Health
Vol.4 No.11A(2012), Article ID:24973,7 pages DOI:10.4236/health.2012.431172
Pharmacological evaluation of catalepsy in low calcium and/or magnesium deficient feeding mice
![]()
1Department of Pharmacology, Tohoku Pharmaceutical University, Sendai, Japan; *Corresponding Author: osamun@tohoku-pharm.ac.jp
2Laboratory of Environmental and Health Sciences, College of Medical Pharmaceutical and Health Sciences, Kanazawa University, Kanazawa, Japan
Received 31 August 2012; revised 3 October 2012; accepted 10 October 2012
Keywords: Calcium; Catalepsy; Dopamine; Magnesium
ABSTRACT
Populations from the Kii peninsula of Japan and Guam present a high incidence of amyotrophic lateral sclerosis and parkinsonism-dementia complex. It is thought that the low levels of calcium (Ca) and magnesium (Mg) in the drinking water are involved in the pathogenesis of these diseases. The present study aimed to test the hypothesis that catalepsy, a behavioral immobility and one of the Parkinsonian symptoms, may result from functionally impaired dopaminergic neurons in low Ca and Mg (LCa/Mg) fed mice. A group of mice fed with an LCa/Mg diet for 6 weeks was compared to a control group on a standard diet. Cataleptic symptoms such as akinesia and rigidity were measured using the bar test. The antiparkinsonian drugs dopamine (DA) precursor L-3, 4-dihydroxy phenylamine (L-DOPA), the selective DA receptor D2 agonist bromocriptine and the DA releaser amantadine were tested for their effects on the induced catalepsy. Mice developped catalepsy after 3 weeks on the LCa/Mg diet. LCa/Mg diet-induced catalepsy was improved by the administration of either L-DOPA (50 - 200 mg/kg i.p.) in combination with benserazide (25 mg/kg i.p.), bromocriptine (0.25 - 4 mg/kg i.p.) or amantadine (5 - 20 mg/kg i.p.). These results suggest that catalepsy in LCa/Mg mice might result from a hypofunction of dopaminergic neurons. Moreover, our results support the hypothesis that LCa/Mg intake may be one etiological factor in neurodegenerative disorders including Parkinson’s disease.
1. INTRODUCTION
Parkinson’s disease (PD) is characterized by a loss of dopaminergic neurons in the substantia nigra (SN) accompanied by a reduction in striatal dopamine (DA). The aetiology of the disease is still unclear, but it is now widely accepted that many factors are involved in the development of the neuropathology. Certain metals have been suggested to play a role as imbalanced levels and abnormal deposits were observed in the brain of patients with PD [1,2]. Of particular interest is the higher incidence of PD and amyotrophic lateral selerosis (ALS) among primitive Auyu and Jakai people on the southern coastal plain of West New Guinea attributed to a marked deficiency of Ca and Mg in the soil and water of this region [3]. Likewise, a survey in Guam also suggested that low Ca, Mg (LCa/Mg) and high aluminum (Al) and manganese (Mn) in river, soil and drinking water might be responsible for the higher incidence of PD in that region [4]. Furthermore, an LCa/Mg diet has been shown to accelerate the deposition of Mn and Al in the brain [5]. Although numerous studies investigated the role of Mg in the etiology of clinical and experimental PD [3,6], reports on animal behavior after LCa/Mg feeding are scarce. The present study aimed to test the hypothesis that catalepsy, a behavioral immobility and one of the parkinsonian symptoms, may result from damage to DA-ergic neurons in LCa/Mg fed mice. Accordingly, we examined the effects of DA-ergic drugs on the catalepsy induced by LCa/Mg in mice.
2. MATERIALS AND METHODS
2.1. Animals
Male ddY mice (Japan SLC, Hamamatsu, Japan) weighing 10 g (post-natal days 20 - 21) at the beginning of the experiment, were used in this study. The animals were housed in plastic cages (31 cm × 21 cm × 13 cm) under conditions of constant temperature (23˚C ± 1˚C), humidity (55% ± 5%), and light-dark cycle (light from 9 to 21 h; dark from 21 to 9 h). The mice had free access to food and water throughout the experimental period. They were divided into seven different dietary groups for 6 weeks: 1) low Ca diet (LCa) group (n = 20); 2) control group (pair-fed (PF) group for LCa group; n = 20); 3) Mg deficient diet (MgD) group (n = 20); 4) control group (PF group for MgD group; n = 20); 5) low Ca and Mg deficient diet (LCa/Mg) group (n = 110); 6) control group (PF group for LCa/Mg group; n = 50); and 7) normal control group (n = 60). The latter group had access to a completely normal diet ad libitum (standard mineral diet: CLEA Japan, Tokyo, Japan). The PF groups were also given the standard diet, with the exception that the amount of food was matched to that of their respective/paired deficient diet group (1, 2 and 5). The PF groups were included to control for the contributing ef-

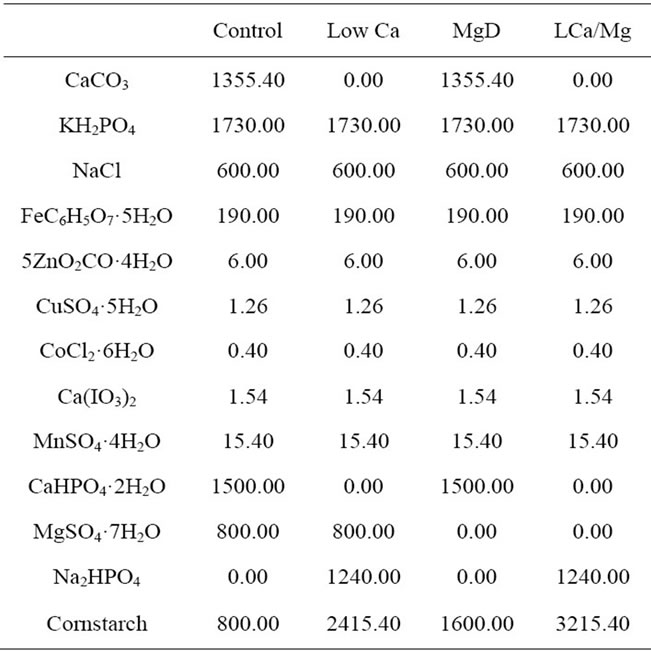
Table 1. Composition of experimental diets.
fects of anorexia. The deficient diet groups 1), 3) and 5) were given distilled and deionized milli-Q water to drink. All diet ingredients (CLEA Japan, Tokyo, Japan) are listed in Table 1. All experiments were performed with the approval of the Ethics Committee of Animal Experiments in Tohoku Pharmaceutical University and according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Efforts were made to minimize suffering and to reduce the number of animals used.
2.2. Drugs
All drugs were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Bromocriptine, L-DOPA and benserazide were dissolved in 0.5% Tween80. Amantadine was dissolved in saline. Bromocriptine (1, 2, 4 mg/kg), L-DOPA (100, 200, 400 mg/kg) and amantadine (5, 10, 20 mg/kg) were injected 30 min before the catalepsy test. Benserazide (25 mg/kg) was injected 30 min prior to L-DOPA. All drug injections were performed intraperitoneally (i.p.).
2.3. Catalepsy Test
The bar test was used to measure cataleptic symptoms such as akinesia and rigidity. Catalepsy was evaluated by placing both forepaws of the mouse on a horizontal bar (diameter: 0.2 cm), elevated 5 cm from the floor. The degree of catalepsy was assessed by measuring the time from placement of the mouse until removal of one of its paws (descent latency), with a cut-off time of 180 s. A score was assigned to each test based on the duration of the cataleptic posture (score 1, between 0 and 29 s; score 2, between 30 and 59 s; score 3, 60 s or more).
2.4. Statistical Analysis
The significance of differences was determined using Student’s t-test for two-group comparison. The score for the catalepsy test was carried out by Mann-Whitney U-test (two-tailed). Analysis of variance (ANOVA) was used for multiple comparisons. Post hoc analysis was performed using Fisher’s PLSD test. The criterion of significance was set at p < 0.05. All results are expressed as mean ± standard error of the mean (SEM).
3. RESULTS
3.1. Growth Curves and the Mortality Rate
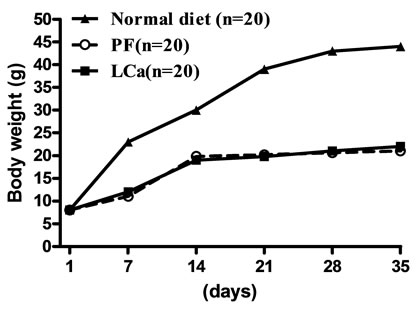
Mice on standard mineral or experimental diets were housed individually for food intake assessment. The body weight of the LCa group did not differ from its PF group during the entire treatment, but was significantly decreased compared with the normal control group (Figure 1(a)). After 1 week on the diet, LCa mice showed signs of aphagia. The amount of food made available to the PF group was reduced accordingly to equal that consumed by the LCa group (Table 2). In the LCa group, death occurred in 1.4% of mice on the 21st day and in 5.6% on the 35th day, while no deaths occurred in the PF group (Table 3). In the MgD group, a profound loss in body weight was seen after 17 days of treatment (Figure 1(b)). MgD mice showed aphagia after 4 days of treatment which persisted until death (Table 2). In the MgD group, death occurred in 1.7% of mice on the 7th day, in 19.3% on the 14th day, in 54.4% on the 21st day, in 77.2% on the 28th day and in 87.7% on the 35th day, while no deaths occurred in their PF group (Table 3). As shown on the growth curves in Figure 1, despite the LCa/Mg diet, the average body weight at the end of the first week had steadily increased to about 14 g, similarly to that observed with mice from the PF group. LCa/Mg mice showed aphagia from the second week onwards (Table 2). Accordingly, the weight of these mice started to plateau and to differ significantly from the PF group, which continued to increase (Figure 1(c)). In the LCa/Mg group, death occurred in 3.3% on the 21st day, in 6.6% on the 28th day and in 8.3% on the 35th - 42nd days, while no deaths occurred in the PF group (Table 3).
3.2. Influence of Ca and/or Mg Deficiency on Catalepsy Test
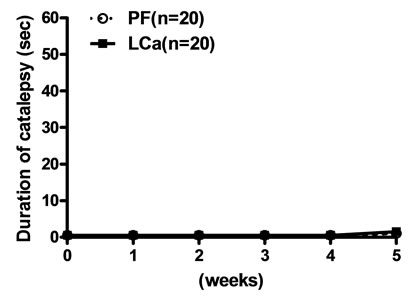
Mice on the LCa diet did not show any signs of catalepsy even after 5 weeks (Figure 2(a)). Catalepsy was not observed either in any of the PF groups. In contrast, mice on the MgD diet showed a significant increase in their descent latency from the bar on the second week but were not further evaluated due to their high death rate (Figure 2(b)). On the other hand, a significant increase in the duration of catalepsy was observed from the 3rd to the 6th week in mice fed with the LCa/Mg diet (Figure 2(c)).
3.3. Effects of Antiparkinsonian Drugs on Catalepsy in LCa/Mg Mice
The antiparkinsonian drugs bromocriptine (DA recaptor D2 agonist), L-DOPA (precursor of DA), and amantadine (DA releaser) were tested on the 6th week in cataleptic LCa/Mg mice. The catalepsy scores significantly improved and peaked by 1 hr after bromocriptine injection (1 - 4 mg/kg) (Figure 3) or 0.5 hr after L-DOPA (50 - 200 mg/kg) (Figure 4) or amantadine injections (10 - 20 mg/kg) (Figure 5) before returning to control levels.
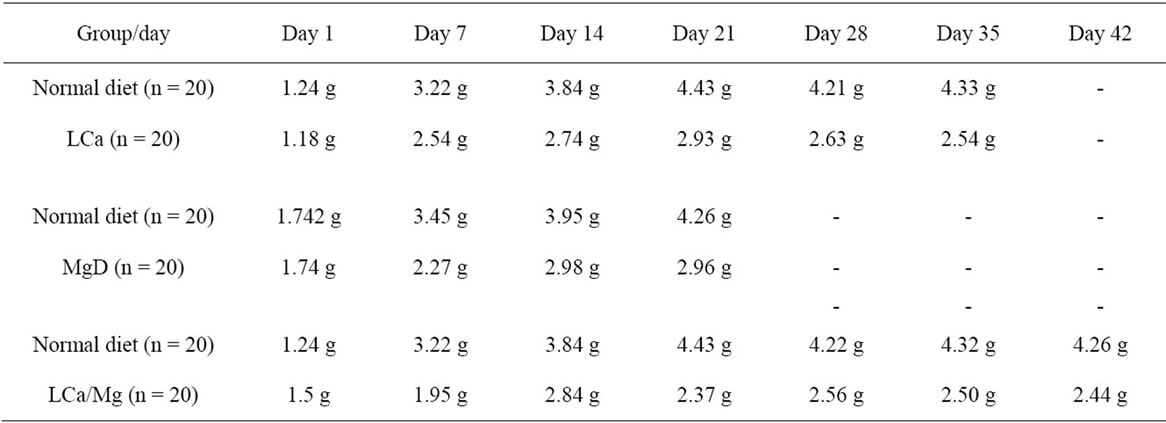
Table 2. Effect of LCa, MgD and LCa/Mg on food intake in mice.
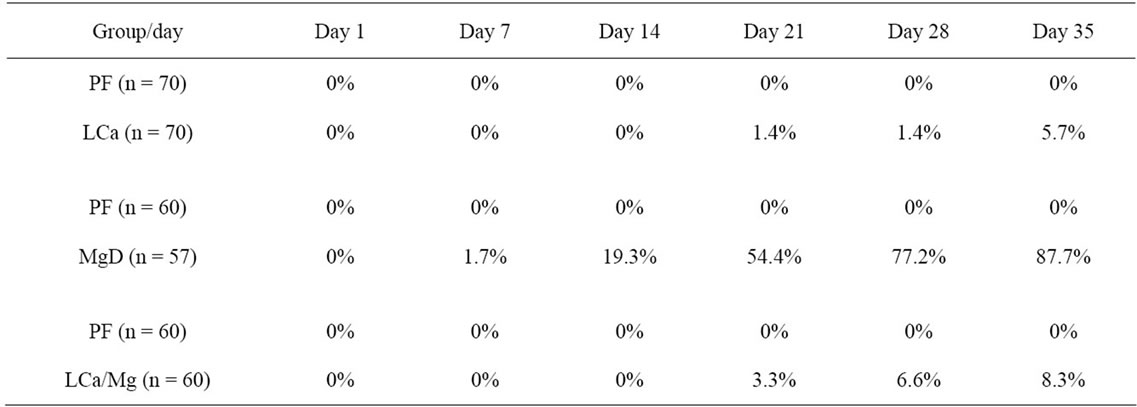
Table 3. Influence of LCa, MgD and LCa/Mg on the death rate during the first 35 days on the diet.
 (a)
(a)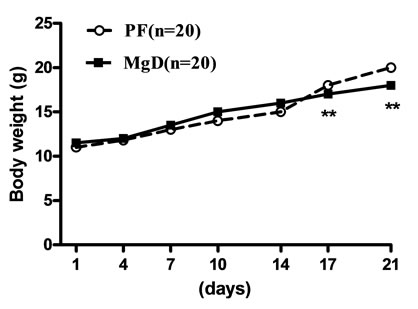 (b)
(b)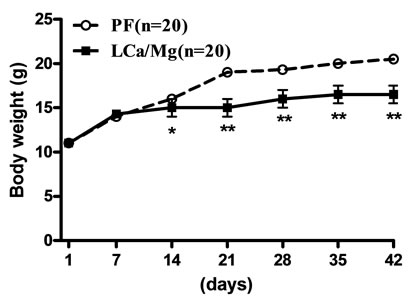 (c)
(c)
Figure 1. Growth Curves for LCa (a), MgD (b) and LCa/Mg (c) diet groups. Vertical bars represent standard errors of the mean (S.E.M.). *: p < 0.05; **: p < 0.01 compared with PF groups.
 (a)
(a)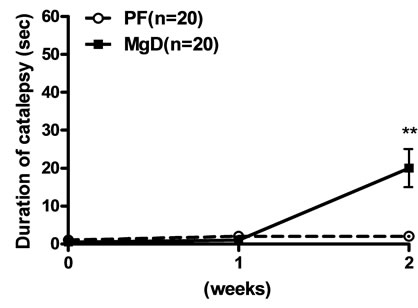 (b)
(b)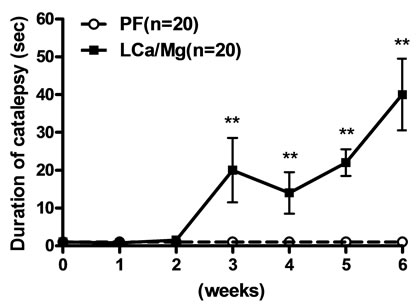 (c)
(c)
Figure 2. Effects of LCa (a), MgD (b) and LCa/Mg (c) diet on catalepsy in mice. Vertical bar represent S.E.M. *: p < 0.05; **: p < 0.01 compared with PF groups.
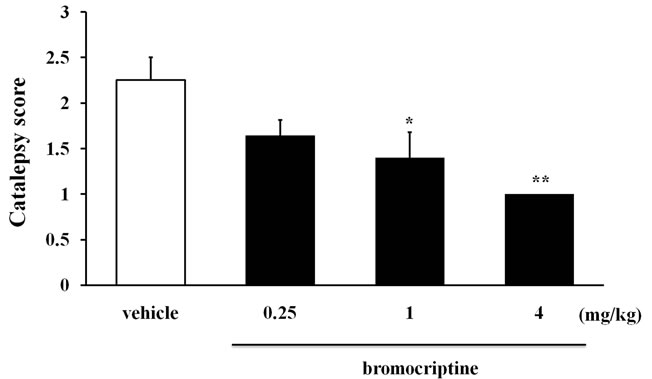
Figure 3. The inhibitory effect of bromocriptine on catalepsy induced after 6 weeks of LCa/Mg diet in mice. Catalepsy score on peak time of effect (1 hr after bromocriptine i.p. injection). Vertical bar represent S.E.M. (n = 10 per group). *: p < 0.05; **: p < 0.01 vs vehicle.
4. DISCUSSION
The present study showed that LCa, MgD and LCa/ Mg diets led to a decrease in the daily food intake in mice and, accordingly, to their markedly reduced growth. The growth curves and food intake were similar between these groups. The mortality rate was more pronounced however in the MgD group than in the LCa and LCa/Mg groups. Under normal physiological conditions, voltage-dependent Mg blockade limits free radical generation promoted by increases in cytosolic Ca levels, and is
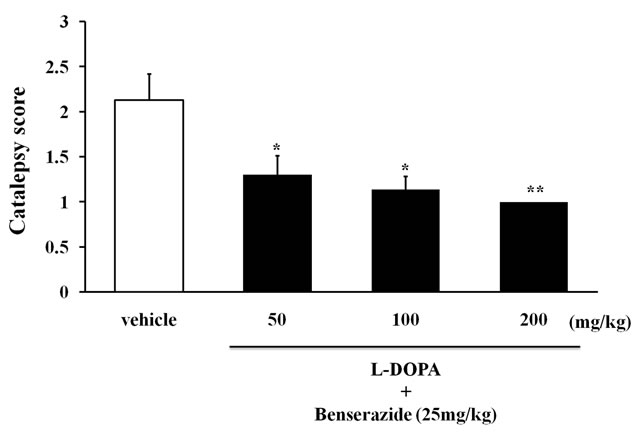
Figure 4. The inhibitory effect of combined L-DOPA and benserazide on catalepsy induced after 6 weeks of LCa/Mg diet in mice. Catalepsy score on peak time of effect (30 min after L-DOPA in combination with benserazide i.p. injection). Vertical bar represent S.E.M. (n = 10 per group). *: p < 0.05; **: p < 0.01 vs vehicle.
thus neuroprotective [1,7]. In support, Oyanagi et al. reported that the degree of neuron loss in the SN, and the decrease in body weight were more evident in the MgD group than in the LCa/Mg group [6]. These findings suggest that an MgD diet is more hazardous to neuron and other cells than an LCa or LCa/Mg diet.
Both the MgD and LCa/Mg groups, but not the LCa group, showed a prolonged duration of catalepsy as com-
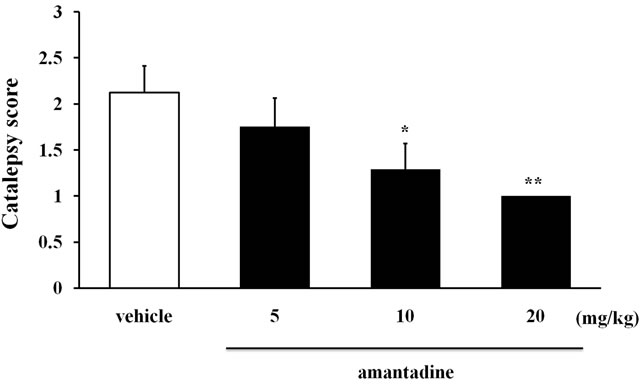
Figure 5. The inhibitory effect of amantadine on catalepsy induced after 6 weeks of LCa/Mg diet in mice. Catalepsy score on peak time of effect (30 min after amantadine i.p. injection). Vertical bar represent S.E.M. (n = 10 per group). *: p < 0.05; **: p < 0.01 vs vehicle.
pared to their PF group on the bar test. The result suggests that the deficiency in Mg may be responsible for the appearance of catalepsy. It is well known that reduced DA activity causes Parkinsonian symptoms such as rigidity and akinesia in human. This state manifests itself as catalepsy in rodents. The high mortality observed in mice on the MgD diet makes it difficult to study the mechanisms leading to catalepsy using this paradigm. Moreover, it was reported that in this model mice showed a more complex state than a simple Mg deficiency that could be reversed with a mere oral Mg supplement [3]. Therefore, we have focused on the LCa/Mg group to investigate potential causes of catalepsy. The duration of catalepsy was previously shown to increase with the degeneration of the nigrostriatal DAergic neuron pathway [8,9]. Here, the cataleptic behavior observed in LCa/Mg mice was transiently inhibited in a dose-dependent fashion by the administration of the antiparkinsonian drugs bromocriptine, L-DOPA or amantadine. Together, these results suggest that the LCa/Mg diet-induced catalepsy may be due to hypo-dopaminergic function. We propose three hypothetical mechanisms by which LCa/Mg diet feeding may lead to DA-ergic hypofunction.
1) It is well established that Ca and Mg are essential for synaptic transmission and of central importance to the biochemistry of the cell and its energy metabolism in particular. These elements are also cofactors for the tyrosine hydroxylase activity that is essential for DA synthesis [10]. Mg was shown to prevent the death of DA-ergic neurons induced by 1-methyl-4-phenylpyridinium (MPP+) in the experimental model of PD [11,12]. Thus, a chronic LCa/Mg diet may alter DA production in the SN, its release in the striatum and lead to motor dysfunction such as catalepsy.
2) It has been reported that an accumulation of pollutant metals such as Mn and Al is caused by some metabolic changes due to a lack of protective factors (e.g. LCa/Mg) [5]. In this respect, a reduction in blood Ca levels was shown to enhance Al absorption [13]. The accumulation of Al in the central nervous system can play a critical role in its degeneration by inducing neurofibrillary tangle formation and cell death [14,15]. Interestingly, in a recent study subjects with Parkinsonism and arthritis showed high levels of Mn and Al accumulated in their brain [16]. Chronic Mn exposure shows many pathological features in common with PD [17,18]. DA deficiency was shown to occur in chronic Mn poisoning in squirrel monkeys and rabbits [19,20]. It is noteworthy that some pathological neurological symptoms observed after long-term Mn intoxication can be diminished by L-DOPA treatment [21]. Mn neurotoxicity has shown selectivity for DA neurons [22]. Recently, it was demonstrated that Mn augments the neurotoxic effect of 6- hydroxydopamine, a treatment used to model PD in animals [23]. Together, these results suggest that LCa/Mg in mice may lead to an indirect accumulation of Mn and Al in the brain with subsequent degeneration of DA neuron.
3) Mg decreases the excitotoxic activity of N-methylD-aspartate (NMDA) [24]. The NMDA receptors are thought to be responsible for this excitotoxicity and the subsequent downstream events like neuroinflammation and apoptosis, and thus have been implicated in many important human pathologies ranging from ALS to Alzheimer’s disease, PD, depression, epilepsy, trauma and schizophrenia [25]. Mg’s neuroprotective effect is mediated via a reduction in the presynaptic release of the neurotransmitter glutamate [26] or blockade of NMDA receptors [27]. In a variety of experimental models of PD, many NMDA receptor blockers have shown anticataleptic activity, including dizocilpine, phencyclidine, memantine, and others [28-31]. In this study, the administration of amantadine, a DA releaser and a non-selective NMDA receptor antagonist, also improved the catalepsy in LCa/Mg mice. Thus, this ameliorating effect may be due not only to the drug’s induction of DA release but also to its NMDA receptor antagonist properties. These results suggest that the LCa/Mg-induced catalepsy may be caused by DA-ergic hypofunction via a supersensitivity of NMDA receptors. Further experiments are needed to examine how LCa/Mg can lead to the degeneration of SN DA-ergic neurons.
In conclusion, these results suggested that catalepsy in LCa/Mg mice might result from a hypofunction of dopaminergic neurons. Moreover, the present study demonstrated that a chronic deficiency in environmental factors, such as dietary Ca and Mg, may contribute to the development of neurodegenerative disorders such as PD.
5. ACKNOWLEDGEMENTS
This study was supported in part by Grants-in-Aid for Scientific Research (22600010) and Program for the Strategic Research Foundation at Private Universities (2010-2014) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
![]()
![]()
REFERENCES
- Olanow, C.W. and Arendash, G.W. (1994) Metals and free radicals in neurodegeneration. Current Opinion in Neurology, 7, 548-558. doi:10.1097/00019052-199412000-00013
- Yasui, M., Kihira, T. and Ota, K. (1992) Calcium, magnesium and aluminum concentrations in Parkinson’s disease. Neurotoxicology, 13, 593-600.
- Durlach, J., Bac, P., Durlach, V., Durlach, A., Bara, M. and Guiet-Bara, A. (1997) Are age-related neurodegenerative diseases linked with various types of magnesium depletion? Magnesium Research, 10, 339-353.
- Purdey, M. (2004) Elevated levels of ferrimagnetic metals in food chains supporting the Guam cluster of neurodegeneration: Do metal nucleated crystal contaminants [corrected] evoke magnetic fields that initiate the progressive pathogenesis of neurodegeneration? Medical Hypotheses, 63, 793-809. doi:10.1016/j.mehy.2004.04.029.
- Yasui, M., Ota, K. and Garruto, R.M. (1995) Effects of calcium-deficient diets on manganese deposition in the central nervous system and bones of rats. Neurotoxicology, 16, 511-517.
- Oyanagi, K., Kawakami, E., Kikuchi-Horie, K., Ohara, K., Ogata, K., Takahama, S., Wada, M., Kihira, T. and Yasui, M. (2006) Magnesium deficiency over generations in rats with special references to the pathogenesis of the Parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Neuropathology, 26, 115-128. doi:10.1111/j.1440-1789.2006.00672.x
- Iseri, L.T. and French, J.H. (1984) Magnesium: Nature’s physiologic calcium blocker. American Heart Journal, 108, 188-193. doi:10.1016/0002-8703(84)90572-6
- Ross, R.T. (1990) Drug-induced parkinsonism and other movement disorders. The Canadian Journal of Neurological Sciences, 17, 155-162.
- Schmidt, W.J. and Kretschmer, B.D. (1997) Behavioural pharmacology of glutamate receptors in the basal ganglia. Neuroscience and Biobehavioral Reviews, 21, 381-392. doi:10.1016/S0149-7634(96)00044-9
- Iuvone, P.M. (1984) Calcium, ATP, and magnesium activate soluble tyrosine hydroxylase from rat striatum. Journal of Neurochemistry, 43, 1359-1368. doi:10.1111/j.1471-4159.1984.tb05395.x
- Hashimoto, T., Nishi, K., Nagasao, J., Tsuji, S. and Oyanagi, K. (2008) Magnesium exerts both preventive and ameliorating effects in an in vitro rat Parkinson disease model involving 1-methyl-4-phenylpyridinium (MPP+) toxicity in dopaminergic neurons. Brain Research, 1197, 143-151. doi:10.1016/j.brainres.2007.12.033
- Nakamura, K., Bindokas, V.P., Marks, J.D., Wright, D.A., Frim, D.M., Miller, R.J. and Kang, U.J. (2000) The selective toxicity of 1-methyl-4-phenylpyridinium to dopaminergic neurons: The role of mitochondrial complex I and reactive oxygen species revisited. Molecular Pharmacology, 58, 271-278.
- Yasui, M., Yano, I., Yase, Y. and Ota, K. (1991) Distribution of calcium in central nervous system tissue and bones of maintained on calcium-deficient diets. Journal of the Neurological Sciences, 105, 206-210. doi:10.1016/0022-510X(91)90146-X
- Garruto, R.M., Shankar, S.K., Yanagihara, R., Salazar, A.M., Amyx, H.L. and Gajdusek, D.C. (1989) Low-calcium, high-aluminum diet-induced motor neuron pathology in cynomolgus monkeys. Acta Neuropathologica, 78, 210-219. doi:10.1007/BF00688211
- Strong, M.J. (1996) Modeling of acute and chronic aluminum neurotoxicity. In: Yasui, M., Strong, M.J., Ota, K. and Verity, M.A., Eds., Mineral and Metal Neurotoxicology, CRC Press, Boca Raton, 99-106.
- Komatsu, F., Kagawa, Y., Kawabata, T., Kaneko, Y., Chimedregzen, U., Purvee, B. and Otgon, J. (2011) A high accumulation of hair minerals in Mongolian people: 2(nd) report; influence of manganese, iron, lead, cadmium and aluminum to oxidative stress, Parkinsonism and arthritis. Current Aging Science, 4, 42-56. doi:10.2174/1874609811104010042
- Mena, I., Marin, O., Fuenzalida, S. and Cotzias, G.C. (1967) Chronic manganese poisoning. Clinical picture and manganese turnover. Neurology, 17, 128-136. doi:10.1212/WNL.17.2.128
- Pentschew, A., Ebner, F.F. and Kovatch, R.M. (1963) Experimental manganese encephalopathy in monkeys. A preliminary report. Journal of Neuropathology & Experimental Neurology, 22, 488-499. doi:10.1097/00005072-196307000-00010
- Mustafa, S.J. and Chandra, S.V. (1971) Levels of 5-hydroxytryptamine, dopamine and norepinephrine in whole brain of rabbits in chronic manganese toxicity. Journal of Neurochemistry, 18, 931-933. doi:10.1111/j.1471-4159.1971.tb12022.x
- Neff, N.H., Barrett, R.E. and Costa, E. (1969) Selective depletion of caudate nucleus dopamine and serotonin during chronic manganese dioxide administration to squirrel monkeys. Cellular and Molecular Life Sciences, 25, 1140-1141. doi:10.1007/BF01900234
- Cotzias, G.C., Papavasiliou, P.S., Ginos, J., Steck, A. and Düby, S. (1971) Metabolic modification of Parkinson’s disease and of chronic manganese poisoning. Annual Review of Medicine, 22, 305-326. doi:10.1146/annurev.me.22.020171.001513
- Verity, M.A. (1999) Manganese neurotoxicity: A mechanistic hypothesis. Neurotoxicology, 20, 489-497.
- Nowak, P., Bojanek, K., Szkilnik, R., Jośko, J., Boroń, D., Adwent, M., Gorczyca, P., Kostrzewa, R.M. and Brus, R. (2011) Ontogenetic exposure of rats to preand post-natal manganese enhances behavioral impairments produced by perinatal 6-hydroxydopamine. Neurotoxicity Research, 19, 536-543. doi:10.1007/s12640-010-9184-0
- Mayer, M.L., Westbrook, G.L. and Guthrie, P.B. (1984) Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature, 309, 261-263. doi:10.1038/309261a0
- Schrattenholz, A. and Soskic, V. (2006) NMDA receptors are not alone: Dynamic regulation of NMDA receptor structure and function by neuregulins and transient cholesterol-rich membrane domains leads to disease-specific nuances of glutamate-signalling. Current Topics in Medicinal Chemistry, 6, 663-686. doi:10.2174/156802606776894519
- Lin, J.Y., Chung, S.Y., Lin, M.C. and Cheng, F.C. (2002) Effects of magnesium sulfate on energy metabolites and glutamate in the cortex during focal cerebral ischemia and reperfusion in the gerbil monitored by a dual-probe microdialysis technique. Life Science, 71, 803-811. doi:10.1016/S0024-3205(02)01738-1
- Nowak, L., Bregestovski, P., Ascher, P., Herbet, A. and Prochiantz, A. (1984) Magnesium gates glutamate-activated channels in mouse central neurons. Nature, 307, 462-465. doi:10.1038/307462a0
- Elliott, P.J., Close, S.P., Walsh, D.M., Hayes, A.G. and Marriott, A.S. (1990) Neuroleptic-induced catalepsy as a model of Parkinson’s disease. II. Effect of glutamate antagonists. Journal of Neural Transmission: Parkinson’s Disease and Dementia Section, 2, 91-100. doi:10.1007/BF02260897
- Mele, A., Thomas, D.N. and Pert, A. (1997) Different neural mechanisms underlie dizocilpine maleateand dopamine agonist-induced locomotor activity. Neuroscience, 82, 43-58. doi:10.1016/S0306-4522(97)00277-7
- Morelli, M., Fenu, S., Pinna, A. and Di Chiara, G. (1992) Opposite effects of NMDA and AMPA receptor blockade on dopaminergic D1- and D2-mediated behavior in the 6-hydroxydopamine model of turning: relationship with c-fos expression. The Journal of Pharmacology and Experimental Therapeutics, 260, 402-408.
- Ossowska, K. (1994) The role of excitatory amino acids in experimental models of Parkinson’s disease. Journal of Neural Transmission: Parkinson’s Disease and Dementia Section, 8, 39-71. doi:10.1007/BF02250917