figure 1. Metabolism of aflatoxin in liver (Adopted from Dhanasekaran, 2011) [8,10].
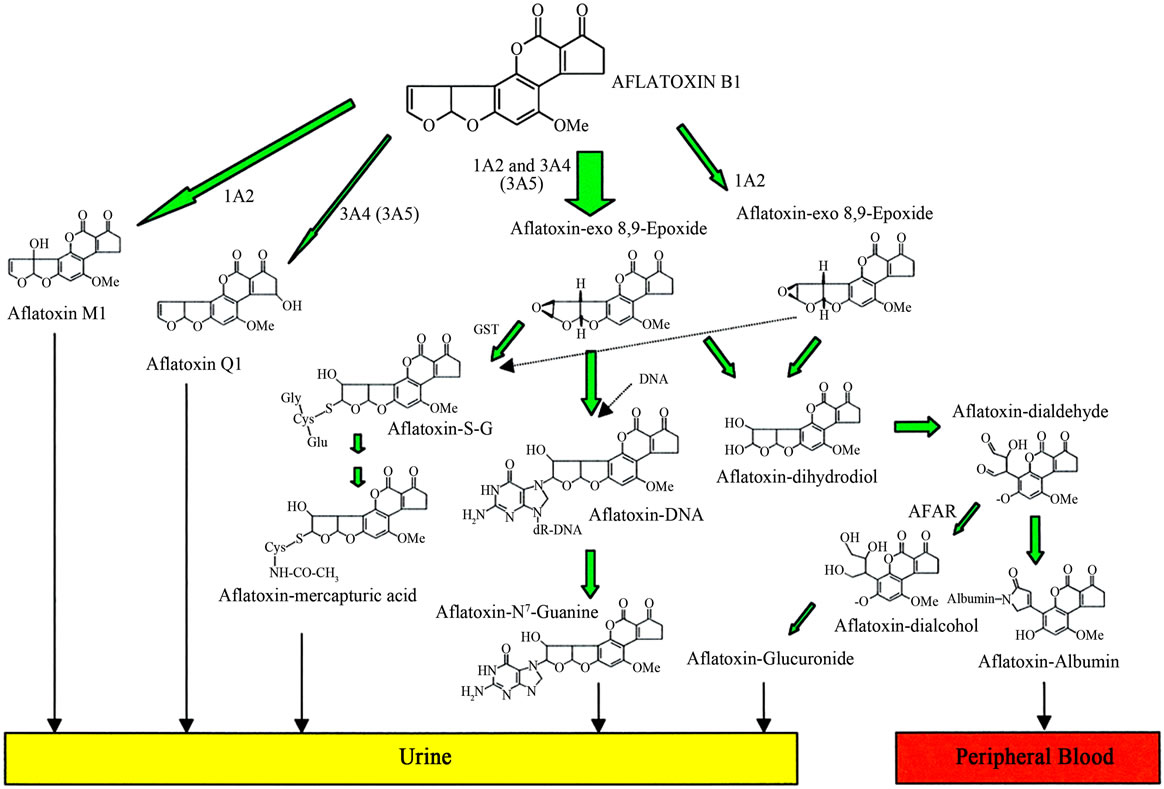
Figure 2. Principle metabolism of aflatoxin B1 leading to reactive metabolites and biomarkers. 1A2, CYP1A2; 3A4, CYP3A4; 3A5, CYP3A5; GST, glutathione S-transferase; AFAR, aflatoxin aldehyde reductase; Aflatoxin-S-G, aflatoxin-glutathione conjugate (Adopted from Wild and Turner, 2002) [13].
fore less mutagenic, carcinogenic and toxic than AFB1 [13,15].
2.2. Effects of AFB on Nuclear Receptors and Induction of CYP450 Enzymes in Aflatoxin Metabolism
During species evolution, living organisms protect themselves from foreign substances (xenobiotics) such as drugs, poisons, toxins and nutrients by use of nuclear receptors such as AhR (aryl hydrocarbon receptor, PXR (pregnane X receptor and CAR (constitutive androstane receptor) that act as sensors (xenosensors) of the chemical substances and the xenobiotic metabolizing and transporter systems [78,79]. Chronic ingestion of aflatoxincontaminated foods leads to increased AFB concentrations especially AFB1 in the body that leads to activation of the synthesis of messenger RNA (mRNA). This increases messenger RNA (mRNA) expression of the nuclear receptors such as pregnane X receptor (PXR), constitutive androstane receptor (CAR), and aryl hydrocarbon receptor (AhR) [18-21]. The PXR and CAR nuclear receptors are critical in protecting the body against environmental chemicals like the xenobiotics. PXR and CAR are activated by a wide range of xenobiotics and regulate cytochrome P450 and other genes whose products are involved in the detoxification of these chemicals [21]. The expression of these receptors leads to the induction of some of their target cytochromes (CYPs) that are involved in the metabolism of xenobiotics like aflatoxins [18,21]. A number of xenobiotics are known to be inducers or inhibitors of CYPs. The induction of CYPs utilizes receptor-mediated mechanisms involving important nuclear receptors such as the PXR, the constitutive CAR and the AhR that increases gene transcription and hence the induction of CYP3A, CYP2B, and CYP1A subfamily isoforms [18-21]. The PXR and CAR forms two important members of the nuclear receptors 1 (NR1) of nuclear receptor family that were previously known as the xenobiotic receptors. They are the master-regulators of the phase I and II chemical substance-metabolizing enzymes as well as their uptake and transportation in the body to prevent their accumulation leading to toxicity. The AhR activation has been reported to control the expression of CYP1A1 and CYP1A2 isoenzymes involved in the metabolism of chemical substances. Its activation by various toxicants has been reported to induce disturbances in cell signaling pathways that regulate cell proliferation, differentiation, apoptosis, immune response and carcinogenesis. Aflatoxin B1 has been reported to induce an overexpression of CYP1A1 and CYP1A2 genes and their mRNAs accompanied by an increase in AhR mRNA expression [18,21]. The AFB1 exposure also has been reported to induce PXR and CAR mRNA by increasing PXR and CAR gene levels [18,21]. The chronic exposure of the individual to AFB1, therefore induce an upregulation of PXR, CAR, and CYP2B6, CYP3A5, CYP2C9, and CYP3A4 genes. The induction of CYP3A, CYP2B, and CYP2C predominantly takes place via the PXR and/or CAR nuclear receptor [18,21]. The CYP3A5 and CYP2B6 isoenzymes and their mRNAs have been reported to be over expressed and more induced by noncytotoxic increasing concentrations of AFB1 in a concentration-dependent manner more than the CYP3A4 and CYP2C9 isoenzymes and their mRNAs [14,18-21]. The induction of these CYP450 isoenzymes and over expression of CYP1A1, CYP1A2, CYP2B6, CYP3A5, CYP2C9, and CYP3A4 and the over expression of AhR, PXR and CAR nuclear receptors facilitate the Phase I and Phase II biotransformation and elimination of the aflatoxins in the body though there may be individual variation (toxicogenetic effects) that may influence the individual’s development of carcinogenicity related to consumption of aflatoxin-contaminated foods [18-21].
2.3. Phase II Metabolism of Aflatoxins: Role of Glutathione Conjugation in Body Detoxification of Aflatoxins
The AFB metabolites of Phase I metabolism undergo Phase II enzymatic metabolism by glutathione S-transferases (GST), that primarily catalyze conjugation reactions. Glutathione (GSH) is regarded as the body’s master antioxidant with three amino acids-cysteine, glycine, and glutamate [8,9,22]. It is found in almost all body cells with the highest concentration found in the liver where it plays a critical role in the body’s detoxification process. Glutathione is also an essential component of the body’s natural defense system. GSH is used as a cofactor by 1) multiple peroxidase enzymes involved in detoxification of peroxides generated from oxygen free radical attack on biological molecules; 2) transhydrogenases that reduce oxidized centers on DNA, proteins, and other biomolecules; and 3) glutathione S-transferases (GST) that conjugate GSH with endogenous substances like estrogens, exogenous electrophiles like aflatoxins and its metabolites, and other various xenobiotics. GSH act as antioxidant and has many functions in membrane maintenance and stability as well as in reducing oxidative stress factors and the high reactive oxygen species (ROS) produced from the metabolic process of lipid peroxidation [17,23,24]. However, the increased depletion of GSH leads to abnormally high levels of ROS in cells. The depletion is also affected by aflatoxin due to uncoupling of metabolic processes due to the lack of GSH for GSH-peroxidase catalysis of O2 to H2O2 thus affecting the integrity of the cell membranes. Its reduction further enhances the damage to critical cellular components (DNA, lipids, proteins) by the AFB-8,9-epoxides that form adducts [10,14,24]. Aflatoxin-B1-8,9-oxide is also a substrate for several isoforms of human glutathione S-transferases (GSTs), which yield a stable, nontoxic, polar product that is excreted in the bile. The aflatoxin-glutathione product also undergoes sequential metabolism in the liver and kidneys in which its excreted as a mercapturic acid (aflatoxin-N-acetylcysteine) in urine [14,25] (Figures 2 and 3).
Glutathione S transferases (GST) are a large family of metabolic enzymes comprised of several members, such as alpha, mu, pi, theta, and others [26-29]. GST in the alpha and pi classes are abundant in hepatocytes and are inducible by exogenous chemical substances and carcinogens like the aflatoxins [14,24]. GSTs catalyze nucleophilic attack by reduced glutathione (GSH) on nonpolar compounds that contain an electrophilic carbon, nitrogen, or sulphur atom. There are three major families of proteins with glutathione transferase activity including 1) cytosolic GST, 2) mitochondrial GST and 3) microsomal GSTs, that are membrane-associated proteins used in eicosanoid and glutathione (MAPEG) metabolism [26- 29]. The cytosolic and mitochondrial GST are soluble enzymes with some resemblances in their 3-dimensional fold though they are distantly related. They are structurally different from the MAPEG enzymes but all contain substrates that conjugate GSH with peroxidase activity (Figure 4) [26-29]. A number of GST sequence polymorphisms exists and include GSTA1, GSTA2, GSTM1, GSTM3, GSTM4, GSTO1, GSTO2, GSTP1, GSTT2, GSTZ1), deletion polymorphisms (GSTM1, GSTT1), and duplication polymorphisms (GSTM1). The nomenclature for MAPEG enzymes include LTC4S, leukotriene C4 synthase (conjugates leukotriene A4 with GSH); FLAP, 5-lipoxygenase-activating protein (arachidonic acid-binding protein required for 5-lipoxygenase to exhibit full activity); PGES1, prostaglandin E2 synthase 1 (catalyzes GSH-dependent isomerization of PGH2 to PGE2) (Figure 4) [22,26-29]. The GSTs are induced by chemical substances such as aflatoxins, butyrate, a product of gut flora-derived from fermentation of plant foods by increasing histone acetylation [22]. Also, the human microsomal epoxide hydrolase (mEH) may also participate in the detoxification of AFBO in the absence of significant GST activity [14,30,31].
2.4. Detoxification of AFB1 in the Extra-Hepatic Organs and Tissues
In the liver hepatocytes, cytoplasmic reductase en-
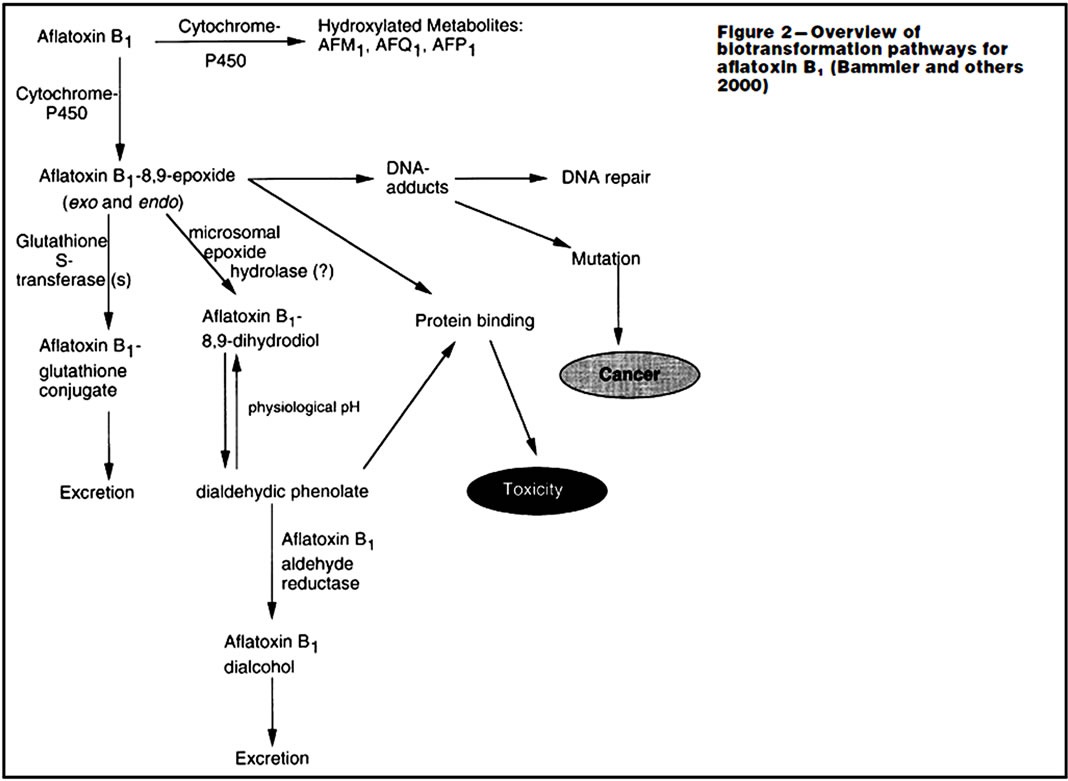
Figure 3. Metabolism and excretion of aflatoxin in liver (Adopted from Dhanasekaran, 2011; Eaton and Gallagher, 1994) [8,14,25,75].
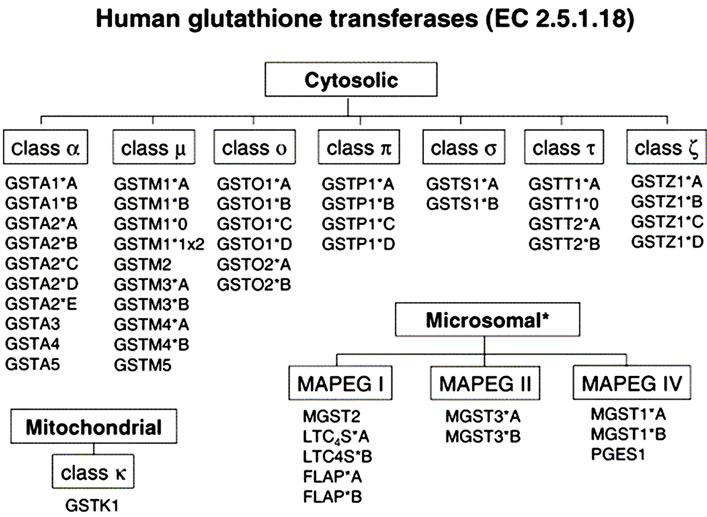
Figure 4. Genes encoding human glutathione transferases used in Phase II metabolism of xenobiotics (EC 2.5.1.18) (Adopted from Hayes et al.) [26]. (*) A higher number of single nucleotide polymorphisms reviewed for individual population [22,26].
zymes especially aflatoxin B1-reductase enzyme also metabolize and convert AFB1 to various AFB metabolites such as aflatoxicol, AFM1, AGFQ1, AFP1 and AFB1-epoxide [11,13,24,75]. The AFBO are carcinogenic while other metabolites are less toxic and are conjugated with other molecules to enhance their rapid elimination in urine [9]. The metabolite AFQ1 has very little cancercausing potential and they are usually excreted in urine with little effect on the body. Aflatoxins are also metabolized extra-hepatically in the enterocytes of the small intestinal epithelium that contain high levels of CYP3A enzymes and this possibly reduces their systemic absorption [11,13]. The lipoxygensase and prostaglandin H synthase also provides an important route of aflatoxin metabolism in other extra-hepatic organs [11,13].
2.5. Metabolic Differences and Carcinogenic Potential of AFB1, AFG1, AFM1 and AFB2
The induction of cancer by aflatoxins has been reported in both humans and animals globally and in experimental studies in various species of animals have provided evidence of the carcinogenic potential of aflatoxins. The carcinogenicity of aflatoxins has been classified based on the evidence observed in experimental animals as “sufficient evidence for carcinogenicity of naturally occurring AFB1, AFG1 and AFM1, limited evidence for AFB2 and inadequate evidence for AFG2” [73]. The AFB1 is considered the main potent chemical of all aflatoxins in inducing cancers especially the liver cancers such as the hepatocellular carcinoma (HCC) and AFB2 being the least potent hepatic carcinogen [73,74]. The carcinogenic capability and toxicity of AFB1, AFG1, AFM1, AFB2 and AFG2 have been linked to the differences in the aflatoxins biotransformation pathways in the various species of animals including humans, rats, mice, ducks and many other animals [74-77]. AFB1 undergo microsomal oxidation in the liver to form reactive AFB1- 8,9-epoxide (AFBO, also referred to as AFB-2,3-epoxide in older literature) to exert its carcinogenic effects to the liver especially the HCC [75]. The carcinogenic potency of AFB1 has been highly correlated to the extent of the covalent binding of AFBO, the metabolite of AFB1 to the cellular DNA, its capability of activating epigenetic mechanisms and the suppression of P53 genes (tumor suppressor genes) [74-77]. The formation of AFBO has been reported to be an important critical determinant of the differences in the susceptibility of aflatoxins to cancers especially the HCC in the various species of animals [75,76]. In ducks, they are susceptible to AFB2 as compared to other species of animals [74-77]. AFB2 causes liver toxicity to ducks and this is due to the ability of the ducks’ liver to biotransform AFB2 into AFB1 which is then further activated by further metabolic reactions through epoxidation pathway as AFB1 [74-77]. AFB2 is activated in the liver by 2,3 desaturation to AFB1 [76,77]. In humans and rodents, the desaturation of AFB2 in the liver does not occur and this reduces its carcinogenic potential by more than 150 times as compared to the AFB1 in humans and rodents [76,77]. In humans and other animals, AFB1 is activated in the liver leading to the formation of the AFB1-2,3-epoxide which is a potent carcinogenic chemical compound. AFB2 does not undergo the same metabolic pathway as AFB1 due to the absence of the 2,3-double bond in the compound. The biotransformation of AFB2 to AFB1-2,3-epoxide occurs through the intermediate formation of AFB1 by desaturation at the 2,3-carbons of AFB2 [74-77]. And therefore, the ratio of nucleic acid adducts formed from the two compounds (AFB1 and AFB2) is reported to be similar to the carcinogenic potencies of the two aflatoxins to approximately 1:100 (AFB2:AFB1) [76,77]. However, the formation of the small amounts of AFB1 from AFB2 is suspected to be the cause of the carcinogenicity of AFB2 in humans and animals. AFB1, AFM1, and AFG1 are reported to cause various types of cancer in different animal species. However, the difference in the carcinogenicity of these aflatoxins depends on their intercellular variation in the formation of adduct levels in DNA and other biomolecules in the body; with AFB1 the most potent. AFG1 binds to DNA and produces chromosomal aberrations in rodents treated in vivo. AFB1 mainly causes liver cancer in humans and rodents while AFG1 in addition cause cancers of the kidney in rodents experimentally.
3. MOLECULAR TARGETS OF AFLATOXIN IN THE BODY
After consumption of aflatoxin-contaminated foods by individuals, aflatoxins and their metabolites targets and interact with many different biomolecules, organs and tissues. There are two types of interaction recognized especially with nucleic acids involving non-covalent, weak and reversal binding and the other involving irreversible covalent binding requiring mammalian metabolizing enzyme systems. Aflatoxins targets protein synthesis pathways especially the DNA template, RNA template (mRNA, tRNA, rRNA), proteins, transcription, translation and cellular metabolic reactions (Figure 5) [10,24,32-34].
3.1. Nucleic Acids as AFB Targets (DNA and RNA)
AFB1 is metabolically activated by cytochrome P450 enzymes in the liver to its reactive metabolite, AFB1- 8,9-epoxide (AFBO), which binds to macromolecules resulting in formation of adducts such as DNA adduct leading to mutations and carcinogenesis [2,10,14,24,33, 35-38]. The AFB1-8,9-epoxide can further be converted to AFB1-8,9-diol that specifically binds to lysine in albumin and form AFB1-lysine adducts (AFB-Lys), which has been validated as a biomarker of human exposure to aflatoxins [2,14,33,35-38]. AFBO metabolites are highly

Figure 5. Targets of Mycotoxins (aflatoxins) and the major target sites in DNA and protein synthesis (Adopted from Kiessling, 1986) [10,33].
reactive and it covalently binds to cellular biomolecules and forms a long-lived lysine adducts with serum albumin and the promutagenic adducts with DNA, damaging it as well as impairing the DNA repair enzymes [2,10,14, 33,35-38]. The major DNA damage is the formation of the AFB-N7-guanine adduct in its structure, which is chemically unstable and undergoes rapid urinary excretion following depurination. Also the AFB-N7-guanine adduct may be stabilized by rearrangement to form a ring-opened formamidopyramidine structure. And if AFBDNA-adducts is not repaired by DNA repair enzymes, somatic alterations may develop especially if they are localized in transcriptionally active DNA regions ([2,14, 33,35-38]).
3.2. Mitochondrial Structure as Target of Aflatoxins
AFB and AFBO causes ultrastuctural changes in mitochondria [2,39]. The reactive aflatoxin-8,9-epoxide preferentially binds to mitochondrial DNA (mitDNA) during hepatocarcinogenesis as compared to nuclear DNA that hinder ATP production and FAD/NAD-linked enzymatic functions. This causes the disruption of mitochondrial functions in the various parts of the body and hence ATP formation [24,34]. Aflatoxin-associated damage to mitochondria may be responsible for aging processes [2, 14,33,35-38]. The AFB1 also binds to DNA to cause its structural alterations. The damage to mitDNA is caused by adduction and mutations of mitochondrial membranes leading to increased cell death (apoptosis) as well as disruption of energy production [2,24].
3.3. Cell Division as Aflatoxin Target
The aflatoxins and its metabolites especially the AFBO affects the telomere length during the cell division and the various check point in the cell cycle thus effecting the signaling pathways and the regulatory processes of the cell cycle [2]. Also the extent of aflatoxin binding to DNA and its damage, affects the level of different protein synthesis in the cell cycle and the apoptotic pathways such as c-Myc, p53, pRb, Ras, protein kinase A (PKA), protein kinase C (PKC), Bcl-2, NF-kB, CDK, cyclins and CKI that contribute to the life or death decision making process of the cell (apoptosis). They may also contribute to the deregulation of the cell proliferation leading to carcinogenesis [2]. The reactive aflatoxin- 8, 9-epoxide can affect the mitotic (M) phase, growth process (G1 and G2 phase) and DNA synthesis (S phase) in the cell cycle leading to carcinogenesis [2].
3.4. Protein Synthesis Pathways as Target of Aflatoxin
Mycotoxin especially aflatoxins leads to induced abnormal protein effects that may be due to primary and/or to secondary disruption of nucleic acid or protein synthesis [34]. Aflatoxins after activation to reactive AFBO, binds to biological molecules including essential enzymes, blocks RNA polymerase and ribosomal translocase thus inhibiting protein synthesis [75]. They also form DNA adducts and therefore affecting protein synthesis. The aflatoxin binds and interferes with enzymes and substrates that are needed in the initiation, transcription and translation processes involved in protein synthesis. They interacts with purines and pyrimidine nucleosides causing impairment of protein synthesis by forming adducts with DNA, RNA and proteins [24,32-34,40]. Aflatoxin also inhibits RNA synthesis by interacting with the DNA-dependent RNA polymerase activity and degranulation of endoplasmic reticulum. Also the reduction in protein content in body tissues like in skeletal muscle, heart, liver and kidney has been associated with the increased liver and kidney necrosis due to the damage caused by the accumulated AFB and its metabolites in the body following the aflatoxin exposure [2]. AFB1 is a potent mutagenic, carcinogenic, teratogenic, and immunosuppressive toxins that may interfere with normal process of protein synthesis as well as inhibition of several important metabolic pathways of the vital organs like the liver, kidney and heart [41]. Aflatoxins may also interfere with the flow of genetic information from DNA to RNA and to protein. It also affects the epigenetic mechanisms such as DNA methylation like methyl-cytosine, histone modifications, and chromatin remodeling thus affecting the cellular signaling pathways, cell proliferation and growth (Figure 6) [40,42].
3.5. Effects of Aflatoxins on Oxidative Phosphorylation of Carbohydrate Metabolism
The effect of mycotoxins especially aflatoxins on carbohydrate metabolism is due to reduced hepatic glycogen and increased blood glucose levels. Aflatoxin B1 and cyclochlorotine, a secondary metabolite of the fungus Penicillium islandicum that causes hepatic necrosis and has carcinogenic properties have been reported to have an effect on oxidative phosphorylation pathways. They also interfere with the cellular metabolism of glucose [33]. They both inhibit glycogen synthesis by decreasing glycogen synthetase and transglycosylase enzyme activities that catalyse elongation and rearrangement of the glycogen molecules [33]. AFB1 decreases the activity of phosphoglucomutase that reversibly converts glucose-6- phosphate into glucose-1-phosphate. It also reduces hepatic glycogen by accelerating glucose-6-phosphate oxidation (Figure 7) [33].
3.6. Effects of Aflatoxins on Electron Transport Chain in Oxidative Phosphorylation of Carbohydrate Metabolism
Aflatoxin B1 inhibits electron transport in mitochondria at both ADP-coupled and dDNP-uncoupled levels. It inhibits electron transport at the cytochrome oxidase level at site II and the inhibition occurs between cytochrome b and c, though the inhibition can be reversed by the electron acceptor N,N,N’,N’-tetramethylphenylenediamine (TMPD) (Figure 8) [33].
4. EFFECTS OF AFLATOXINS ON EPIGENETIC MECHANISMS
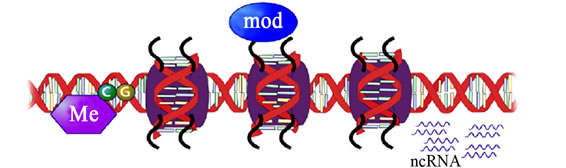
Epigenetics is the study of changes in gene expression or cellular phenotype, caused by mechanisms such as chromatin remodeling, non-coding RNAs (microRNAs), DNA methylation and histone modification that serve to regulate gene expression without altering the underlying DNA sequence or nucleotide sequence (Figures 9 and 10) [43-49]. These mechanisms affect protein synthesis required in gene expression (Figure 11 [14]). Gene expres-
Figure 6. Targets of xenobiotics like aflatoxins on various sites of protein synthesis and their effects on genetic and epigenetic mechanisms (Adopted from Li et al., 2011) [40,42].
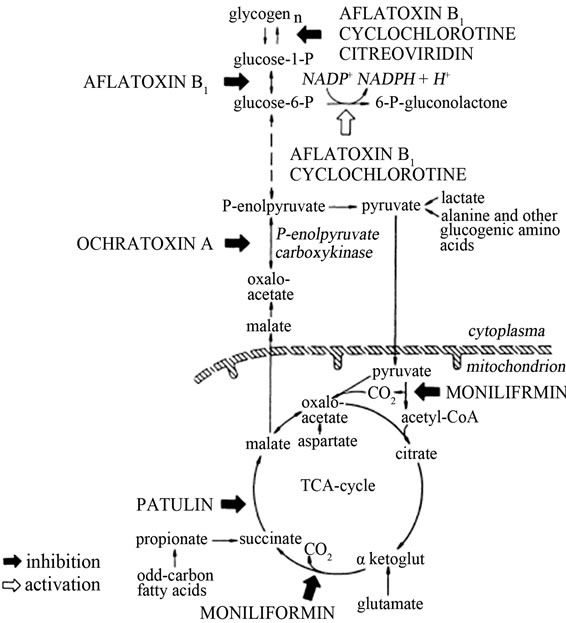
Figure 7. Aflatoxins inhibits enzymes involved in oxidative phosphorylation (Adopted from Kiessling, 1986) [33].
sion can be controlled through the actions of repressor proteins that attach to silencer regions of the DNA. These changes may remain all through cell divisions, for the rest of the cell’s life and may also last for multiple generations. Specific epigenetic processes involved in epigenetic mechanisms include paramutation, bookmarking, imprinting, gene silencing, X chromosome inactivation, position effect, reprogramming, transvection, maternal effects, and the progress of carcinogenesis, teratogenic effects, regulation of histone modifications and heterochromatin, and technical limitations affecting parthenogenesis and cloning effects. All these processes may be affected by aflatoxins exposure especially during a prolonged period of exposure [43-48].
4.1. DNA Methylation
DNA methylation is a biochemical process that involves addition of a methyl group to the cytosine or adenine of the DNA nucleotides to the 5’ position of the cytosine on pyrimidine ring or the number 6 nitrogen of the adenine purine ring. DNA methylation represses transcription mainly at Cytosine-phosphate-Guanine (CpG) rich “islands” where most promoters are embedded [45, 46,50-53]. DNA methylation stably alters the expression of genes in dividing cells and during the cell differentiation from embryonic stem cells into specific tissues. Some DNA methylation modifications that regulate gene expression are inheritable and cause genomic imprinting. It suppresses the expression of endogenous retroviral genes and other harmful stretches of DNA that have been
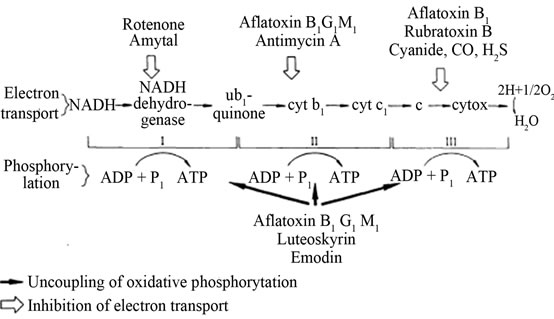
Figure 8. Aflatoxin inhibition of electron transport chain in oxidative phosphorylation (Adopted from Kiessling, 1986) [33].
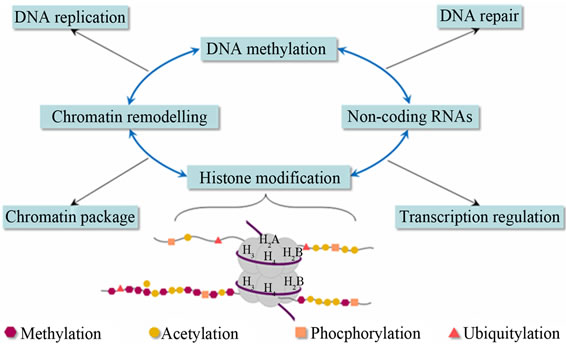
Figure 9. Epigenetic mechanisms gene regulation and expression (Adopted from Balogh and Engelmann, 2011) [43].
 (a)
(a)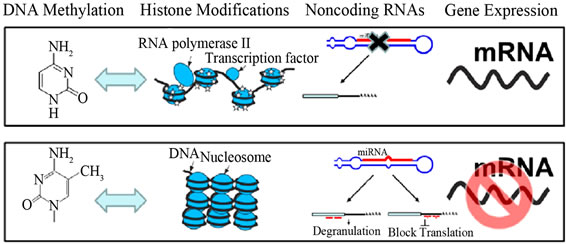 (b)
(b)
Figure 10. Epigenetic mechanisms in Eukaryotes—DNA methylation, modifications of histone tails, and non-coding RNAs. (b) Effect of epigenetic marks on gene expression. Blue arrows indicate cross-talk between DNA methylation and histone modifications (Adopted from Yang and Schwartz, 2011) [47].
incorporated into the genome of the host for a prolonged time. DNA methylation also forms the basis of chromatin structure, which enables a single cell to grow into multi-
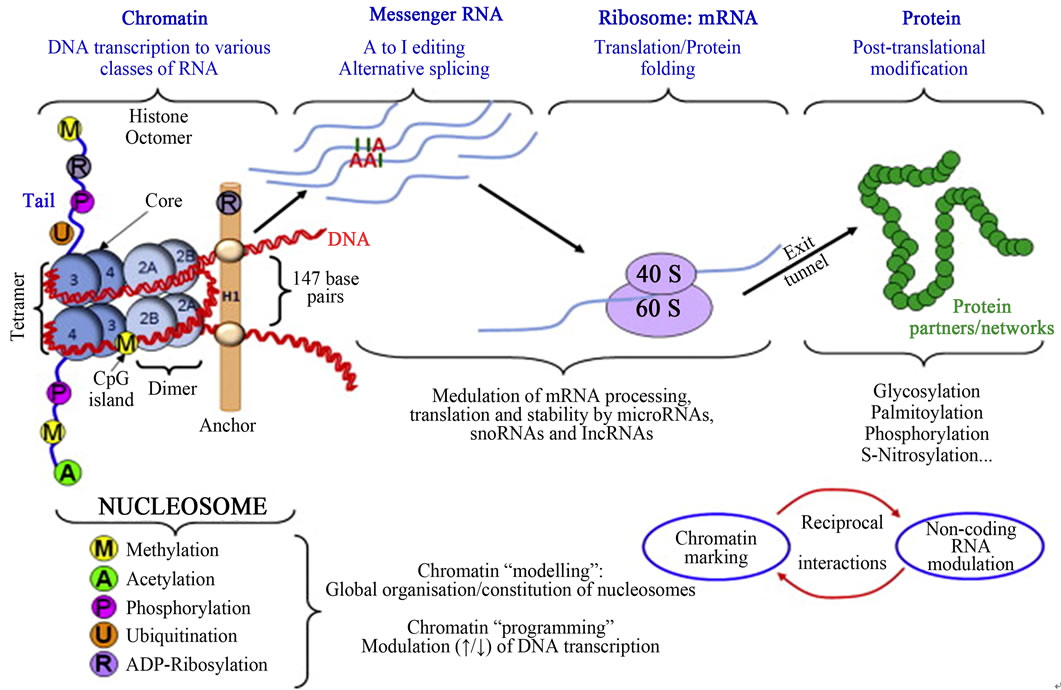
Figure 11. Epigenetic mechanisms from gene to protein: epigenetic mechanisms modulating gene expression in the absence of alterations in DNA sequence (Adopted from Millan, 2013) [50].
ple organs or perform multiple functions [45,46,50-53]. During the period of cell proliferation, the established pattern of DNA methylation is maintained by the action of a maintenance methylase enzyme following DNA replication. However, changes in the methylation status of gene expression by xenobiotics such as aflatoxins, provides a mechanism by which its potential for expression can be altered in an epigenetic heritable manner, and it is expected that modifications in DNA methylation may result from threshold-exhibition events [51,52]. The maintenance of the methylation activity is necessary to preserve DNA methylation after every cellular DNA replication cycle. Absence of DNA methyltransferase (DNMT) enzyme, the replication machinery itself would produce daughter strands that are unmethylated and, over time, would lead to passive demethylation [45,46,50-53]. The DNMT1 enzyme is thought to play a significant role in the maintenance of methyltransferase enzyme that is responsible for copying DNA methylation patterns to the daughter strands during DNA replication. And this process can be interfered with the chronic exposure by the individual to aflatoxins and it could be the mechanism by which aflatoxins cause cancers. Since many tumor suppressor genes are silenced by DNA methylation during carcinogenesis, the 5-Aza-2’-deoxycytidine (decitabine) has been used to re-express these genes by inhibiting the DNMTs [45,46,50]. The DNA methylation plays a critical role in the development of almost all cancer types. Both the DNA hypermethylation and hypomethylation have been associated with a large number of human malignancies (Figures 12 and 13) [45,46,50]. During tumor initiation and progression, the epigenome goes through multiple alterations, including a genome-wide loss of DNA methylation (hypomethylation-low levels of DNA 5-methylcytosine), frequent increase in promoter methylation of CpG islands, changes in nucleosome occupancy, and modification profiles of which these processes can be facilitated by mycotoxins especially aflatoxins [48]. Global hypomethylation of DNA is one of the most common molecular alterations identified in human cancer cells. The hypermethylation occurs at CpG islands in the promoter region and are associated with gene inactivation. The alteration by hypermethylation of selected promoters causes silencing of critical genes such as tumor suppressor genes (p53 gene) and hypomethylation of other DNA sequences. Genomic hypermethylation in cancer has been observed most often in CpG islands of the gene. In cancer, DNA methylation is an important regulator of gene transcription and a large body of evidence has demonstrated that genes with high levels of 5-methylcytosine in their promoter region are transcriptionally silent, and that DNA methylation gradually accumulates upon long-term gene silencing [49,54]. Altered DNA methylation patterns in hepatocellular carcinoma (HCC) due to AFB are closely related to the disruption of the DNA methylation machinery. Several
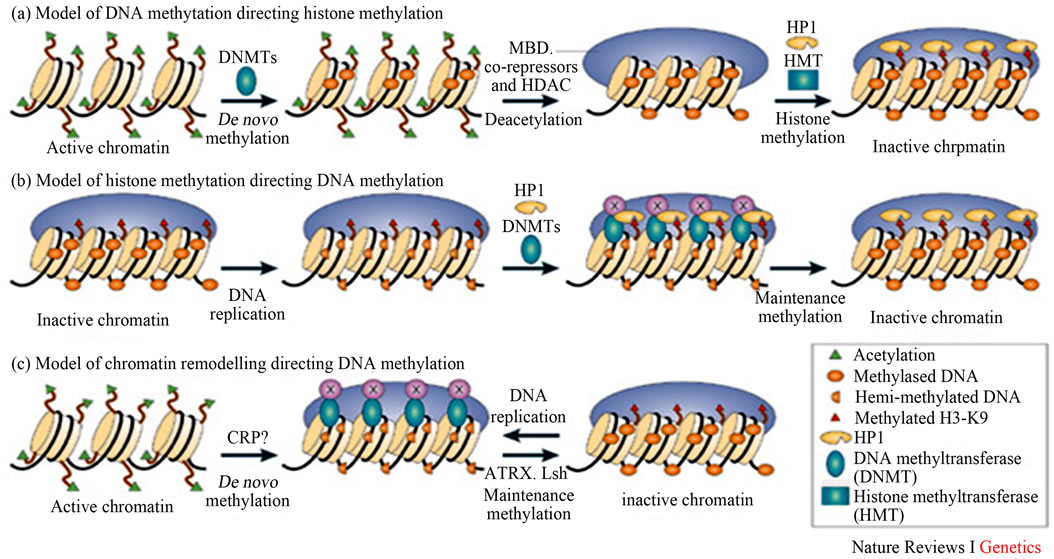
Figure 12. Epigenetic marks (DNA methylation, modifications of histone tails, and noncoding RNAs) work in concert with other components of the cellular regulatory machinery to control the spatial and temporal levels of expressed genes (Adopted from Li, 2002) [55].
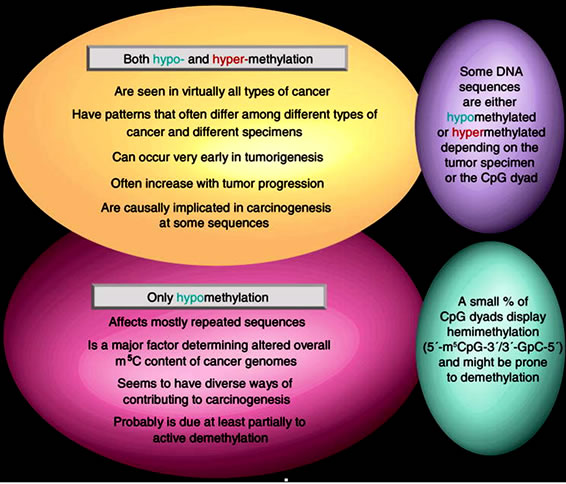
Figure 13. Similarilities and differences in cancer-associated hypoand hyper-methylation of DNA (Adopted from Ehrlich, 2009) [51].
studies have demonstrated involvement of altered expression of the maintenance DNMT1 enzyme, de novo DNA methyltransferases 3A and 3B, and methyl-CpGbinding proteins in the initiation, establishment, and maintenance of aberrant DNA methylation patterns during the development and progression of HCC [49,54,55]. Cell proliferation is a fundamental component of carcinogenesis associated with AFB exposure.
4.2. Histone Posttranslational Modifications
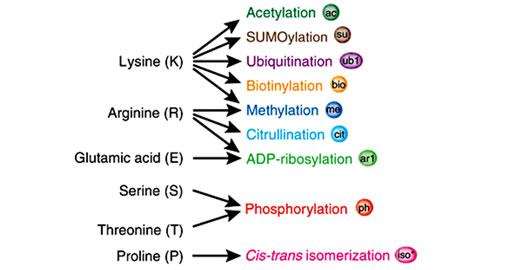
Histones are highly alkaline proteins that forms the main component of chromatin found in Eukaryotic cell nuclei that package and order the DNA into structural units called nucleosomes. Histones wind DNA in the process of packaging since the unwound DNA in chromosomes would be very long and this plays a role in gene regulation. Histone posttranslational modifications are important in the regulation of chromatin as well as in the initial formation of its structure [50,56,57]. Chromatin consists of nucleosomes which are made up of DNA wrapped around histone octamers containing dimers of core histone H2A, H2B, H3, and H4. Histone modifications have been found in the structured regions of histones including the histone-DNA and histone-histone interactions within the nucleosome core [50,56,57]. Modifications such as acetylation or phosphorylation lowers the charge of the globular histone core and its thought to “loosen” the core-DNA association, and the strength of the effect depends on location of the modification within the core. Some modifications have been shown to be correlated with gene silencing while others are involved in gene activation [50,53,56-59]. The commonest histone modifications occur to specific amino acids in nucleosome and they include acetylation, methylation, or ubiquitination of lysine; methylation of arginine; and phosphorylation of serine, ubiquitylation, sumoylation, biotinylation, citrullination, ADP-ribosylation and cis-transisomerization (Figure 14(a)) [50,53,56-59]. Histone posttranslational modifications are also linked to chromatin remodelling such as H4K8ac and H4K16ac that delineate the doughnut structure marked by H3K9me2. Histone modifications such as H3 lysine-9 methylation (H3K9me) or H3 lysine-27 methylation (H3K27me), are generally repressive marks, while H3 lysine-9/14 acetylation (H3KAc) and H3 lysine-4 methylation (H3K4me) are generally activation marks (Figure 14(b)) [50,53,56-59]. The H3Kme is regulated by histone methyltransferases/demethylases (HMTs/HDMs) and H3KAc is regulated by histone acetylases/histone deacetylases (HATs/HDACs) enzymes. Histone modifications coupled with DNA methylation at CpG islands by DNMTs/DNA demethylases (DNAdeMeth) determine the active or inactive state of chromatin leading to gene expression or repression, respectively (Figure 15). This dynamic state of the chromatin is subject to alteration by external stimuli like exposure to aflatoxins via the regulation of epigenetic machinery and miRNAs leading to gene expression and pathophysiological phenotypes [50,53,56-59]. Generally, histone marks are described as “permissive” (active promoters), “repressive” (inactive promoters), or “poised” (accessible promoters). Although histone modifications occur throughout the entire DNA sequence, the unstructured N-terminals of histones (called histone tails) are particularly highly modified. And these modifications may occur to individuals exposed to chronic consumption of aflatoxincontaminated foods, causing histone modifications leading to active chromatin and hence increased expression of the gene contributing to carcinogenesis associated with AFB exposure.
4.3. MicroRNAs
MicroRNAs (miRNA) belong to a class of endogenously expressed, noncoding single-stranded RNAs (ncRNAs) that contain 17 to 25 nucleotides and they include microRNAs, small RNAs and long or large RNAs that regulate gene expression posttranscriptionally by binding to 3’-untranslated regions (3’-UTRs), coding sequences, or 5’-UTRs of target messenger RNAs (mRNAs), leading to inhibition of translation or mRNA degradation [44,47,60,61]. The ncRNAs cause epigenetic modification mechanisms by cytosine methylation and histone modifications that are related to gene expression [44,60, 61]. The miRNAs represent the most abundant class of gene expression regulators that bind complementarily to transcripts to repress their translation or mRNA degradation (Figure 16). They are currently recognized to be involved in posttranscriptional gene regulation and contributes to cellular processes in cell cycle, cell proliferation, development, differentiation, cell signaling and immune responses, pathological and physiological processes such as disease development, drug responses, and carcinogenesis [44,60-62]. Many CYP450s and nuclear receptors are reported to be regulated posttranscriptionally by miRNAs. They are important in modulating detoxification mechanisms of xenobiotics, drug and chemical metabolic activation processes. Many miRNAs are epigenetically regulated and about 60% of human pro-
 (a)
(a)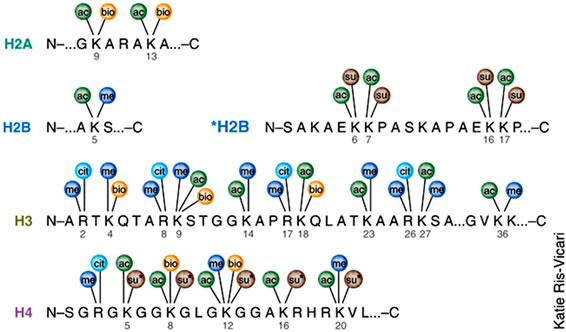 (b)
(b)
Figure 14. Histone modification choices (a) Known posttranslational modifications and the amino acid residues they modify. (b) Residues that can undergo several different forms of posttranslational modification or cross-talk in situ. (Adopted from Latham & Dent, 2007) [58].
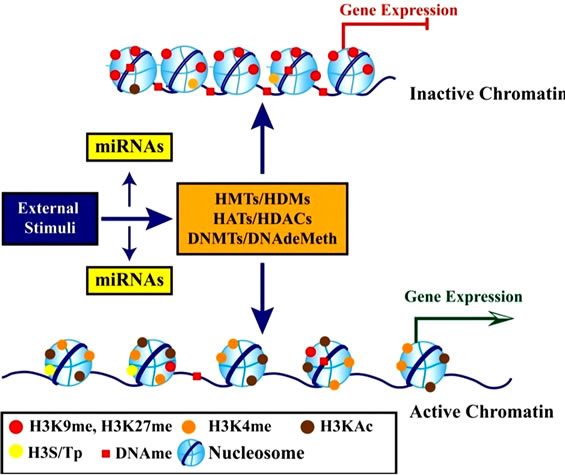
Figure 15. Schematic diagram showing the role of epigenetic mechanisms in transcription regulation (Adopted from Reddy and Natarajan, 2011) [53].
teins coding genes are regulated by miRNAs and about 50% of miRNA genes are associated with CpG islands that may be repressed by epigenetic methylation, histone modifications or by combined DNA methylation and histone modifications. Depending on the genes they target, miRNAs can function as oncogenes or tumour
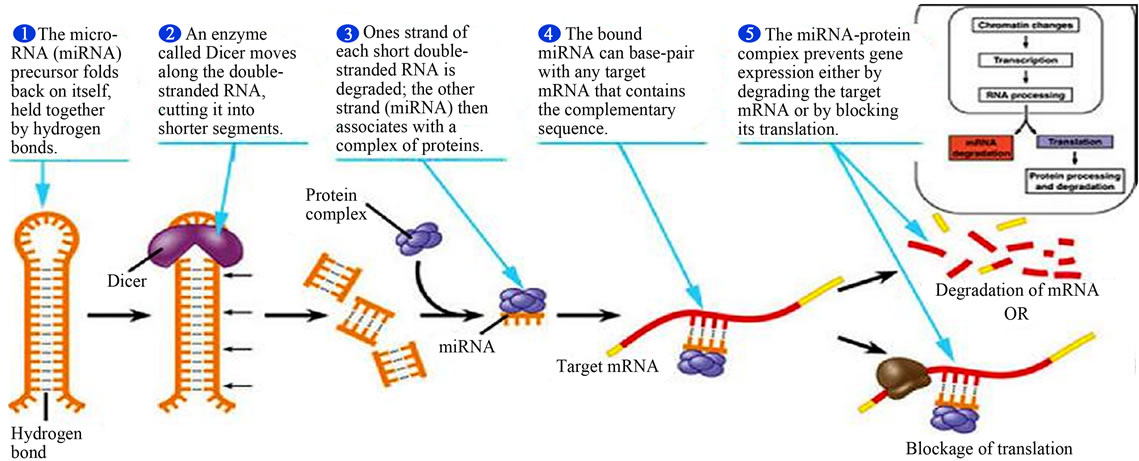
Figure 16. Major steps in miRNA processing and function (Adopted from Adam, 2005) [64].
suppressor genes and AFB may play a significant contribution in the oncogenes development. In hepatocarcinogenesis, many miRNAs may be dysregulated by aflatoxins in hepatocellular carcinoma (HCC), resulting in aberrant gene expression [44,60-62]. A few miRNAs (miR-1, miR-191, miR-124, miR-125b and miR-203) are silenced in HCC tissues, through DNA methylation mechanism [48,50,63]. There is decreased miR-10a expression in cancer cells or tumor tissues due to host gene’s hypermethylation by aflatoxins, supporting a tumor suppressor role for miR-10a in the repression of downstream target oncogenes. Its reported that microRNA-17~92 cluster (miR-17~92) are oncogenic miRNAs that promotes tumorigenesis, angiogenesis, invasion, and metastasis of HCC cells and that p53 gene regulates transcriptional expression and the maturation of a group of miRNAs especially miR-34 family. The miR-34 can regulate p53 gene activity and function by directly repressing p53 gene or p53 regulators in cells [48,50,63]. The p53 hot spot, located at codon 249 of exon 7, provides a molecular signature for aflatoxin exposure [48, 50,63]. Ingested aflatoxin B1, which is metabolized in the liver, can bind to DNA, where it may cause a characteristic inactivation and mutation of codon 249 of the p53 gene and this is modulated by p53-induced miRNAs [48, 50,61,63]. This mutation has been found in 30% - 60% of hepatocellular carcinomas in aflatoxin-endemic areas [48,50,61,63,64].
4.4. Single Nucleotide Polymorphism (SNPS)
A single nucleotide polymorphism (SNP) is a DNA sequence variation that occur when a single nucleotide— adenine (A), thymine (T), cytosine (C) or guanine (G)— in the genome (or other shared sequence) differs between members of a biological individuals or paired chromosomes in an individual such as two sequenced DNA fragments from different individuals, AAGCCTA to AAGCTTA, contain a difference in a single nucleotide [61,65,66]. SNPs may occur within coding sequences of genes, non-coding regions of genes, or in the intergenic regions (regions between genes) [44,61,66]. The SNPs within a coding sequence may not change the amino acid sequence of the protein that is produced due to degeneracy of the genetic code. The SNPs in the coding regions are of two types, synonymous and nonsynonymous SNPs [44,61,66]. Synonymous SNPs do not affect the protein sequence while nonsynonymous SNPs change the amino acid sequence of protein. The nonsynonymous SNPs are of two types: missense and nonsense. SNPs that are not in protein-coding regions may still affect gene splicing, transcription factor binding, messenger RNA degradation, or the sequence of non-coding RNA. The gene expression affected by this type of SNP is referred to as an eSNP (expression SNP) and may be upstream or downstream from the gene. It’s reported that about 15 million SNPs occur in the human genome, many of which affect gene function in promoter regions or cause a change in cisor trans-regulatory elements by modifying sequences of proteins [61,65,66]. SNPs may affect activities of various enzymes including the CYP450 enzyme systems and are associated with differences in drug response as well as the adverse effects of drugs and responses to toxic chemicals. The SNPs in the primary miRNAs (primiRNAs), precursor miRNAs (pre-miRNAs), or mature miRNAs can modify various biological processes by influencing the processing of miRNAs or causes target selection of miRNAs [61,65,66]. SNPs may affect activities of various enzymes and are associated with differences in drugs, poisons and toxins responses as well as the adverse effects of drugs and toxic chemicals like aflatoxins [61,65,66]. AFB1-associated HCCs, promotes methylation to cause alterations in p53 pathway where the G-- > T point mutation of p53 gene at codon 249 or p14(ARF) promoter methylation occurs (Figure 17) [61, 65-67]. Generally the RB1, p53 and Wnt pathways are commonly affected by aflatoxins and AFBO in HCCs [61,65,66].
DNA damage occurs naturally in about 10,000 times a day per cell of the human body. The damages are normally repaired; though the sites of DNA repair may remain causing epigenetic changes. The damages can be exaggerated by the AFB1, AFBO and many of its metabolites by forming AFB-DNA adducts [62,68,69]. The adducts can cause a double strand break in DNA and this can initiate unprogrammed epigenetic gene silencing both by causing DNA methylation as well as promoting the silencing of histone modifications types (chromatin remodeling) and maturation of the miRNAs and SNPs [62,68,69]. The epigenetic modifications lead to various health endpoints like cancers, autoimmune diseases and many other disease conditions (Figures 18 and 19). However in most cases the DNA repair mechanisms occur in the altered points thus preventing the deleterious effects that would occur due to the modifications by the epigenetic mechanisms (Figure 18).
Hepatocellular carcinoma (HCC) is one of the most prevalent life-threatening human cancers globally. It is commonly associated with increased consumption of aflatoxin-contaminated foods. The AFB and its metabolites in the body interact with various biochemicals, cells and tissues and various organs to cause organ toxicity. They also cause genotoxicty and affect many various biochemical pathways where they cause metabolic activation, interact directly with DNA and causes mutations with eventual tumor formation [65,67]. The metabolic activation by AFB causes DNA modifications, histone modifications, SNPs and maturation of miRNAs and all these causes cell deregulation leading to cell death and/or cellular transformation and hence cancer development. Aflatoxins may lead to DNA hypermethylation, leading to gaining of repressive histone methylation and loss of active histone modifications and up-regulation of microRNAs and thus inactivation of tumor suppressor genes and development of HCC [65,67]. While on the other hand, DNA hypomethylation leads to loss of repressive histone methylations and gaining of active histone modifications leading to down-regulation of T5 miRNAs and this increases the activation of the oncogenes or cancer-promoting genes and hence the development of HCC (Figure 20) [65,67,68]. Also aflatoxin especially AFB1 and AFBO exposure causes multiple genetic and epigenetic changes that are involved in the molecular pathogenesis of HCC including somatic mutations in the p53 tumor suppressor gene (TP53) and the activation of the WNT signal transduction pathway. AFB1 frequently induces G:C to T:A transversions at the
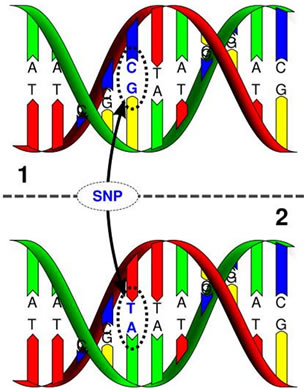
Figure 17. Single nucleotide polymorphism and point mutation of G-- > T at codon 249 in p53 gene epigenetic mechanisms of aflatoxins in carcinogenesis [65,67].
third base in codon 249 of TP53 causing p53 mutations in HCC (Figures 17 and 20) [65,67]. However, the AFB exposure by individuals infected by hepatitis B virus (HBV) leads to their interaction and hence promoting the development of hepatocelllular carcinoma (HCC) (Figures 19 and 20) [65,67,71,72].
5. CONCLUSION
The AFB, AFBO and other metabolites interact with various biomolecules including nucleic acids like DNA and RNA as well as the various body metabolic pathways such as protein synthesis, glycolytic pathway and electron transport chain involved in ATP production in body cells. The AFB interacts with DNA to form AFB-DNA adducts causing DNA breakages. The AFB and its metabolites induce the upregulation of nuclear receptors such as the PXR, CAR and AhR through gene expression as well as induction of CYP450 isoenzymes leading to over expression of CYP1A, CYP1A2, CYP2B6, CYP2C9 and CYP3A4 that are involved in the Phase I metabolic reactions that increase the production of AFBO and the glutathione (GSH) conjugation reactions especially GST in Phase II metabolic reactions. The accumulation of AFB and its metabolites in the body, especially the AFB1-exo- 8,9-epoxide (AFBO), depletes the GSH due to the formation of high amounts of epoxides and other reactive oxygen species (ROS). The AFB, AFB1-exo-8,9-epoxide and other metabolites also affect the epigenetic mechanisms including the DNA methylation (hypermethylation and hypomethylation), histone modifications (acetylation, SUMOylation, methylation, phosphorylation and others), maturation of miRNAs as well as single nucleotide polymorphism (SNP) where AFB induces G:C to T:A transversions at the third base in codon 249 of TP53 causing p53 mutations in HCC. The changes in epigenetic me-
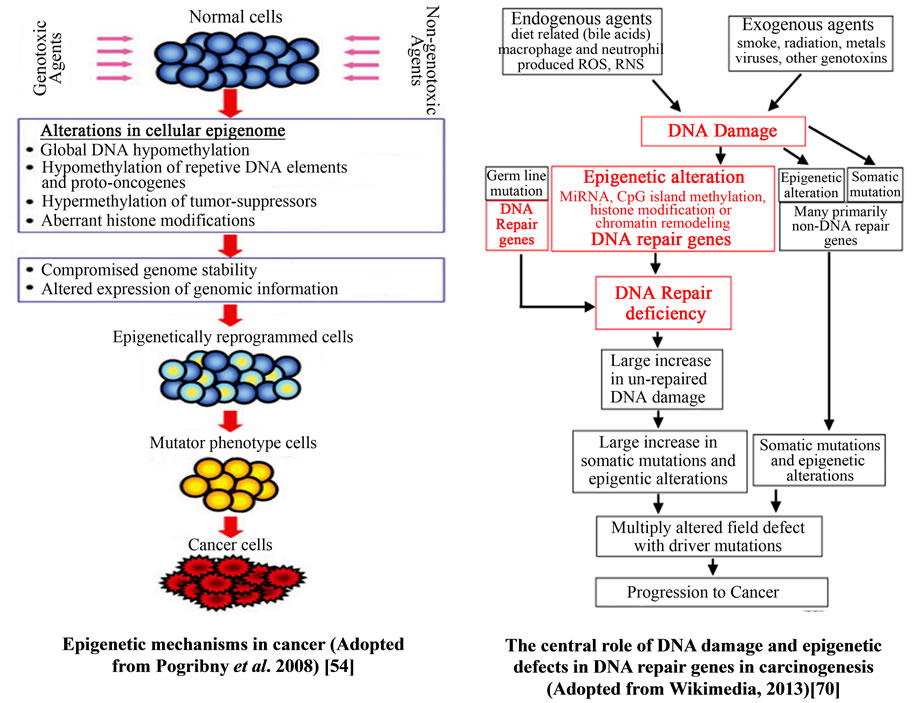
Figure 18. Epigenetic mechanisms in cancer progression.
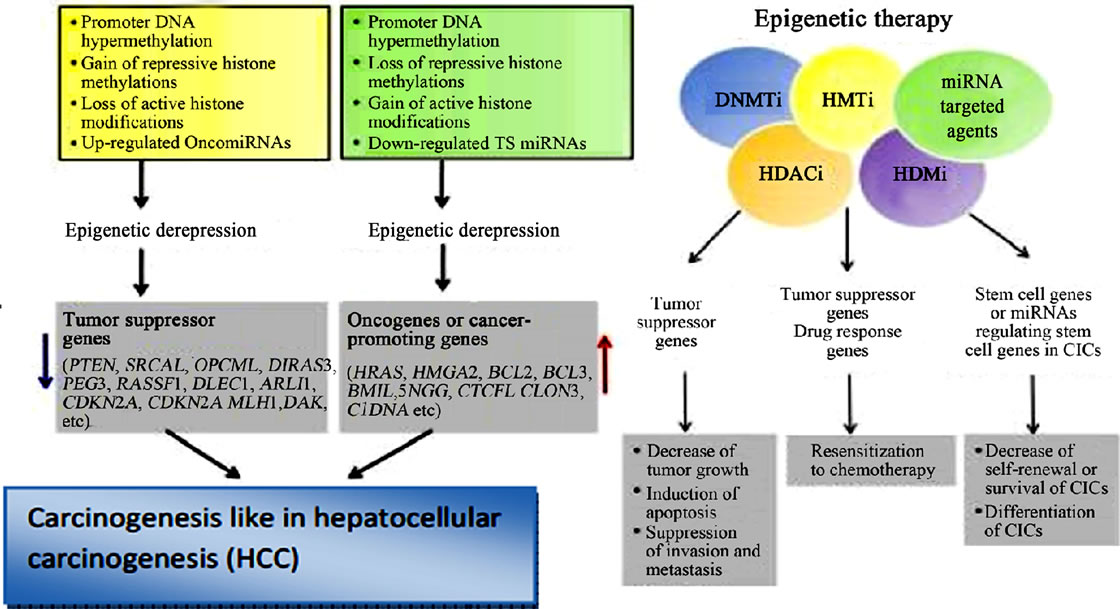
Figure 19. Epigenetic inactivation and derepression, and the role of epigenetic therapy in HCC (Adopted from Kwon, 2011) [68].
chanisms lead to either epigenetic inactivation or epigenetic derepression and all these affect the gene expression, cellular differentiation and growth. AFB also through epigenetic mechanisms promotes tumorigenesis, angiogenesis, invasion and metastasis in hepatocellular carcinoma. The AFB2 has the lowest potency as compared to all other aflatoxins in humans and rodents’ carcinogenesis and this is due to the differences in the metabolic pathways. The biotransformation of AFB2 to AFB1-2,3- epoxide occurs through the intermediate formation of
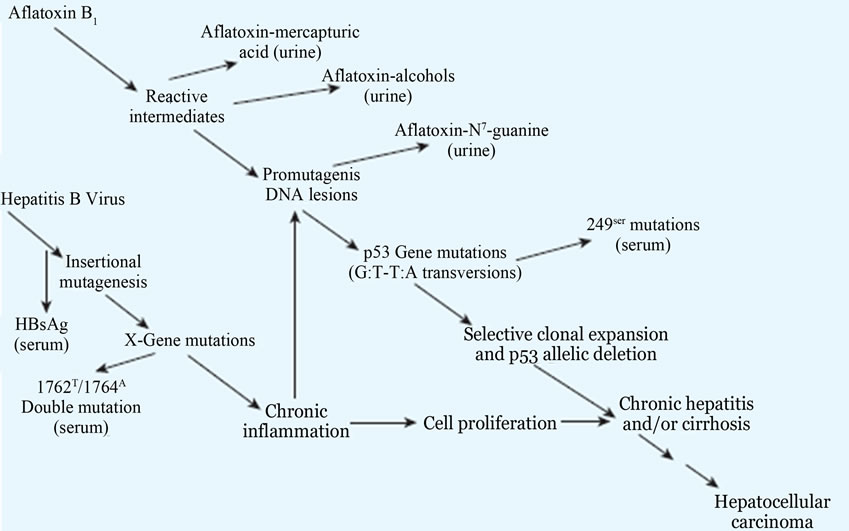
Figure 20. Potential molecular mechanisms of interaction between HBV and aflatoxin in the development of hepatocellular carcinoma, as elucidated by several biomarkers. HBsAg, hepatitis B surface antigen Adopted from Groopman et al., 2005 and Feingold et al., 2010) [65,67,71,72].
AFB1 by desaturation at the 2,3-carbons of AFB2. However, the formation of the small amounts of AFB1 from AFB2 is suspected to be the cause of the carcinogenicity of AFB2 in humans and animals.
6. PERSPECTIVE VIEW
Aflatoxin exposure in the population is a silent and long-term process and many people are unaware of its health consequences. Exposure to aflatoxins, in contaminated foods especially grains is a slow and continuous process and in most cases individual are not aware of the exposure problem until they develop the liver cancers (hepatocellular carcinoma). There is scarce knowledge in the global population and various communities about the source, exposure risks of mycotoxins especially aflatoxins and their associated health problems. Aflatoxin-related cancers are continuing to be a global problem especially in poor developing countries where famine and conflicts are continuing to be a problem. In these populations, individuals are exposed to aflatoxin contaminated foods as their daily meals with no choice of alternative foods especially people living in camps. The poor methods of harvesting of grains, storage and processing of the various foods; leads to food contamination with aflatoxin-producing fungi. The aflatoxins in foods are a current and future problem especially with the increasing global population and maximization of food production using poor methods of harvesting, processing and storage of foods. Therefore there is need to sensitize the population on the dangers of aflatoxins and the various available methods of preventing the contamination of foods with aflatoxin producing fungi. There is also need to increase the regulation and monitoring of aflatoxins levels in foods by various regulatory authorities globally.
7. ACKNOWLEDGEMENTS
Special thanks to International Brain Research Organization (IBRO), Society for Neuroscientists for Africa (SONA) and the Cold Spring Harbor Laboratory (CSHL), New York, USA for their support.
REFERENCES
- Bennett, J.W. and Klich, M. (2003) Mycotoxins. Clinical Microbiology Reviews, 16, 497-516. http://dx.doi.org/10.1128/CMR.16.3.497-516.2003
- Bbosa, G.S., David Kitya, D., Lubega, A., Ogwal-Okeng, J., Anokbonggo, W.W. and Kyegombe, D.B. (2013) Review of the biological and health effects of aflatoxins on body organs and body systems: Aflatoxins—Recent advances and future prospects. Intechopen Publisher, 12, 239-265. http://cdn.intechopen.com/pdfs/38731/InTechReview_of_the_biological_and_health_effects_of_aflatoxins_on_body_organs_and_body_systems.pdf
- Mushtaq, M., Sultana, B., Anwar, F., Khan, M.Z. and Ashrafuzzaman, M. (2012) Occurrence of aflatoxins in selected processed foods from Pakistan. International Journal of Molecular Sciences, 13, 8324-8337. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3430236/pdf/ijms-13-08324.pdf http://dx.doi.org/10.3390/ijms13078324
- Reddy, S.V. and Waliyar, F. (2012) Properties of aflatoxin and its producing fungi: Aflatoxins, 2012. http://www.icrisat.org/aflatoxin/aflatoxin.asp
- Monosson, E. (2012) Biotransformation. National Library of Medicine (NLM): The Encyclopeadia of earth. http://www.eoearth.org/view/article/150674/
- Eaton, L.D., Evan, P.G., Theo, K.B. and Kent, L.K. (1995) Role of cytochrome P4501A2 in chemical carcinogenesis: Implications for human variability in expression and enzyme activity. Pharmacogenetics, 5.
- Vondracek, M., Xi, Z., Larsson, P., Baker, V., Mace, K., Pfeifer, A., Tjalve, H., Donato, M.T., Gomez-Lechon, J. and Grafstrom, R.C. (2001) Cytochrome P450 expression and related metabolism in human buccal mucosa. Carcinogenesis, 22, 481-488. http://carcin.oxfordjournals.org/content/22/3/481.full.pdf http://dx.doi.org/10.1093/carcin/22.3.481
- Dhanasekaran, D., Shanmugapriya, S., Thajuddin, N. and Panneerselvam, A. (2011) Panneerselvam, aflatoxins and aflatoxicosis in human and animals. In: Guevara-Gonzalez, R.G., Ed., Aflatoxins—Biochemistry and Molecular Biology, InTech, 221-254.
- Zhang, B.C., Zhu, Y.R., Wang, J.B., Wu, Y., Zhang, Q.N., Qian, G.S., et al. (1997) Oltipraz chemoprevention trial in Qidong, Jiangsu Province, People’s Republic of China. Journal of Cellular Biochemistry, Suppl. 28-29, 166-173. http://dx.doi.org/10.1002/(SICI)1097-4644(1997)28/29+<166::AID-JCB20>3.0.CO;2-E
- Omar, H.E. (2013) Mycotoxins-induced oxidative stress and disease. INTECH, Chapter 3. http://cdn.intechopen.com/pdfs/44091/InTech-Mycotoxins_induced_oxidative_stress_and_disease.pdf
- Gallagher, E.P., Wienkers, L.C., Stapleton, P.L., Kunze, K.L. and Eaton, D.L. (1994) Bioactivation of aflatoxin B1DNA-expressed cytochromes P4501A2 and P4503A4 in the role of human. Cancer Research, 54, 101-108. http://cancerres.aacrjournals.org/content/54/1/101.full.pdf
- Code, E.L., Crespi, C.L., Penman, B.W., Gonzalez, F.J., Chang, T.K.H. and Waxman, D.J. (1997) Human cytochrome P4502B6 interindividual hepatic expression, substrate specificity, and role in procarcinogen activation. Drug Metabolism and Disposition, 25, 985-993.
- Wild, C.P. and Turner, P.C. (2002) The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis, 17, 471-481. http://mutage.oxfordjournals.org/content/17/6/471.full.pdf http://dx.doi.org/10.1093/mutage/17.6.471
- Gross-Steinmeyer, K. and Eaton, D.L. (2012) Dietary modulation of the biotransformation and genotoxicity of aflatoxin B(1). Toxicology, 299, 69-79. http://ac.els-cdn.com/S0300483X12001722/1-s2.0-S0300483X12001722-main.pdf?_tid=9b0beef8-eeb7-11e2-9c9a-00000aacb35d&acdnat=1374048492_6eb572064347d54a06695eb561bbe44d http://dx.doi.org/10.1016/j.tox.2012.05.016
- Wild, C.P., Yin, F., Turner, P.C., Chemin, I., Chapot, B., Mendy, M., Whittle, H., Kirk, G.D. and Hall, A.J. (2000) Environmental and genetic determinants of aflatoxin-albumin adducts in The Gambia. International Journal of Cancer, 86, 1-7. http://onlinelibrary.wiley.com/doi/10.1002/%28SICI%291097-0215%2820000401%2986:1%3C1::AID-IJC1%3E3.0.CO;2-I/pdf http://dx.doi.org/10.1002/(SICI)1097-0215(20000401)86:1<1::AID-IJC1>3.0.CO;2-I
- Lampe, J.W., King, I.B., Li, S., Grate, M.T., Barale, K.V., Chen, C., Feng, Z. and Potte, J.D. (2000) Brassica vegetables increase and apiaceous vegetables decrease cytochrome P450 1A2 activity in humans: Changes in caffeine metabolite ratios in response to controlled vegetable diets. Carcinogenesis, 21, 1157-1162. http://carcin.oxfordjournals.org/content/21/6/1157.full.pdf http://dx.doi.org/10.1093/carcin/21.6.1157
- Farombi, E.O. and Nwaokeafor, I.O. (2005) Anti-oxidant mechanisms of kolaviron: Studies on serum lipoprotein oxidation, metal chelation and oxidative membrane damage in rats. Clinical and Experimental Pharmacology and Physiology, 32, 667-674. http://dx.doi.org/10.1111/j.0305-1870.2005.04248.x
- Ayed-Boussema, I., Pascussi, J., Maurel, P., Bacha, H. and Hassen, W. (2012) Effect of aflatoxin B1 on nuclear receptors PXR, CAR, and AhR and their target cytochromes P450 mRNA expression in primary cultures of human hepatocytes. International Journal of Toxicology, 31, 86-93. http://ijt.sagepub.com/content/31/1/86.full.pdf http://dx.doi.org/10.1177/1091581811422453
- Droździk, A., Kowalczyk, R., Urasińska, E. and Kurzawski, M. (2013) Expression of nuclear receptors (AhR, PXR, CAR) and transcription factor (Nrf2) in human parotid gland. Acta Poloniae Pharmaceutica, 70, 215-219.
- Li, H. and Wang, H. (2010) Activation of xenobiotic receptors: Driving into the nucleus. Expert Opinion on Drug Metabolism & Toxicology, 6, 409-428. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2842451/pdf/nihms-169048.pdf http://dx.doi.org/10.1517/17425251003598886
- Maglich, J.M., Stoltz, C.M., Goodwin, B., HawkinsBrown, D., Moore, J.T. and Kliewer, S.A. (2001) Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Molecular Pharmacology, 62, 638-648. http://molpharm.aspetjournals.org/content/62/3/638.full.pdf
- Pool-Zobela, B., Veeriaha, S. and Böhmer, F.D. (2005) Modulation of xenobiotic metabolising enzymes by anticarcinogens—Focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colorectal carcinogenesis. Mutation Research, 591, 74-92. http://ac.els-cdn.com/S0027510705003179/1-s2.0-S0027510705003179-main.pdf?_tid=1c76459c-eed6-11e2-bd46-00000aab0f27&acdnat=1374061594_85d96b33ac97b1513863e6623919d826 http://dx.doi.org/10.1016/j.mrfmmm.2005.04.020
- Halliwell, B. (2007) Oxidative stress and cancer: Have we moved forward? Biochemistry Journal, 401, 1-11. http://dx.doi.org/10.1042/BJ20061131
- Verma, R.J. (2004) Aflatoxin cause DNA damage. International Journal of Human Genetics, 4, 231-236.
- Murphy, P.A., Hendrich, S., Landgren, C. and Bryant, C.M. (2006) Food mycotoxins: An update. Journal of Food Science, 71, P51-P65. http://www.ift.org/Knowledge-Center/Read-IFT-Publications/Science-Reports/Scientific-Status-Summaries/~/media/Knowledge%20Center/Science%20Reports/Scientific%20Status%20Summaries/mycotoxins_0606.pdf http://dx.doi.org/10.1111/j.1750-3841.2006.00052.x
- Hayes, J.D., Flanagan, J.U. and Jowsey, I.R. (2005) Glutathione transferases. Annual Reviews in Pharmacology and Toxicology, 45, 51-88. http://www.annualreviews.org/doi/pdf/10.1146/annurev.pharmtox.45.120403.095857 http://dx.doi.org/10.1146/annurev.pharmtox.45.120403.095857
- Sherrat, P.J. and Hayes, J.D. (2002) Glutathione S-transferases. Enzyme systems that metabolize drugs and other xenobiotics. Cell Biochemistry, 9, 319-352. http://www.uniroma2.it/didattica/cellbiochem/deposito/Glutathione_S-transferases.pdf
- Thomson-Reuters (2013) Glutathione metabolism: Pathway map details. Thomson Reuters. http://pathwaymaps.com/maps/896/
- Ye, Z., Song, H., Higgins, J.P.T., Pharoah, P. and Danesh, J. (2006) Five glutathione S-transferase gene variants in 23,452 cases of lung cancer and 30,397 controls: Metaanalysis of 130 studies. PLoS Medicine, 3, e91. http://dx.doi.org/10.1371/journal.pmed.0030091
- Kensler, T.W., Qian, G., Chen, J. and Groopman, J.D. (2003) Molecular pathway of aflatoxin detoxification: Translational strategies for cancer prevention in liver. Nature Reviews Cancer, 3, 321-329. http://dx.doi.org/10.1038/nrc1076
- Kensler, T.W., Roebuck, B.D., Wogan, G. N. and Groopman, J.D. (2010) Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicological Sciences, 120, S28-S48. http://toxsci.oxfordjournals.org/content/120/suppl_1/S28.full.pdf
- Clifford, J.I. and Rees, K.R. (1967) The interaction of aflatoxins with purines and purine nucleosides. Biochemistry Journal, 103, 467-471.
- Kiessling, K.H. (1986) Biochemical mechanism of action of mycotoxins. Pure & Applied Chemistry, 58, 327-338. http://dx.doi.org/10.1351/pac198658020327
- Bhat, N.K., Emeh, J.K., Niranjan, B.G. and Avadhani, N.G. (1982) Inhibition of mitochondrial protein synthesis during early stages of aflatoxin b-induced hepatocarcinogenesis. Cancer Research, 42, 1876-1880. http://cancerres.aacrjournals.org/content/42/5/1876.full.pdf
- Qian, G., Wang, F., Tang, L., Massey, M.E., Mitchell, N.J., Su, J., Williams, J.H., Phillips, T.D. and Wang, J. (2013) Integrative toxicopathological evaluation of aflatoxin B1 exposure in f344 rats toxicologic pathology. http://tpx.sagepub.com/content/early/2013/02/15/0192623313477256.full.pdf
- Riley, R.T. (1998) Mechanistic interactions of mycotoxins: Theoretical consideration. In: Sinha, K.K. and Bhatanagar, D., Eds., Mycotoxins in Agriculture and Food Safety. Marcel Dekker, Inc, Basel, New York, 227-254.
- Riley, R.T. and Norred, W. P. (1996) Mechanisms of mycotoxicity. In: Howard, D.H. and Miller, J.D., Eds., The Mycota, Springer, Berlin, 194-195.
- Speijers, G.J.A. and Speijers, M.H.M. (2004) Combined toxic effects of mycotoxins. Toxicology Letters, 153, 91- 98. http://ac.els-cdn.com/S0378427404002486/1-s2.0-S0378427404002486-main.pdf?_tid=4a0d6cf4-eeec-11e2-a112-00000aacb35d&acdnat=1374071119_27c29902ddd1f844d877d6e8220026de http://dx.doi.org/10.1016/j.toxlet.2004.04.046
- Vermeulen, K., Berneman, Z.N. and Bockstaele, D.R.V. (2003) Cell cycle and apoptosis. Cell Proliferation, 36, 165-175. http://dx.doi.org/10.1046/j.1365-2184.2003.00267.x
- Li, Y., Daniel, M. and Tollefsbol, T.O. (2011) Epigenetic regulation of caloric restriction in aging: Metabolism, diet and disease. BMC Medicine, 9, 98. http://www.biomedcentral.com/content/pdf/1741-7015-9-98.pdf http://dx.doi.org/10.1186/1741-7015-9-98
- Mohammed, A.M. and Metwally, N.S. (2009) Antiaflatoxicogenic activities of some aqeous plant extracts against AFB1 induced renal and cardiac damage. Journal of Pharmacology and Toxicology, 4, 1-16. http://dx.doi.org/10.3923/jpt.2009.1.16
- Chahwan, R., Wontakal, S. and Roa, S. (2011) The multidimensional nature of epigenetic information and its role in disease. Discovery Medicine. http://www.discoverymedicine.com/Richard-Chahwan/2011/03/17/the-multidimensional-nature-of-epigenetic-information-and-its-role-in-disease/
- Balogh, P. and Engelmann, P. (2011) Epigenetic factors in transdifferentiation. Transdifferentiation and regenerative medicine, University of Pécs. http://www.tankonyvtar.hu/hu/tartalom/tamop425/0011_1A_Transzdifferenciation_en_book/ch01s03.html
- Costa, F.F. (2008) Non-coding RNAs, epigenetics and complexity. Gene, 410, 9-17. http://ac.els-cdn.com/S0378111907006208/1-s2.0-S0378111907006208-main.pdf?_tid=eb96b322-ef01-11e2-851c-00000aacb35f&acdnat=1374080410_c1ee123065f1641d8ea049172feb7b85 http://dx.doi.org/10.1016/j.gene.2007.12.008
- Craig, J.M. and Wong, N.C. (2011) Epigenetics: A reference manual. Caister Academic Press.
- Day, J.J. and Sweatt, J.D. (2011) Review of epigenetic mechanisms in cognition. Neuron, 70, 813-829. http://dx.doi.org/10.1016/j.neuron.2011.05.019
- Yang, I.V. and Schwartz, D.A. (2012) Epigenetic mechanisms and the development of asthma. Journal of Allergy and Clinical Immunology, 130, 1243-1255. http://dx.doi.org/10.1016/j.jaci.2012.07.052
- You, J.S. and Jones, P.A. (2012) Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell Review, 22, 9-20.
- Kim, G.H., Ryan, J.J., Marsboom, G. and Archer, S.L. (2011) Epigenetic mechanisms of pulmonary hypertension. Pulmonary Circulation, 1, 347-358. http://www.pulmonarycirculation.org/temp/PulmCirc13347-4509573_123135.pdf
- Millan, M.J. (2013) An epigenetic framework for neurondevelopmental disorders: From pathogenesis to potential therapy. Neuropharmacology, 68, 2-82. http://ac.els-cdn.com/S002839081200562X/1-s2.0-S002839081200562X-main.pdf?_tid=d2620e4e-ef04-11e2-93c0-00000aab0f6b&acdnat=1374081657_7c4d7a4fa9e05f5637c2ba2a36a58003 http://dx.doi.org/10.1016/j.neuropharm.2012.11.015
- Ehrlich, M. (2009) DNA hypomethylation in cancer cells. Epigenomics, 1, 239-259. http://dx.doi.org/10.2217/epi.09.33
- Goodman, J.I. and Counts, J. L. (1993) Hypomethylation of DNA: A possible nongenotoxic mechanism underlying the role of cell proliferation in carcinogenesis. Environmental Health Perspectives, 101, 169-172.
- Reddy, M.A. and Natarajan, R. (2011) Epigenetic mechanisms in diabetic vascular complications. Cardiovascular Research. http://dx.doi.org/10.1093/cvr/cvr024
- Pogribny, I.P., Rusyn, I. and Beland, F.A. (2008) Epigenetic aspects of genotoxic and non-genotoxic hepatocarcinogenesis studies in rodents. Environmental Molecular Mutagenicity, 49, 9-15. http://dx.doi.org/10.1002/em.20342
- Li, E. (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nature Reviews Genetics, 3, 662-673. http://dx.doi.org/10.1038/nrg887
- Cosgrove, M.S., Boeke, J.D. and Wolberger, C. (2004) Regulated nucleosome mobility and the histone code. Nature Structural & Molecular Biology, 11, 1037-1043. http://dx.doi.org/10.1038/nsmb851
- Fenley, A.T., Adams, D.A. and Onufriev, A.V. (2010) Charge state of the globular histone core controls stability of the nucleosome. Biophysical Journal, 99, 1577-1585. http://dx.doi.org/10.1016/j.bpj.2010.06.046
- Latham, J.A. and Dent, S.Y.R. (2007) Histone modification choices: Cross-regulation of histone modifications. Nature Structural & Molecular Biology, 14, 1017-1024. http://dx.doi.org/10.1038/nsmb1307
- Ye, J., Ai, X., Eugeni, E.E., Zhang, L., Carpenter, L.R., Jelinek, M.A., Freitas, M.A. and Parthun, M.R. (2005) Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Molecular Cell, 18, 23-30.
- [61] Su, H., Yang, J., Xu, T., Huang, J., Xu, L., Yuan, Y. and Zhuang, S. (2009) MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Journal of Cancer Research, 69, 1135. http://dx.doi.org/10.1158/0008-5472.CAN-08-2886
- [62] Yokoi, T. and Nakajima, M. (2013) microRNAs as mediators of drug toxicity. Annual Review of Pharmacology and Toxicology, 53, 377-340. http://www.annualreviews.org/doi/pdf/10.1146/annurev-pharmtox-011112-140250 http://dx.doi.org/10.1146/annurev-pharmtox-011112-140250
- [63] Ng, I. (2012) Molecular pathogenesis of hepatocellular carcinoma. Hong Kong Journal of Radiology, 15, S23- S28.
- [64] Shen, J., Wang, S., Zhang, Y., Kappil, M.A., Wu, H., Kibriya, M.G., et al. (2012) Genome-wide aberrant DNA methylation of microRNA host genes in hepatocellular carcinoma. Epigenetics, 7, 1230-1237. http://dx.doi.org/10.4161/epi.22140
- [65] Adam, F. (2005) microRNA: A revolution in gene expression. The Journal of Young Investigators. http://www.jyi.org/issue/microrna-a-revolution-in-gene-expression/#sthash.ZRYJb02d.TSAvzrT6.dpuf
- [66] Park, U.S., Su, J.J., Ban, K.C., Qin, L., Lee, E.H. and Lee, Y.I. (2000) Mutations in the p53 tumor suppressor gene in tree shrew hepatocellular carcinoma associated with hepatitis B virus infection and intake of aflatoxin B1. Gene, 251, 73-80. http://dx.doi.org/10.1016/S0378-1119(00)00183-9
- [67] Zheng, A. (2005) Molecular genetic and epigenetic mechanisms of hepatocarcinogenesis. Chinese Journal of Cancer, 24, 757-768.
- [68] Hussain, S.P., Schwank, J., Staib, F., Wang, X.W. and Harris, C.C. (2007) TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene, 26, 2166-2176. http://dx.doi.org/10.1038/sj.onc.1210279
- [69] Kwon, M.J. and Shin, Y.K. (2011) Epigenetic regulation of cancer-associated genes in ovarian cancer. International Journal of Molecular Sciences, 12, 983-1008. http://dx.doi.org/10.3390/ijms12020983
- [70] Wikipedia (2013) Epigenetics. Wikipedia, the free encyclopedia. http://en.wikipedia.org/wiki/File:Epigenetic_mechanisms.jpg
- [71] Wikimedia (2013) The central role of DNA damage and epigenetic defects in DNA repair genes in carcinogenesis. https://en.m.wikipedia.org/wiki/File:Diagram_Damage_to_Cancer_Wiki_300dpi.svg
- [72] Groopman, J.D., Johnson, D. and Kensler, T.W. (2005) Aflatoxin and hepatitis B virus biomarkers: A paradigm for complex environmental exposures and cancer risk. Cancer Biomarkers, 1, 5-14.
- [73] Feingold, B.J., Vegosen, L., Davis, M., Leibler, J., Peterson, A. and Silbergeld, E.K. (2010) A niche for infectious disease in environmental health: Rethinking the toxicological paradigm. Environmental Health Perspective, 118, 1165-1172. http://dx.doi.org/10.1289/ehp.0901866
- [74] WHO (2002) Aflatoxins (Group 1): Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. World Health Organization International Agency for Research on Cancer: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 82, 171. http://monographs.iarc.fr/ENG/Monographs/vol82/volume82.pdf
- [75] Butler, W.H., Greenblatt, M. and Lijinsky, W. (1969) Carcinogenesis in Rats by aflatoxins B1, G1 and B2. Cancer Research, 29, 2206-2211. http://cancerres.aacrjournals.org/content/29/12/2206.full.pdf
- [76] Eaton, D.L. and Gallagher, E.P. (1994) Mechanisms of aflatoxin carcinogenesis. Annual Review of Pharmacology and Toxicology, 34, 135-172. http://toxicology.usu.edu/endnote/mechanisms-of-alfatoxin.pdf http://dx.doi.org/10.1146/annurev.pa.34.040194.001031
- [77] Roebuck, B.D., Siegel, W.G. and Wogan, G.N. (1978) In vitro metabolism of aflatoxin B2 by animal and human liver. Cancer Research, 38, 999-1002. http://cancerres.aacrjournals.org/content/38/4/999.full.pdf
- [78] Agag, B.I. (2004) Mycotoxins in foods and feeds 1-aflatoxins. Ass. Univ. Bull. Environ. Res., 7, 173-205. http://www.aun.edu.eg/env_enc/env%20mar/173-206.PDF
- [79] Pascussi, J., Gerbal-Chaloin, S., Duret, C., Daujat-Chavanieu, M., Vilarem, M. and Maurel, P. (2008) The tangle of nuclear receptors that controls xenobiotic metabolism and transport: Crosstalk and consequences. Annual Review of Pharmacology, 48, 1-32. http://hal.archives-ouvertes.fr/docs/00/16/21/44/PDF/ARPT_Text_23_04.pdf
- [80] Xie, W., Barwick, J.L., Simon, C.M., Pierce, A.M., Safe, S., Blumberg, B., Guzelian, P.S. and Evans, R.M. (2000) Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes and Development, 14, 3014-3023. http://genesdev.cshlp.org/content/14/23/3014.full.pdf http://dx.doi.org/10.1101/gad.846800
{kind=link}
{kind=link}