Health
Vol.5 No.7A1(2013), Article ID:34601,15 pages DOI:10.4236/health.2013.57A1003
Intravascular infusion of autologous delipidated plasma induces antiatherogenic lipoproteins and causes regression of atherosclerosis
—Studies in non-primates, monkeys and humans
![]()
Vanuatu Research Centre, Port Vila, The Republic of Vanuatu; *Corresponding Author: bill.cham@gmail.com
Copyright © 2013 Bill E. Cham, Tania R. Chase. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 16 May 2013; revised 17 June 2013; accepted 1 July 2013
Keywords: Atherosclerosis; Delipidation; Regression; Pre-β HDL; Delipidated LDL
ABSTRACT
Atherosclerosis is the primary pathophysiological cause of heart disease and cerebrovascular disease. It is responsible for more than 20% of deaths worldwide each year. Treatments for atherosclerosis may include lifestyle changes, drugs, and medical procedures or surgery. There is a need for a rapid and effective treatment for this disease. In 1976, it was hypothesized that a multifunctional plasma delipidation process when applied to hyperlipidemic patients would lead to rapid regression of atherosclerosis. The procedure has now been applied to a variety of nonprimates, primates and humans. In all models studied, large quantities of antiatherogenic lipoprotein particles were generated that led to the mechanisms of reverse cholesterol transport. Trends to regression and actual regression of atherosclerosis have now been reported using a specific plasma delipidation process consisting of lipid extraction from plasma with mixtures of butanol and ethers.
1. INTRODUCTION
1.1. Atherosclerosis
Atherosclerosis is a slowly progressive disease of arteries that begins early in life but rarely produces symptoms until middle age. Atherosclerosis is characterized by focal fatty thickening in the inner aspects of vessels supplying blood to the heart, brain and other vital organs. These lesions obstruct the lumen of the vessel and result in ischaemia of the tissue supplied by the vessel (Figure 1) [1].

Figure 1. Atherogenesis: decades of build up to acute coronary event.
Prolonged or sudden ischaemia may result in a clinical heart attack or stroke from which the patient may or may not recover. Atherosclerosis (Figure 2) [2] and its primary clinical manifestation coronary heart disease (CHD) are the major causes of death in developed countries.
The association between cholesterol and atherosclerosis has been almost universally accepted and cholesterol is a major component of atherosclerotic plaques. Cholesterol accumulation within atherosclerotic plaque occurs when cellular cholesterol synthesis and/or influx into the arterial wall (from ApoB-containing lipoproteins) exceeds cholesterol efflux [3]. Parenchymal cells maintain cholesterol balance by downregulation of de novo cholesterol synthesis and low-density lipoprotein (LDL) receptor expression whereas macrophages continue to acquire cholesterol from ApoB-containing lipoproteins via pathways that are not subject to sterol-mediated feedback control. This results in a continuing cholesterol uptake by macrophages and their eventual conversion into lipidladen atherosclerotic foam cells.
Macrophages have specific receptors to identify normal and modified (oxidized, acetylated) lipoprotein particles. These include LDL receptors as well as several scavenger receptors. Pro-inflammatory macrophages in the vascular smooth muscle bind and transduce signals from oxLDL and differ from adipose or hepatic tissues. Macrophages ingest oxLDL to become lipid-rich foam cells. These foam cells differ markedly from normal resident macrophages in healthy vessels. Foam cells are less mobile and secrete more inflammatory mediators than normal macrophages.
Macrophages differentiation and lipid accumulation are not unidirectional. Macrophages can also export cholesterol, secreting it through ATP-binding cassette transporters such as ABCA1, donating cholesterol to ApoA1 to form high-density lipoprotein(HDL). Consequently, if there is sufficient ApoA1, macrophages can offset the influx of cholesterol from LDL captured by scavenger receptors and have a net loss of cellular cholesterol. Prevention of the accumulation of cellular cholesterol in the macrophages reduces the secretion of monocyte chemoattractants resulting in decreased tissue monocyte/macrophage numbers and thus prevents further lipid accumulation. Increasing ApoA1, consequently, is anti-inflammatory, reducing lipid accumulation as well as keeping the number of lipid-accumulating cells low.
A combination therapy decreasing the influx and increasing the efflux of cellular cholesterol would be effective in treating patients with atherosclerosis. To reduce the cholesterol influx and thus to prevent or retard coronary artery diseases the current therapeutic modalities are aimed at reducing the concentrations of cholesterol-rich ApoB-containing lipoproteins by dietary means, hypolipidemic drugs or extracorporeal removal of plasma lipoproteins.
Clinical trials of LDL-C lowering drugs have estab-
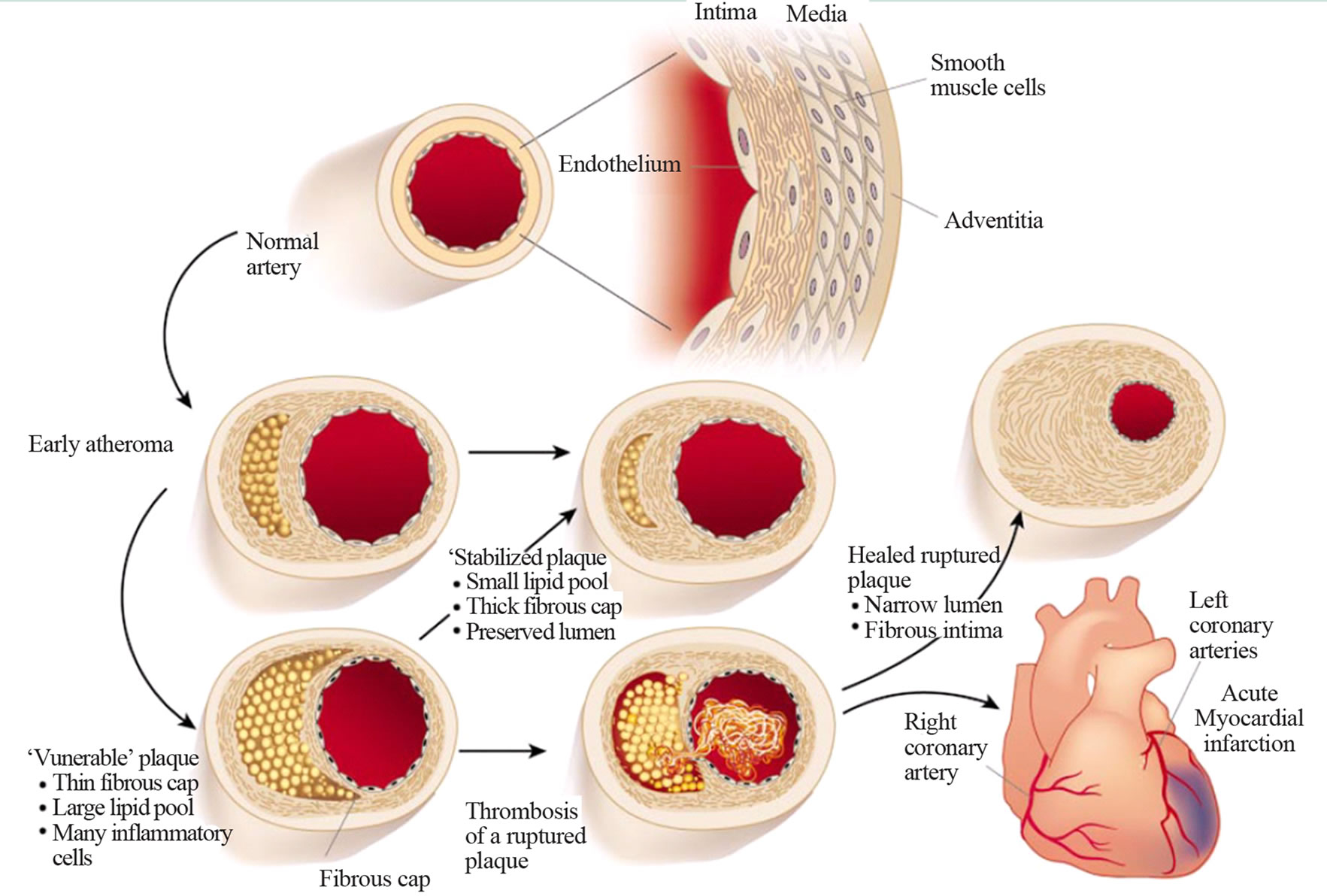
Figure 2. Atherosclerosis.
lished that reductions in LDL-C are associated with a 30% to 45% decrease in clinical cardiovascular disease (CVD) events. However, in many subjects with lowered LDL-C cardiac events still occur.
Low high-density lipoprotein cholesterol (HDL-C) is often present in high-risk subjects with CVD [4]. Epidemiological studies have identified HDL-C as an independent risk factor in CVD and it has been suggested that raising HDL-C reduces the risk of atherosclerosis [5].
This review centres on a procedure that achieves rapid regression of atherosclerosis by modulating the LDL and HDL particles resulting in decreased influx and increased efflux of cellular cholesterol in plaques.
1.2. Atherogenic Lipoproteins
Cholesterol is an important biological molecule that has roles in membrane structure as well as being a precursor for the synthesis of the steroid hormones and bile acids. Both dietary cholesterol and that synthesized de novo are transported through the circulation in lipoprotein particles.
Dietary cholesterol is transported from the small intestine to the liver within chylomicrons. Cholesterol synthesized by the liver, as well as any dietary cholesterol in the liver that exceeds hepatic needs, is transported in the serum within LDLs. The liver synthesizes very low-density lipoproteins (VLDLs) and these are converted to LDLs through the action of endothelial cell-associated lipoprotein lipase. LDL transports a major cholesterol load from the liver to the peripheral cells of the body.
Under conditions of impaired LDL catabolism in the periphery, LDL particles persist in the circulation, their physicochemical characteristics are modified, and the physiological pathway of LDL degradation—via LDL receptors—fails. The consequence of this sequence of events is the formation of an alternative metabolic pathway of LDL degradation through scavenger receptors and the formation of cholesterol deposits in the subendothelial space of the arterial wall. In this way, the process of atherogenesis and atherothrombosis begins (Figures 1 and 2); and LDL particles play a crucial role in the beginning and in the development of this injury process in the vessel walls.
All lipoproteins that contain ApoB, such as LDL, VLDL, and intermediate-density lipoprotein, tend to promote atherosclerosis. Each of these lipoprotein fractions is heterogeneous regarding their lipid and protein contents. Even a narrow LDL fraction with a density of 1.027 - 1.050, initially considered as lipoprotein B consisting reputedly of only ApoB, was subsequently shown to be heterogeneous in its protein contents as shown with ultracentrifugation, gel permeation chromatography, polyacrylamide gel electrophoresis and isoelectric focusingstudies [6].
Several cross-sectional studies have reported differences in LDL particle size, density, and composition between patients with CHD and healthy controls. The presence of small, dense LDL particles is reportedly associated with more than 3-fold increase in CHD risk.
Lipoprotein (a) (Lp(a)) particles contain Apo(a) and ApoB in a 1:1 molar ratio and is considered an independent risk factor for CHD.
1.3. Antiatherogenic Lipoproteins
Numerous epidemiologic and interventional studies have shown that HDL is an important risk factor for coronary heart disease. There are several well-documented HDL functions that may explain the antiatherogenic effects of this lipoprotein. The foremost is the capacity of HDL to transport cholesterol from the periphery to the liver, and therefore prevent cholesterol deposition in the arterial wall. Further antiatherogenic properties of HDL include its potent antioxidative and antiinflammatory actions.
Plasma HDL is a heterogeneous group of discoid and spherical particles that differ in physicochemical properties, intravascular metabolism and biological activity. Discoid HDL particles include small, lipid-poor, nascent particles made up of apolipoproteins embedded in a lipid monolayer of phospholipids and free cholesterol. The plasma concentrations of these small lipid-poor HDL particles are low and constitute about 5% of total HDL. Spherical HDL particles are larger, mature particles that additionally contain a hydrophobic core of cholesteryl esters and triglycerides. The plasma concentration of spherical HDL constitutes approximately 93% of total HDL. Spherical HDL particles are generated from lipidfree ApoA1 or discoid HDL, which is mainly produced by the liver and intestine or can be released as surface fragments from triglyceride-rich lipoproteins during lipolysis. HDL particles possess multiple antiatherogenic activities, which include reverse cholesterol transport (RCT) from the arterial wall to the liver for excretion, and antioxidative, anti-inflammatory, antiapoptotic, antithrombotic, anti-infectious and vasodilatory properties.
Cholesterol efflux from peripheral blood cells, particularly macrophages, constitutes the initial step of RCT. Lipid-free HDL apolipoproteins, primarily ApoA1, cause specific efflux of cellular cholesterol and phospholipids via ABCA1 receptor. Decrease in the lipid content of HDL increases its capacity to remove cellular cholesterol; small, lipid-poor HDL particles thus represent moreefficient cholesterol acceptors than their large, lipid-rich counterparts.
1.4. Apolipoproteins
The importance of apolipoproteins in lipid metabolism has long been recognized [7].
LDL and HDL are composed of roughly 22% and 50% protein, respectively. The protein components of these particles are largely responsible for carrying out the various functions. Tremendous advances in the field of mass spectrometry have allowed the identification of a vast array of proteins, many of which were previously not known to be associated with lipoproteins. The proteomic studies have identified over fifty different proteins in the HDL particle. Many of these proteins mediate the atheroprotective functions of HDL. Much more work is required to identify the functions of these proteins in all the lipoprotein classes.
Nevertheless, at this stage of our understanding it appears, at face value, that reducing lipoproteins containing ApoB and increasing the concentration of lipid-free or lipid deficient HDL particles will be beneficial for the treatment of atherosclerosis.
1.5. Study Aims
In this report it is shown that plasma or serum delipidation with organic solvents resulting in reduction of lipid components of all, or specific, lipoprotein classes results in rapid regression of atherosclerosis.
2. METHODS
2.1. Development of the Delipidation Process
It was hypothesized to develop a plasma or serum delipidation procedure that would result in:
• removal of lipid from biological fluids without protein denaturation.
• simultaneous extraction of different classes of lipids but at different rates.
• extraction of partial or total lipids from lipoprotein fractions but at different rates.
Different classes of lipids associated with different apolipoproteins circulate in biological fluid as lipoproteins. These lipids and apolipoproteins differ in their binding affinities. For example, the neutral lipids triglycerides and cholesterylesters are very hydrophobic and are associated at hydrophobic cores within the lipoproteins. Phospholipids, on the other hand, are amphiphilic lipids that possess both hydrophilic and lipophilic (hydrophobic) properties that bind differently than the neutral lipids to apolipoproteins.
In order to obtain delipidation of lipoproteins without protein denaturation, molecules were required to compete and displace the lipids from the apolipoproteins in a mild non-denaturing manner.
The binding affinity of lipids to apolipoproteins is dependent to some extent on their dielectric constants (DEC). DEC gives an idea of the hydrophilic, hydrophobic and amphiphilic nature of the lipids. For example triglycerides have a DEC of about 3.1; cholesterylesters 2.2; cholesterol 2.9 and for phospholipids such as lecithin the DEC is 30 for the head group (hydrophilic) water interface and a DEC of 10 for the ester group (hydrophobic) region. For a reference purpose water has a DEC of 80.
It was considered advantageous to develop a biphasic organic/aqueous delipidation system, so that after the delipidation procedure the lipid moieties could be conveniently separated from the aqueous phase, which contain the apolipoproteins.
Various organic solvents were investigated to determine their suitability for delipidation. These organic solvents had to meet crucial conditions. In particular, they should not denature proteins or even inactivate enzymes.
Eventually, diisopropyl ether (DIPE) or a related halogenated form of this ether (sevoflurane) [8,9] was shown to be a good delipidator for neutral lipids. DIPE has a DEC of 3.8 and could displace triglycerides (DEC3), cholesterol esters (DEC 2.2) and cholesterol (DEC 2.9) from the lipoprotein particles. However, the DEC of DIPE was too low to compete with the binding of phospholipids to apolipoproteins. Butanol with a DEC of 17.8 was shown to be adequate to release phospholipids from their lipoproteins.
It was also crucial to be aware that mixed solvent systems, especially in ternary mixtures consisting of a biphasic system with biological fluids would have an effect on the DEC of the individual components. For example butanol mixed singly with plasma will result in protein denaturation and even precipitation. However, butanol present in DIPE then mixed with plasma will not denature proteins. These phenomena depend on the ratio of butanol and DIPE.
In 1976 it was first reported that a mixture of butanol and DIPE attained removal of triglycerides, cholesterol, cholesteryl esters, phospholipid and unesterified fatty acids from plasma without protein denaturation. The plasma proteins, including the apolipoproteins remained in solution in the aqueous phase, while the organic phase contained the dissolved lipids. The phases could easily be separated by low speed centrifugation. Different lipids were simultaneously extracted, but the rate of extraction was most rapid for unesterified fatty acids, followed by triglycerides, esterified cholesterol, cholesterol and phospholipids at, respectively, decreasing rates. Selective extraction of unesterified fatty acids, triglycerides and total cholesterol was achieved by DIPE alone. Ionic strength, pH, enzyme activities, and other serum constituents were not altered by these procedures [8].
Delipidation of plasma removes only lipids. After delipidation the apolipoproteins including ApoB remain in solution in the aqueous phase [8-10]. Figure 3 shows an
 (a)
(a) (b)
(b) (c)
(c)
Figure 3. An example of the appearance of an undelipidated turbid plasma sample (a), a delipidated sample, the upper organic solvent phase contains the lipids, the lower aqueous phase contains all plasma constituents but no lipids (b), and the recovered lipids extracted from the plasma sample (c).
example of the appearance of an undelipidated plasma sample, a delipidated sample and the recovered lipids extracted from the plasma sample by a butanol-DIPE mixture. Lipids are extracted from the turbid plasma sample (a) into the upper organic phase, resulting in a clear delipidated plasma (b). When the organic phase containing the extracted lipids is evaporated, the lipids originally present in the undelipidated plasma are recovered (c).
The DIPE-butanol delipidation procedure has been applied to plasma or lipoprotein fractions for the assay of many apolipoproteins [11-22].
Other lipid-associated proteins in biological fluids were also unaffected when their lipids were removed by the delipidation procedure [23-26]. Removal of lipids from such proteins did not affect their physicochemical roles and in fact this delipidation procedure has been used to estimate cholesteryl ester transfer/exchange activity in serum or plasma [27,28].
It was also shown that delipidation of plasma with butanol/DIPE mixtures yielded ordered delipidation of HDL, VLDL and LDL in plasma. HDL was more readily delipidated than were LDL and VLDL fractions [8-10].
2.2. Relipidation of Apolipoproteins
2.2.1. Extracellular Lipids
In vitro studies showed that the apolipoproteins in delipidated plasma could combine with extracellular lipids to form lipoprotein complexes in the HDL, LDL and VLDL fractions. Reconstitution of the HDL particle with extracellular lipids was more complete than with the LDL and VLDL fractions [29].
2.2.2. Intracellular Lipids—In Vitro Cell Culture Studies
Partial delipidated LDL containing virtually no cholesterol exhibits a binding activity to cultured human fibroblasts identical to that obtained with untreated LDL. This very important observation means that receptors on cells will recognize LDL even if the LDL contains virtually no cholesterol. The recognition of ligand by a receptor is very specific and confirms immunological studies that the delipidation procedure does not cause denaturetion [8-29]. Delipidated LDL did not stimulate cellular cholesteryl ester (ACAT) synthesis indicating that no cholesterol was presented to those cells by delipidated LDL [30,31].
Gangliosides are amphiphilic molecules consisting of very polar sialic acid-containing glycosphingolipids and have relatively high DEC of approximately 33 to 47. The DEC of these lipids are much higher than the DEC of the selected organic solvents and accordingly, gangliosides are not extracted from plasma with the butanol-DIPE mixture under the conditions studied [32]. Gangliosides constitute approximately 0.7% of total lipid in plasma and they are bound to lipoprotein particles. Lipoprotein bound gangliosides in plasma have been implicated with the binding of the LDL particle to the specific high affinity LDL receptor and may have some relevance to the binding of delipidated LDL to its specific receptors [33].
Partial delipidation of HDL with DIPE-butanol mixtures converted the spherical lipoprotein particles to disc and amorphous forms but this did not alter receptorbinding activity [31,34,35].
Delipidated serum effluxes cholesterol from human mononuclear leukocytes. When delipidated serum substantially depletes the contents of cellular cholesterol, stimulation of intracellular sterol synthesis occurs by a factor of eight to eventually meet the needs of cholesterol for the survival of the cells. However, the intracellular sterol synthesis can be inhibited by the addition of nondelipidated LDL. This indicates that cells preferentially utilize cholesterol given to them rather than synthesizing their own cholesterol [30]. The activity of HMGCoA reductase (the rate limiting enzyme for the synthesis of cholesterol) in skin and lung fibroblasts increased 5 to 30 fold in media containing delipidated serum AFTER substantial removal of endogenous cellular cholesterol causing cellular cholesterol depletion when compared with media supplemented with non-delipidated serum [31]. Addition of non-delipidated LDL to the cells resulted in the inhibition of cholesterol synthesis within the cells [34]. It was subsequently established that the component in the delipidated serum responsible for the removal of cholesterol from cells grown in culture was delipidated HDL and that delipidated HDL was a much better (more than 6 fold) cholesterol acceptor than non-delipidated HDL [34].
3. RESULTS
3.1. Whole Animal Studies
3.1.1. LA Procedure
Plasma delipidation therapy when applied in vivo to whole animals was termed lipid apheresis (LA) [36-39]. LA, an extracorporeal procedure, when applied to diet and drug induced hyperlipidemic roosters resulted in a rapid reduction of plasma cholesterol and triglyceride concentrations. The LA procedure consisted of the removal of approximately 25% of the calculated blood volume. The blood was collected in heparinized tubes and the blood cells were separated from plasma by centrifugation. The plasma was delipidated with a mixture of butanol and DIPE. The delipidated plasma, free of lipids and solvents, was remixed with the original blood cells, which was then reinfused through a vein back into the identical donor animals. Sham treatments served as placebos, were also performed, but in this case no delipidation was applied to the plasma.
3.1.2. Effect of Infused Delipidated Autologous Plasma (LA) on In Vivo Plasma Lipids over a Period of 16 h
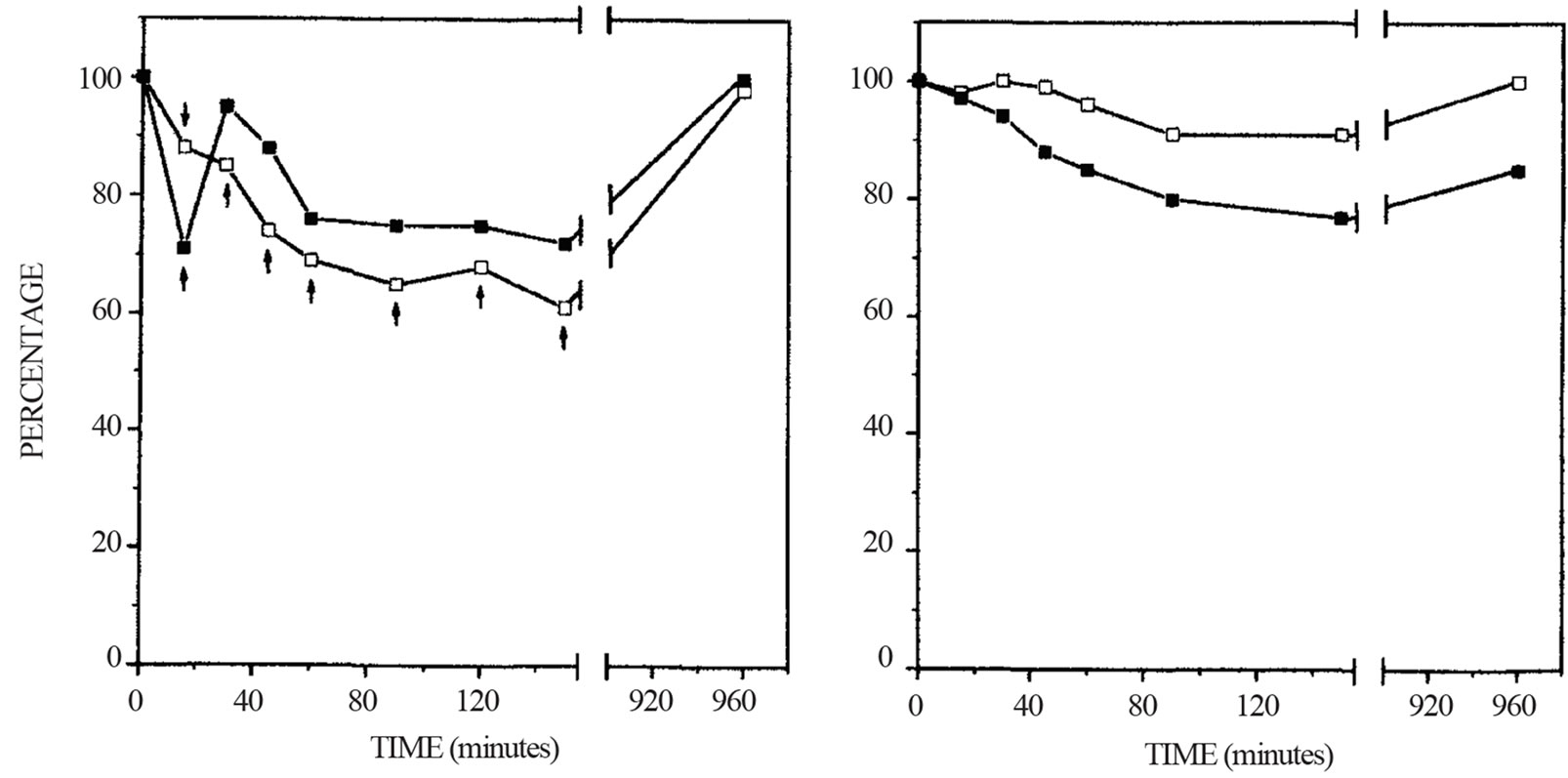
One LA treatment resulted in immediate reduction of plasma cholesterol and triglyceride concentrations in the vascular pool of the animals. These changes were highly significant when compared with the sham treated groups [36]. Two LA treatments resulted in more effective removal of lipids from the plasma in the animals. Lipid was also removed from lipid stores in the liver and aorta of hyperlipidemic animals [38]. One LA treatment resulted in rapid reduction of plasma cholesterol. In the normolipidemic animals the reduction of total cholesterol was immediate and was sustained for up to 150 minutes. Sixteen hours after LA, the concentrations of total cholesterol in plasma had returned to pretreatment values. In contrast, with the hypercholesterolemic animals, LA reduced the plasma level of total cholesterol only very transiently. Fifteen minutes after LA, total cholesterol was significantly reduced in the plasma of the hypercholesterolemic animals. Thirty minutes and thereafter, following LA treatment, the concentration of total cholesterol in plasma was generally lower than the pretreatment value but were not statistically different from the pretreatment value (Figure 4). Sham treatment resulted in no statistical changes in plasma total cholesterol.
These data strongly suggest that the reason for the different responses to LA treatment observed in hypercholesterolemic animals when compared with normocholesterolemic animals is that more of an excessive cholesterol pool in the hypercholesterolemic animals exist that
 (a) (b)
(a) (b)
Figure 4. Changes of plasma total cholesterol concentrations after lipid apheresis or sham treatment. The plasma cholesterol concentrations of roosters on control diet (☐-☐) and hypercholesterolemic diet (n-n) were determined before and at various times after reinfusion of lipid apheresed blood or sham treated blood. The results are expressed as percent changes. The data of the two groups of animals were pooled to determine statistical differences. The concentrations of plasma cholesterol before lipid apheresis (a) were 3.25 ± 0.87 (n = 7) and 14.10 ± 4.30 (n = 7) mmol/L for the normocholesterolemic and hypercolesterolemic roosters, respectively. The concentrations of plasma cholesterol before sham treatment (B) were 3.18 ± 0.66 (n = 3) and 11.60 ± 2.53 (n = 3) mmol/L for the normocholesterolemic and hypercholesterolemic roosters, respectivley. Arrows indicate significant differences (P < 0.05) of those values compared to the pre-treatment values (100%).
is rapidly mobilizable by the LA treatment [36] and this supports the observations with cell culture studies.
3.1.3. In Vitro Effect of the Delipidation Procedure on Plasma Components
Delipidation of plasma in vitro did not affect the concentrations of total protein, albumin, immunoglobulins, and other ionic and chemical constituents. The activities of AP, LDH, AST, GGT and ALT were also unaffected; only lipids were removed from the plasma by the delipidation procedure [8-10].
3.1.4. Effect of Infusion of Delipidated Autologous Blood (LA) and Sham Treated Blood (Placebo) on In Vivo Haematological and Biochemical Parameters
Biochemical and haematological parameters obtained from each animal prior to and after infusion of delipidated autologous plasma and sham treated plasma remixed with the original blood cells indicated that these parameters remained within the recommended normal range. There were no significant changes in these parameters as a consequence of LA or sham treatment [36].
3.1.5. Effect of LA on the Electrophoretic Pattern of In Vivo Plasma Lipoproteins. Stimulation of Antiatherogenic HDL particles
Analysis of the lipoprotein particles after LA treatment of hypercholesterolemic and normocholesterolemic animals resulted in conspicuous changes in electrophoretic patterns of plasma lipoproteins. The electrophoretic mobilites of all the lipoprotein fractions had changed. Lipid was present in at least three bands in the α globulin area. In particular, changes in the electrophoretic region of HDLs were observed. LA markedly induced the antiatherogenic pre-ßHDL. The observed changes induced by LA were more pronounced in the hypercholesterolemic animals compared with the normocholesterolemic animals. A novel pre-αHDL-lipoprotein band was observed soon after LA. This lipoprotein band had a density larger than 1.21. At approximately 150 minutes after LA the electrophoretic pattern had almost returned to the original pre-LA treatment pattern (Figure 5).
LA treatment resulted in immediate reduction in plasma unesterified cholesterol concentrations, which was sustained for 150 minutes. The reduction in unesterified cholesterol concentration was higher in the normocholesterolemic animals than in hypercholesterolemic animals. LA induced changes in the ratio of plasma unesterified to total cholesterol in normocholesterolemic animals but not in hypercholesterolemic animals. In hypercholesterolemic animals, lecithin-cholesterol acyltransferase (LCAT) activity was not affected by LA, whereas in normocholesterolemic animals LCAT activity, but not concentration, was acutely reduced for 150 minutes after LA. Saturated LCAT kinetics occurred in the hypercholesterolemic animals but not in the normocholesterolemic animals (Figure 6).
LCAT obeyed Michaelis-Menten kinetics (Figure 7).
It was shown that after LA, there was a pool of unesterified cholesterol that was available as substrate for LCAT to a greater extent in hypercholesterolemic animals than in normolipidemic animals [39]. These observations are very important because it shows that LA has desirable effects on LCAT activities in hypercholesterolemic and normocholesterolemic animals. In hypercholesterolemic animals there is a much larger pool of total body cholesterol, including intracellular and extracellular plaque cholesterol [38], under these conditions LCAT is
 (a)
(a)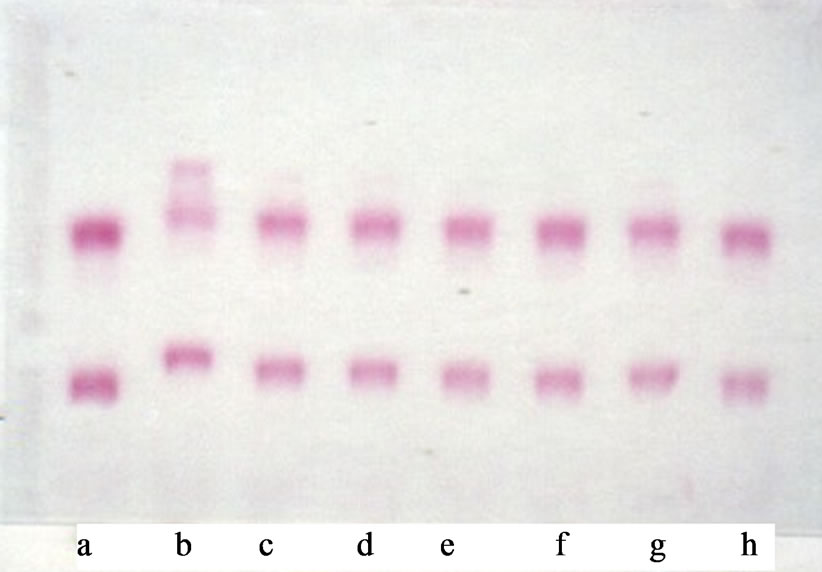 (b)
(b)
Figure 5. Representative agarose electropherograms of plasma before and various times after introduction of delipidated autologous plasma to the vascular pool in a hypercholesterolemic rooster. Lipid and protein were stained with Fat Red 7B (a) and Amido Black 10B (b) respectively: a, before: b-h respective time in minutes. 15, 30, 45, 60, 90, 120 and 150 after introduction of delipidated autologous plasma (approximately 25% of plasma volume) into roosters. These findings are representative of the results obtained from seven experiments.

Figure 6. Changes in plasma LCAT activity after lipid apheresis. The plasma LCAT activities (μmol L−1 h−1) of roosters on control diet (☐) and hypercholesterolemic diet (n) were determined before and at various times after reinfusion of lipid apheresed blood. The LCAT activities before LA were 279.5 ± 152.8 (n = 10) and 327.1 ± 40.5 (n = 10) mmol L−1 h−1 for the normocholesterolemic and hypercholesterolemic animals, respectively. The results are expressed as percentage changes. The data from the two groups of animals were pooled to determine statistical differences. Arrows indicate significant differences (P < 0.05) in those values compared with the pre-lipidapheresed values (100%).

Figure 7. Double-reciprocal plot of LCAT activity and unesterified cholesterol concentration. The plasma LCAT activities (μmol·L−1·h−1) of roosters on control diet (☐) and hypercholesterolemic diet (n) were determined before and seven times after reinfusion of lipid apheresed blood. Results are the mean values at each time point. For the plot (r = 0.95) the calculated Km and Vmax are 1.89 mmol·L−1 and 676 μmol·L−1 h−1 respectively.
working under Vmax kinetics. Unesterified cholesterol in subfractions of plasma HDL is the preferred substrate for LCAT [40]. Separate preparations of HDL3 differ markedly in their reactivity with LCAT. This difference has been shown to be inversely related to lipoprotein particle size. The activity of LCAT depends on the structure of ApoA1 at the interface with lipid [41]. LA induces changes in HDL to pre-ß-like HDL particles and these particles are regarded as good acceptors of unesterified cholesterol of cellular origin and the preferred substrates for LCAT [37].
Five LA treatments in normolipidemic and hyperlipidemic animals with increased concentration of plasma cholesterol, triglycerides and phospholipids resulted in the removal of considerably more lipid from hyperlipidemic animals than in normolipidemic animals. LA treatments of hyperlipidemic animals markedly reduced atherosclerosis in the arterial wall and also led to dramatic reduction of lipid in the adipose tissue [38,42]. Five LA treatments also increased the concentrations of ApoA1 in both normolipidemic and hyperlipidemic animals [42,43]. It is not known whether these increases were due to increased synthesis or decreased catabolism. Histological analyses of the aortic sections of animals fed the control diet developed minimal aortic fatty streaks. In the hyperlipidemic animals, it was shown that there was a marked atherosclerotic lesion in the aorta with part occlusion. The LA treatment of the hyperlipidemic animals resulted in a remarkable reduction of the intensity of lipid in the arterial wall as well as marked increases in the lumen diameter. Figure 8 shows a representative section of a lesion with significant narrowing of the lumen of the thoracic aorta of a hyperlipidemic animal and a representative section of a similar site of the thoracic aorta of a hyperlipidemic animal that had undergone five LA treatments.
Higher magnification of the media sections of the abdominal aorta in hyperlipidemic animals shows the presence of intimal accumulation of extracellular lipid, whereas LA treatment of hyperlipidemic animals had little such lipid accumulation (Figure 9).
Internal elastic lamina disruption and medial damage occurred in the sections of the intima of abdominal aorta in the hyperlipidemic animals, which was not seen in normolipidemic animals. The intima of the abdominal aorta of LA treated hyperlipidemic animals showed much less damage (Figure 10).
The intima-media thickness of the aorta and its lipid content in the hyperlipidemic animals were overall greater than in the corresponding LA-treated hyperlipidemic animals (Figure 11).
Substantive mesenterical adipose tissue was clearly present in the abdomen of hyperlipidemic animals. This pool of lipid was drastically diminished when the hyper-


Figure 8. Histological sections (magnified ×50) of thoracic aortas stained with Oil Red O. (a) Aorta from hyperlipidemic roosters showing lipid-rich atherosclerotic plaque and significant luminal narrowing. (b) Aorta from hyperlipidemic rooster after five LA treatments showing no evidence of atherosclerosis.
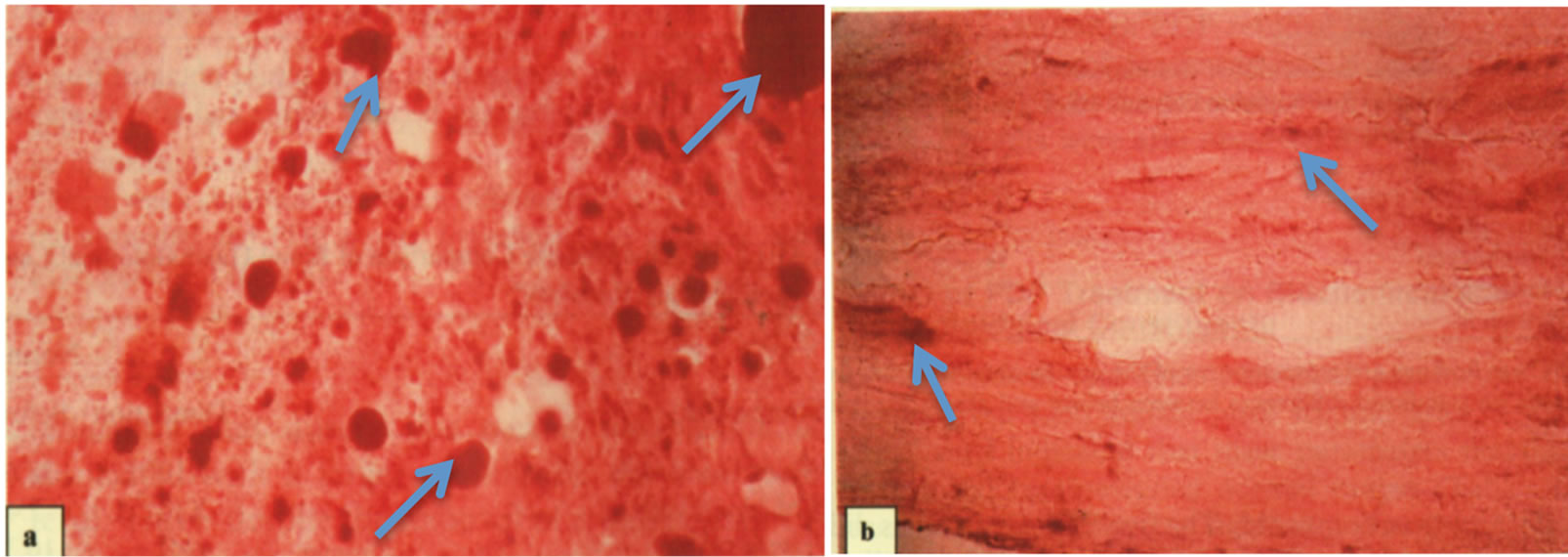
Figure 9. Histological sections (magnified ×800) of abdominal aortas stained with Oil Red O. (a) Aorta from hyperlipidemic roosters showing extensive intimal accumulation of extracellular lipid (see arrows). (b) Aorta from hyperlipidemic roosters after five LA treatments showing little lipid accumulation (see arrows).


Figure 10. Histological sections (magnified ×800) of abdominal aortas. (a) Aorta from hyperlipidemic rooster showing internal elastic lamina disruption and medial damage. (b) Aorta from hyperlipidemic rooster after five LA treatments showing much less elastic lamina and medial damage.
lipidemic animals were treated by five LA procedures (Figure 12).
3.1.6. LA Treated Monkeys
Multiple LA treatments of diet-induced hyperlipidemic non human primates such as the African Green monkey gave similar results to those obtained with the avian model. With the study of the monkeys, plasma was delipidated with butanol-ether to yield selective delipidation of HDL. As with the previous studies in the avian


Figure 11. Histological sections (magnified ×25) of the abdominal aortas. (a) Thick intima media aortas from hyperlipidemic roosters showing lipid stainable material. (b) Thinner intima media aortas from hyperlipidemic roosters after five LA treatments showing less lipid stainable material.
 (a)
(a) (b)
(b)
Figure 12. Abdominal cavities of (a) hyperlipidemic rooster showing substantive mesenterical adipose tissue and (b) hyperlipidemic rooster after five LA treatments showing markedly diminished adipose tissue.
model, cholesterol-rich αHDL particles were converted to a mixture of cholesterol-poor small α1 and pre-ß-like HDLs. Pre-ßHDL had a plasma residence time of 8 ± 6 h and was converted entirely to large αHDL, this was slightly longer than the residence time of pre-ßHDL observed in the LA treated avian model. It was shown that treatment with delipidated plasma reduced diet-induced aortic atherosclerosis in monkeys measured by intravascular ultrasound (IVUS) [44].
3.1.7. LA Treated Humans
Plasma delipidation studies with the butanol-ether mixture, resulting in selective HDL delipidation using autologous infusions in man, have shown to be feasible and well tolerated in patients with acute coronary syndromes (ACS). The levels of pre ß-like HDL and αHDL in the delipidated plasma transiently converted from 5.6% to 79.1% and 92.8% to 20.9%, respectively and confirms the observation of LA treatment with the avian and monkey models. Patients underwent IVUS evaluation of the target vessel during the catheterization for ACS and up to 14 days following the final LA session. The IVUS data demonstrated a numeric trend toward regression in the total atheroma volume of −12.18 ± 36.75mm3 in the delipidated group versus an increase of total atheroma volume of 2.80 ± 21.25mm3 in the control group (p = 0.268, N.S.) [45].
4. DISCUSSION
Reverse cholesterol transport is a multi-step process resulting in the net movement of cholesterol from peripheral tissues back to the liver via the plasma. Cholesterol from non-hepatic peripheral tissues is transferred to HDL by the ABCA1 receptor. Different forms of ApoA1 complexes with different binding affinities for cholesterol coexist. The ApoA1 containing HDL particles act as sinks for mobilizable cholesterol. The cholesterol is converted to cholesteryl esters by the enzyme LCAT. With the help of CETP the cholesteryl esters are exchanged for triglycerides with other lipoproteins such as LDL and VLDL. The LDL cholesteryl esters end up in the liver where they are partly excreted as bile salts.
It is not surprising that ApoA1 containing HDL particles with high capacity for cholesterol and high affinity for ABCA1 receptors are being sought to obtain optimum reverse cholesterol transport. Investigational particles consist of HDL mimetics. HDL mimetics have been constructed from a number of peptides and proteins with varying structures, all of which bind lipids found in HDL. Reconstituted HDL consisting of normal human ApoA1 when combined with soybean phosphatidyl choline initially showed promise as a RCT agent. However, the Erase trial with this recombinant HDL candidate was discontinued because of liver function toxicity [46].
Although cell culture work may show promise for ApoA1 peptide mimetics, it should not be overlooked that these peptides when associated with lipid in the vascular pool may aggregate to a larger new molecule with conformational novelties that the body may consider as a foreign lipid-protein complex which could result in consequential rejection of the body and creation of antibodyies which would be disastrous [9].
Plasma delipidation studies presented in this review show that it is possible to delipidate lipoproteins in plasma without apolipoprotein denaturation. The versatility of this biphasic organic solvent system has tremendous possibilities for the delipidation of biological fluids. The composition of the specific ether-alcohol mixture and the energy applied to the delipidation process can be adjusted to obtain a particular outcome. For example, different lipids can be extracted simultaneously but the rates of extraction differ for the particular lipids and indeed it is possible to extract all neutral lipids but no phospholipid from plasma. The amount of energy applied to the delipidation process determines whether delipidation occurs within minutes or seconds. The ratio of organic solvents to plasma can be very variable. Ordered delipidation can be achieved in plasma. HDL is firstly delipidated by this solvent system, followed by VLDL and LDL. Thus by choosing the appropriate organic solvent system and the amount of energy applied for delipidation it is possible to remove a particular class or classes of lipid from plasma. Using these attributes it was shown that plasma delipidation resulting in the removal of over 90% of cholesterols and triglycerides but retaining over 60% of phospholipids achieved rapid regression of atherosclerosis in a hypercholesterolemic avian model, a porcine model (in preparation) and a bovine model (in preparation). It was shown in a hyperlipidemic avian model that adipose tissue could also be mobilized.
Using this delipidation procedure it is possible to obtain delipidation of HDL only with little or no altered LDL [8-10] and the partially delipidated plasma contains modified HDL particles with no cholesterol or other lipids. These attributes have led to studies with African Green monkeys in which selective delipidation of HDL in plasma was applied to diet-induced hypercholesterolemic animals. Treatment with delipidated plasma tended to reduce atherosclerosis measured by IVUS. A similar study in humans with acute coronary syndrome using delipidated HDL in plasma confirmed previous studies in all the animal models, that the procedure was safe and the levels of pre ß-like HDL increased significantly. The human studies also showed that the IVUS data demonstrated a numeric trend toward regression in the total atheroma volume.
The regression of atherosclerosis, although not significant, in the humans and monkeys which were similar to the results reported in ApoA1 Milano studies [47] were reported to be encouraging. Regression of atherosclerosis was more pronounced in the avian model. This may be explained by the different precise conditions of the delipidation conditions used with the various models.
In this regard, it is very possible that delipidated VLDL and LDL also play roles in RCT.
Figure 13 [48] depicts a simplistic view of the current understanding of the process of reverse cholesterol transport. In all the in vivo studies it was shown that delipidation converted spherical HDL to more antiatherogenic apolipoprotein complexes such as pre-ßHDL. It was further shown that in hyperlipidemia more of these protein complexes were generated compared with normolipidemic animals. These protein complexes had a transitory residence time in the plasma after LA and were rapidly converted to αHDL. This was dependent on LCAT, which showed that in hypercholesterolemic animals this enzyme was operating at maximum capacity, thus rapidly converting unesterified cholesterol in the pre-ßHDL to esterified cholesterol in the αHDL. This did not apply to normocholesterolemic animals in which it was shown that there were reductions in LCAT activities but for concentrations up to 16 h after LA and this was dependent on the availability of the LCAT substrates lecithin and unesterified cholesterol. Km and Vmax values of LCAT determined that such changes were not due to the protein content of LCAT but due to its activity. The cholesteryl esters domiciled in the spherical αHDL molecule may be transported and delivered to the liver through interaction with the Scavenger Receptor class B-type-1 (SR-B1). The cholesteryl esters are then hydrolysed by the enzyme Cholesterol Ester Hydrolase (CEH) in the liver to unesterified cholesterol some of which is ultimately converted to bile salts and eliminated in the feces. In addition, the cholesteryl esters in the spherical αHDL can be transferred to VLDL and LDL particles especially if those
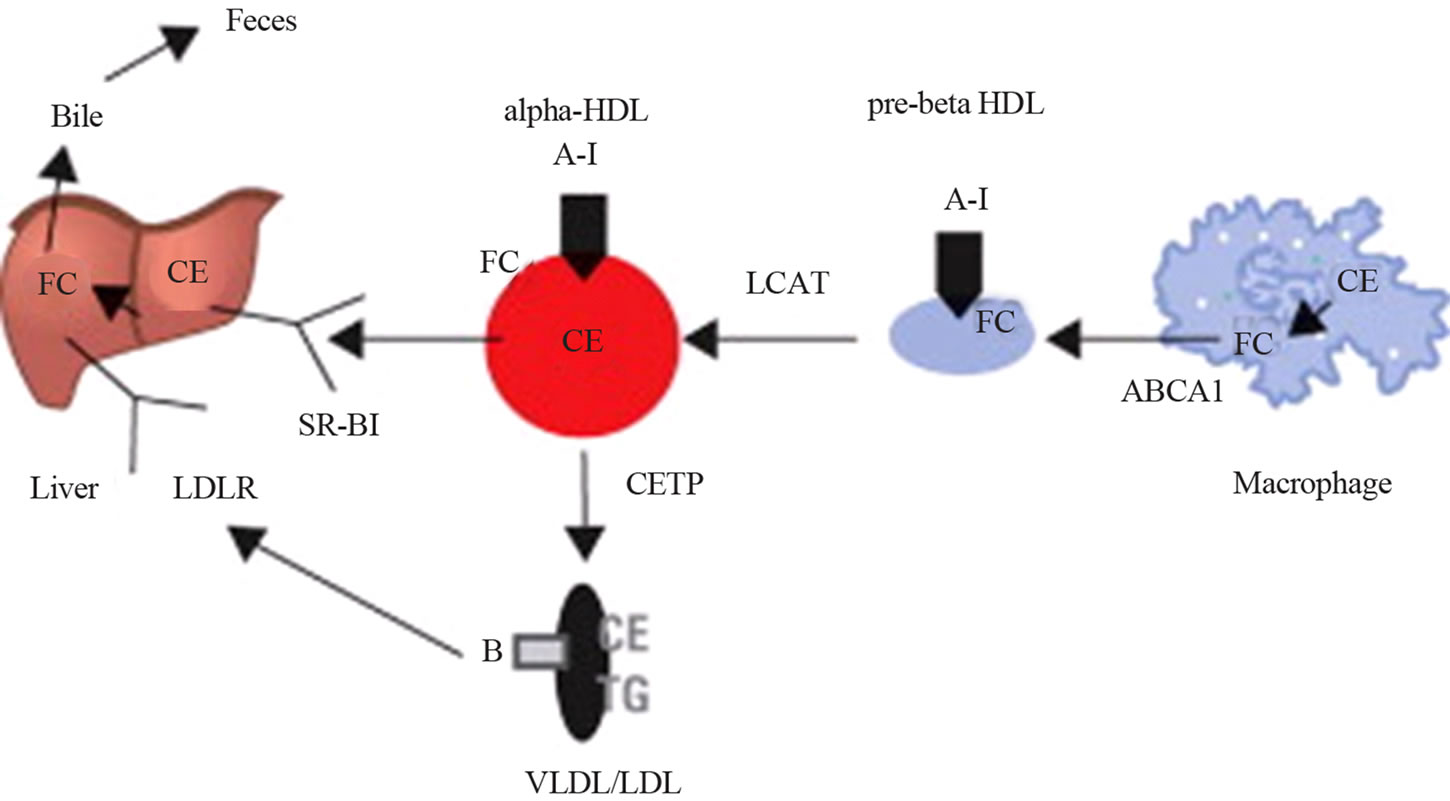
Figure 13. The Process of Reverse Cholesterol Transport Discoidal pre-beta-high-density lipoprotein (HDL) is converted to spherical alpha-HDL as cholesterol ester (CE) accumulates and is returned to the liver by scavenger receptor B1 (SR-B1) or transfers the cholesteryl ester to apolipoprotein B lipoproteins by the cholesteryl ester transfer protein (CETP). ABCA1 = adenosine triphosphate-binding cassette-A1; A-I = apolipoprotein A-I; B = apolipoprotein B; FC = free cholesterol; LCAT = lecithin cholesterol acyltransferase; LDL = low-density lipoprotein; LDLR = low-density lipoprotein receptor; TG = triglycerides; VLDL = very low-density lipoprotein.
lipoproteins are cholesterol deficient in the case of delipidated plasma with the aid of Cholesterol Ester Transfer Protein (CETP), which is a plasma component.
It was shown that the delipidation solvents described in this review did not affect cholesteryl ester transfer activity (CETA) and this activity was about two-fold higher in delipidated plasma of hyperlipidemic subjects than in delipidated plasma of normolipidemic controls [27,28]. Thus delipidated VLDL and LDL may act as cholesterol sinks due to the action of CETP on αHDL. Cholesterol enriched LDL and cholesterol depleted LDL induced by plasma delipidation both bind to the LDL receptors evenly [30,31].
With the RCT model (Figure 13) the cholesteryl esters in VLDL and LDL are transported to the liver where they interact with LDL receptors and subsequently contribute to the pool of unesterified cholesterol to be excreted in the feces or avail themselves for de novo synthesis of VLDL for excretion into the plasma compartment.
Reverse Cholesterol Transport is induced by delipidation of lipoproteins in plasma as shown in the following in vitro and in vivo studies with animals and in human subjects.
Delipidation of plasma introduced into the vascular pool results in:
• Generation of a large number of pre-β HDLs.
• These pre-β HDLs avidly bind and remove cholesterol from macrophages through the ABCA1 pathway (direct RCT).
• LCAT, at maximum capacity with hypercholesterolemia, converts unesterified cholesterol derived from the macrophages now in pre-β HDL repleted cholesterol, to form esterified cholesterol in a spherecal αHDL particle.
• The CETA from the enzyme CETP is increased and transfers cholesteryl esters from α-HDL to delipidated VLDL and LDL particles that act as sinks for the cholesterol initially present in the macrophages (indirect RCT).
• These LDL particles in plasma when presented to the liver interact with the LDL receptors (indirect RCT)
• The unesterified and esterified cholesterol in the αHDL also interacts with SR-B1 receptors in the liver (direct RCT).
• Cholesteryl esters presented to the liver by LDL through LDL receptors and from αHDL by SR-B1 receptors are hydrolysed by CEH to unesterified cholesterol which is subsequently excreted through the biliary system into the feces as bile salts and unesterified cholesterol.
• Substantial pools of cholesterols are physically removed from plasma into the organic phase by plasma delipidation (Figure 3).
These interactions and reactions are stimulated by the delipidation of all the lipoproteins in the delipidated plasma. Therefore, there is an increased shift in cholesterol initially present in the macrophage to the liver for excretion from the body, the hallmark of RCT (Figure 13).
Delipidation and the Effects of Forward Transport of Lipids (from the Liver and Intestine to the Periphery)
Mechanisms by which LDL causes atherosclerosis are multifactorial. It is proposed that delipidation of plasma, including the LDL particle, may result in the decrease of atherosclerosis formation.
Cell culture studies have established that delipidated LDL particles in plasma containing virtually no cholesterol exhibits a binding activity identical to that obtained with undelipidated LDL. Thus, LDL receptors recognize and bind delipidated LDL particles. Delipidated LDL does not stimulate cellular ACAT synthesis or activity indicating that no cholesterol is presented to those cells by delipidated LDL. Thus less cholesterol is presented to the already cholesterol overburden macrophages. It was further shown that cells preferentially utilize cholesterol given to them rather than synthesizing their own. After substantial removal of endogenous cellular cholesterol by delipidated serum when compared with media supplemented with non-delipidated serum, HMGCoA reductase (the rate limiting enzyme for the synthesis of cholesterol) is increased considerably [31]. Addition of non-delipidated LDL to the cells resulted in the inhibition of cellular cholesterol synthesis. These observations suggest the delipidated LDL particle presents less cholesterol to cells compared with native undelipidated LDL possibly resulting in reduced rates of atherosclerosis formation.
These observations for Direct RCT and Indirect RCT induced by delipidated plasma with specific solvents, put together, would seem to be an attractive method for the treatment of atherosclerosis.
Ultimately “The proof of the pudding is in the eating” as exemplified by the observation that LA causes regression of atherosclerosis in various in vivo models.
To the adage “LDL-C is bad and HDL-C is good” it maybe added, “during, and for a period after Lipid Apheresis, some LDL-C is good and HDL-C is super good”.
With the advent of modern proteomic technologies a multitude of proteins have been shown to be present in all the lipoprotein fractions. For example, more than 50 distinct proteins associated with HDL particles have been discovered with many of these proteins being involved in nonlipid transport processes including complement activation, acute phase response and innate immunity. It is important that these newly and, yet to be discovered, proteins associated with lipoprotein particles are not damaged during manipulation of these particles. Already it has been shown that the delipidation with specific organic solvents described in this review does not adversely affect these new proteins associated with the lipoprotein fractions. In fact, the delipidation procedure has been applied to help identify the novel proteins, which are associated with the lipoprotein fractions in plasma [49-51] whereas, a different solvent system consisting of diethyl ether:methanol caused denaturation and precipitation of the delipidated HDL particles [52].
5. CONCLUSIONS
In conclusion, LA of plasma results in removal of lipids from lipoprotein particles without denaturing plasma constituents. The LA process can be adjusted to obtain delipidation of lipid classes, or lipoprotein classes. There is compelling evidence to show that the procedure causes egress of cholesterol from macrophages with cell culture studies. Evidence is also available, when applied in vivo to animals that LA causes reversal of atherosclerosis and, in humans successful shrinking of atherosclerotic lesions in the coronary arteries are observed.
Optimization of the LA procedure in terms of the composition of the mixture of the butanol-ether and conditions for plasma delipidation to determine which lipid(s) to extract from what lipoprotein particles in the plasma, remains to be explored to obtain the most rapid regression and safe method for patients with atherosclerosis and the prevention of atherosclerosis.
REFERENCES
- CSL Company Reports. 2012. Citi Research.
- Libby, P. (2002) Inflammation in atherosclerosis. Nature, 420, 858-874. doi:10.1038/nature01323
- Spady, D.K. (1999) Reverse Cholesterol transport and atherosclerosis regression. Circulation, 100, 576-578. doi:10.1161/01.CIR.100.6.576
- Heart Protection Study Collaborative Group (2002) MRC/ BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomized placebo-controlled trial. Lancet, 360, 7-22. doi:10.1016/S0140-6736(02)09327-3
- Castelli, W.P., Anderson, K., Wilson, P.W. and Levy, D. (1992) Lipids and risk of coronary heart disease: The Framingham Study. American Epidemiology, 2, 23-28. doi:10.1016/1047-2797(92)90033-M
- Cham, B.E., Owen, P., Roeser, H.P., Gaffney, T. and Stanley, B.C. (1981) Heterogeneity of lipoprotein B. Biochemical and Biophysical Research Communications, 103, 196-206. doi:10.1016/0006-291X(81)91679-X
- Cham, B.E. (1978) Importance of apolipoproteins in lipid metabolism. Chemico-Biological Interactions, 20, 263- 277. doi:10.1016/0009-2797(78)90105-9
- Cham, B.E. and Knowles, B.R. (1976) A solvent system for delipidation of plasma or serum without protein precipitation. Journal Lipid Research, 17, 176-181.
- Cham, B.E. (2007) Manipulation of reverse cholesterol transport system—An exploration for rapid regression of atherosclerosis. Research Journal Biological Sciences, 2, 291-300.
- Cham, B.E. and Knowles, B.R. (1976) Changes in electrophoretic mobilities of alpha and beta lipoproteins as a result of plasma delipidation. Clinical Chemistry, 22, 305-309.
- Curry, M.D., Alaupovic, P. and Suenram, C.A. (1976) Determination of apolipoprotein A and its constitutive A-I and A-II polypeptides by separate electroimmunoassays. Clinical Chemistry, 22, 315-322.
- Avogaro, P., Cazzolato, G., Bittolo Bon, G., Quinci, G.B. and Chinello, M. (1978) HDL cholesterol apolipoproteins A1 and B. Atherosclerosis, 31, 85-91. doi:10.1016/0021-9150(78)90040-0
- Kostner, G.M., Avogaro, P., Bittolo Bon, G., Cazzolato, G and Quinci, G.B. (1979) Determination of high-density lipoproteins; screening methods compared. Clinical Chemistry, 25, 939-942.
- Jaukiainen, M.S., Laitinen, M.V., Pentilla, I.M. and Pukakainen, E.V. (1982) Separation of the apoprotein components of human serum high-density lipoprotein: Chromatofocusing a new simple technique. Clinica Chimica Acta, 122, 85-91. doi:10.1016/0009-8981(82)90099-7
- Koren, E., Puchois, P., McConathy, W.J., Fesmire, J.D. and Alaupovic, P. (1985) Quantitative determination of human plasma apolipoprotein A-I by noncompetitive enzyme-linked immunosorbent assay. Clinica Chimica Acta, 147, 85-95. doi:10.1016/0009-8981(85)90068-3
- Koren, E., Puchois, P., Alaupovic, P., Fesmire, J., Kandorissi, A. and Fruchart, J.C. (1987) Quantification of two different types of apolipoprotein A-I containing lipoprotein particles in plasma by enzyme-linked differentialantibody immunosorbent assay. Clinical Chemistry, 33, 38-43.
- Trigatti, B.L., Rigotti, A. and Braun, A. (2000) Cellular and physiological roles of SR-B1 a lipoprotein receptor which mediates selective lipid uptake. Biochimica Biophysica Acta, 1529, 276-286. doi:10.1016/S1388-1981(00)00154-2
- Staprans, I. and Felts, J.M. (1977) The effect of alphaacid glycoprotein (orosomucoid) on triglyceride metabolism in the nephrotic syndrome. BiochemicalBiophysical Research Communications, 79, 1272-1278. doi:10.1016/0006-291X(77)91143-3
- Curry, M.D., McConathy, W.J., Fesmire, J.D. and Alaupovic, P. (1980) Quantitative determination of human apolipoprotein C-III by electroimmunoassay. Biochimica Biophysica Acta, 617, 503-513. doi:10.1016/0005-2760(80)90016-8
- Curry, M.D., McConathy, W.J., Fesmire, J.D. and Alaupovic, P. (1981) Apolipoprotein C-I and C-II levels in human plasma by separate electroimmunoassays. Clinical Chemistry, 27, 543-548.
- Curry, M.D., McConathy, W.J. and Alaupovic, P. (1977) Quantitative determination of human apolipoprotein D by electroimmunoassay and radial immune-diffusion. Biochimica Biophysica Acta, 491, 232-241. doi:10.1016/0005-2795(77)90059-9
- Meunier, S., Gambert, P. and Desgres, J. (1986) Preparative electrophoresis of human apolipoprotein E: An improved method. Journal Lipid Research, 27, 1324-1327.
- Pattnaik, N.M., Montes, A., Hughesand, L.B. and Zilversmit, D.M. (1978) Cholesteryl ester exchange protein in human plasma isolation and characterization. Biochimica Biophysica Acta, 530, 428-438. doi:10.1016/0005-2760(78)90163-7
- Fex, G. and Hansson, B. (1978) Purification of retinolbinding protein from serum and urine by affinity chromatography. Biochimica Biophysica Acta, 537, 358-365. doi:10.1016/0005-2795(78)90519-6
- Rustow, B., Kunze, D., Hodi, J. and Egger, E. (1979) A fatty acid binding peptide of rat liver cytosol. FEBS Letters, 108, 469-472. doi:10.1016/0014-5793(79)80590-6
- Ockner, R.K., Manning, J.A. and Kane, J.P. (1982) Fatty acid binding protein. Journal Biological Chemistry, 257, 7872-7878.
- Groener, J.E.M., Van Rozen, A.E. and Erkelens, D.W. (1984) Cholesteryl ester transfer activity. Localization and role in the distribution of cholesteryl ester among lipoproteins in man. Atherosclerosis, 50, 261-271. doi:10.1016/0021-9150(84)90074-1
- Groener, J.E.M., Pelton, R.W. and Kostner, G.M. (1986) Improved estimation of cholesteryl ester transfer/exchange activity in serum or plasma. Clinical Chemistry, 32, 283-286.
- Cham, B.E. and Knowles, B.R. (1976) In vitro partial relipidation of apolipoprotein in plasma. Journal Biological Chemistry, 251, 6167-6171.
- Slater, H.R. and Robertson, F.W. (1979) A comparison of delipidated sera used in studies of sterol synthesis by human mononuclear lenkocytes. Journal Lipid Research, 20, 413-416.
- Shakespeare, V. and Postle, A.D. (1979) Regulation of cholesterol synthesis in skin fibroblasts derived from old people. Atherosclerosis, 33, 359-364. doi:10.1016/0021-9150(79)90187-4
- Wong, C.G. and Ladisch, S. (1983) Retention of gangliosides in serum delipidated by diisopropyl ether-butanol extraction. Journal Lipid Research, 24, 666-669.
- Prokazova, S.V., Mikhailenko, I.A., Preobrazhensky, S.N., Ivanov, V.O., Prokrovsky, S.N., Timofeeva, N.G., et al. (1986) Interaction of ganliosides with plasma low-density lipoproteins. Glycoconjugate Journal, 3, 273-286. doi:10.1007/BF01051777
- Slater, H.R., Smith, E.B. and Roberston, F.W. (1980) The effect of delipidated high-density lipoprotein on human leukocyte sterol synthesis. Atherosclerosis, 35, 41-49. doi:10.1016/0021-9150(80)90026-X
- Innerarity, T.L. and Mahley, R.W. (1978) Enhanced binding by cultured human fibroblasts of apo-E-containing lipoproteins as compared with low-density lipoproteins. Biochemistry, 17, 1440-1447. doi:10.1021/bi00601a013
- Cham, B.E., Kostner, K.M., Dwivedy, A.K., Shafey, T.M., Fang, N.X. and Mahon, M.G. (1995) Lipid apheresis: An in vivo application of plasma delipidation with organic solvents resulting in acute transient reduction of circulating plasma lipids in animals. Journal Clinical Apheresis, 10, 61-69. doi:10.1002/jca.2920100202
- Cham, B.E., Kostner, K.M., Dwivedy, A.K., Shafey, T.M., Fang, N.X. and Mahon, M.G. (1996) Lipid apheresis in an animal model causes in vivo changes in lipoprotein electrophoretic patterns. Journal Clinical Apheresis, 11, 61- 70. doi:10.1002/(SICI)1098-1101(1996)11:2<61::AID-JCA2>3.0.CO;2-8
- Cham, B.E. and Smith, J.L. (1994) Lipid apheresis in an animal model causes acute reduction in plasma lipid concentrations and mobilization of lipid from liver and aorta. Pharmacology Life Science Advances, 13, 25-32.
- Kostner, K.M., Smith, J.L., Dwivedy, A.K. and Shafey, T.M., et al. (1997) Lecithin cholesterol acyltransferase activity in normocholesterolemic and hypercholesterolemic roosters: modulation by lipid apheresis. European Journal Clinical Investigation, 27, 212-218. doi:10.1046/j.1365-2362.1997.960643.x
- Barter, P.J., Hopkins, G.J. and Gorjatschko, L. (1985) Lipoprotein substrates for plasma cholesterol esterification. Influence of particle size and composition of the lipoproteinsubfraction 3. Atherosclerosis, 58, 97-107. doi:10.1016/0021-9150(85)90058-9
- Jonas, A. and McHugh, H.T. (1984) Reaction of lecithin: Cholesterol acyltransferase with micellar substrates. Effect of particle size. Biochimica Biophysica Acta, 794, 361-372. doi:10.1016/0005-2760(84)90002-X
- Cham, B.E., Kostner, K.M., Shafey, T.M., Smith, J.L. and Colquhoun, D.M. (2005) Plasma delipidation process induces rapid regression of atherosclerosis and mobilization of adipose tissue. Journal Clinical Apheresis, 20, 143-153. doi:10.1002/jca.20060
- Kostner, K.M., Cham, B.E., Dwivedy, A.K., Shafey, T.M. and Fang, N.X. (1992) Increase of apo A1 concentration in hypercholesterolemic chickens after treatment with newly developed extracorporeal lipid elimination. 11th International Symposium on Drugs Affecting Lipid Metabolism, 13-16 May, Italy.
- Sacks, F.M., Rudel, L.L., Conner, A., Keefe, A., Kostner, G., Baki, T., et al. (2009) Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. Journal Lipid Research, 50, 894-907. doi:10.1194/jlr.M800622-JLR200
- Waksman, R., Torguson, R., Kent, K.M., Pichard, A.D., Suddath, W.O., Satler, L.F., et al. (2010) A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. Journal American College Cardiology, 55, 2727-2735. doi:10.1016/j.jacc.2009.12.067
- Tardif, J.C., Grégoire, J., L’Allier, P.L., Ibrahim, R., Lespérance, J. and Heinonen, T.M., et al. (2007) Effect of rHDL on atherosclerosis-safety and efficacy (ERASE). Effects of reconstituted high-density lipoprotein infusion on coronary atherosclerosis: A randomized controlled trial. JAMA, 297, 1675-1682. doi:10.1001/jama.297.15.jpc70004
- Nissen, S.E., Tsunoda, T., Tuzcu, E.M., Schoenhagen, P., et al. (2003) Effect of recombinant apo A-I Milano on coronary atherosclerosis in patients with acute coronary syndromes. A randomized controlled trial. JAMA, 290, 2292-2300. doi:10.1001/jama.290.17.2292
- Davidson, M.H. (2011) Apolipoprotein A-1 therapy. Promise, challenges and disappointment. Journal American College Cardiology, 57, 1120-1121. doi:10.1016/j.jacc.2010.11.025
- Gordon, S.M., Deng, J., Lu, L.J. and Davidson, W.S. (2010) Proteomic characterization of human plasma highdensity lipoprotein fractionated by gel filtration chromatography. Journal Proteome Research, 9, 5239-5249. doi:10.1021/pr100520x
- Nguyen, A.D., Nguyen, T.A., Cenik, B., Yu, G., Herz, J., Walther, T.C., et al. (2013) Secreted progranulin is a homodimer and is not a component of high-density lipoproteins (HDL). Journal Biological Chemistry, 288, 8627- 8635. doi:10.1074/jbc.M112.441949
- Kim, H.Y., Tallman, K.A., Liebler, D.C. and Porter, N.A. (2009) An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Molecular Cell Proteomics, 8, 2080-2089. doi:10.1074/mcp.M900121-MCP200
- Rezaee, F., Casetta, B., Levels, J.H.M., Speijer, D. and Meijers, J.C.M. (2006) Proteomic analysis of high-density lipoprotein. Proteomics, 6, 721-730. doi:10.1002/pmic.200500191