Journal of Biophysical Chemistry
Vol.5 No.2(2014), Article ID:45218,11 pages DOI:10.4236/jbpc.2014.52005
A Unique Conformational Behaviour of Glutamine Peptides
Munusamy Baskar, Mohd Shafique, Fateh Singh Nandel*
Department of Biophysics, Panjab University, Chandigarh, India
Email: *fateh_nandel@yahoo.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 20 February 2014; revised 20 March 2014; accepted 27 March 2014
Abstract
Trinucleotide repeat expansions (CAG) lead to increase in glutamine residues and hence increase in glutamine stretch. This leads to number of neurodegenerative diseases. Therefore, the conformation of poly Q of varying chain lengths has been investigated by quantum mechanical and molecular dynamics approaches. Glutamine contains amide linkage in the side chain. It is the interaction between side chain amide linkage and the peptide bond of the backbone which dictates the conformational behaviour. Some of the glutamine residues adopt phi psi values corresponding to poly-L-proline type II structure. Not more than three glutamine residues are found to have the same set of ϕ, ѱ values and hence polyglutamine is adopting random coil structure. Carbonyl-carbonyl, CH-O interactions and hydrogen bond formation involving backbone and side chain amide chain linkages are found to contribute to the stability of the adopted structure. In simulation studies due to interaction of water molecules with the amide linkages the ϕ, ѱ values undergo change and this leads to weakening of carbonyl-carbonyl interactions and hydrogen bonds. The conformational behaviour of polyglutamine peptides is shown to be chain length dependent and this may provide some insight regarding the aggregation behaviour of proteins containing poly Q stretch. Possibly this is the first systematic study of the conformational behaviour of polyglutamine peptides.
Keywords:Poly Q, Unordered Structure, Peptide and Amide Bond Interactions, Carbonyl-Carbonyl Interactions

1. Introduction
The basic principle of genetics (classical genetics) is transfer of stable mutations to the offspring. For the first time, inheritance of myotonic dystrophy of different nature with increased expressivity and thus earlier onset and more severity of the disease in the patients was reported [1] . Similarly, this mode of inheritance was noticed for huntington’s chorea, spinal and bulbar muscular atrophy and many types of ataxias. These inherited disorders in the proteins are due to expansions of cytosine adenine guanine (CAG) repeats in the corresponding genes and approximately 30 inheritable disorders results from an increase in the number of copies or the stretch; in case of trinucleotide repeat expansions [2] . Nine polyglutamine expansions in the proteins have been related to protein misfolding resulting in neurodegenerative disorders [3] . The speculation concerning pathogenesis has only focussed on the poly Q expansion [4] . Detailed structural information on glutamine peptides has not been feasible due to poor solubility of both long and short chain peptides [5] . It is rational to think that expansion of glutamine stretch may alter the structure of other domains in proteins i.e., misfolding, aggregation and hence fibril formation.
The results of conformational studies of polyglutamine peptides containing only glutamine residues or with flanking residues at either or both the terminal positions are found to be at variance [6] [7] . Perutz et al., 1994 claimed that poly Q forms polar zipper structure and suggested its role in binding with the transcription factors [8] . Multiple flexible conformations have been proposed from the crystallographic studies [9] . But there are some commonalities among these diseases caused by homopolymeric tract expansions i.e., Parkinson’s and Alzheimer’s diseases [10] . Generally, the homopolymeric expansions are at the terminal positions of the concerned proteins [11] . Therefore, it is necessary to understand and investigate the conformational behaviour of poly Q stretches of varying chain length and their aggregation behaviour of peptides containing only glutamines and poly Q having flanking residues at either terminal positions. This problem can be addressed by computational studies. Here, we report the conformational behaviour of poly Q peptides of chain length 1to16 by both quantum mechanical and molecular dynamics simulation approaches with the objectives, 1) do the poly Q stretches adopt regular secondary structure or not, 2) is the conformation of the is chain length dependent, 3) what is the role of amide linkages in the side chain, 4) what interaction stabilize the adopted structure, v) can the network of stabilizing interaction be related to the solubility.
2. Methods
2.1. Quantum Mechanical
To have the knowledge of global, local and low energy minima, the ϕ, ѱ maps and χ maps/potential energy curves for the designed peptide of the Ac-Gln-NHMe were constructed by using standard bond lengths and bond angles [12] [13] . The energy calculations were carried out using the semi-empirical quantum mechanical method PCILO [14] (pertubative configuration interaction using localized molecular orbitals). Energy minimization was done by the systematic variation of torsion angles, keeping bond lengths and angles constant.
The conformational states for higher peptides Ac-(Gln)2-7-NHMe were generated from the knowledge of the global, local and low energy minima in the ϕ, ѱ maps and χ curve and their energies computed. The minimization was further refined by varying ϕ, ѱ and χ values in the neighbourhood of the minima so obtained in steps of 5 and then in 2 degree steps. It may be mentioned that the PCILO result for the peptides containing usual and unusual amino acids is in conformity with ab initio results [15] [16] and knowledge-based crystallographic data [17] [18] .
2.2. Molecular Dynamic (MD) Simulations
Simulation in water provides good deal of information regarding the stability of peptides in terms of non-covalent interactions. Glutamine stretches are present in the both in cytoplasmic and some nuclear proteins surrounded by water. Therefore, MD simulations in water can throw light on the conformational behaviour of these peptides [19] [20] . The simulations have been performed with GROMACS software (Version 4.0.7) [21] by using force field ffG43a1 [22] which is also suitable for peptides containing unusual amino acids [21] .
Conformational results by QM calculations were used as the starting geometries for MD simulation studies. Peptide was placed centrally and box was created at a distance of 1 nm from the surface of the protein orthogonally. To perform calculations in an NVT (Number of molecules, Volume and Temperature) ensemble [23] with spc water model [24] , maximum box size and minimum water molecules were selected amongst all the conformations of a molecule and afterwards, numbers of water molecules were made to be equivalent with –maxsol command in all systems. Convergence value (emtol) was set to 1000 kJ∙mol−1 in energy minimization. The peptide was solvated with water and simple point charge (SPC) water model was used [23] .
In order to allow equilibration of solvent around the model sequence, position of all residues was restrained for 20 ps at 300 K. MD simulation at 300 K, without any restraints was performed at a constant temperature under NVT conditions. The periodic boundary conditions were applied in all the three dimensions. Time step used was 2 fs [25] using Leap Frog Algorithm [26] and Berendsen scheme [27] was used for temperature control using a coupling time constant: τt of 0.1 ps and a reference temperature T0 of 300 K. LINCS algorithm [28] was used to restrict all bonds to their equilibrium lengths and the centre of mass motion of the system was removed at every step to maintain T0. Pressure was controlled using weak coupling with a time constant of 0.5 ps and a reference pressure of 1 Bar.
For evaluation of coulomb and van der Waals interactions, a cut-off of 0.9 and 1.0 nm, respectively was applied. Long-range forces were updated every 10 fs during generation of the neighbor list. Particle Mesh-Ewald summation method was used to calculate long-range electrostatic interactions [29] . Initial velocities of all atoms were taken from a Maxwellian distribution at the desired initial temperature. Same force field and other parameters were used in methanol. All atoms of the system were considered explicitly [21] . All simulations were performed using the NVT ensemble [30] .
3. Results and Discussion
Initially the conformation of glutamine model peptides of varying chain lengths of the type Ac-(Gln)n-NHMe with n = 1 to 7 have been carried out by quantum mechanical and the results are summarized in table 1(a) and table 1(b).
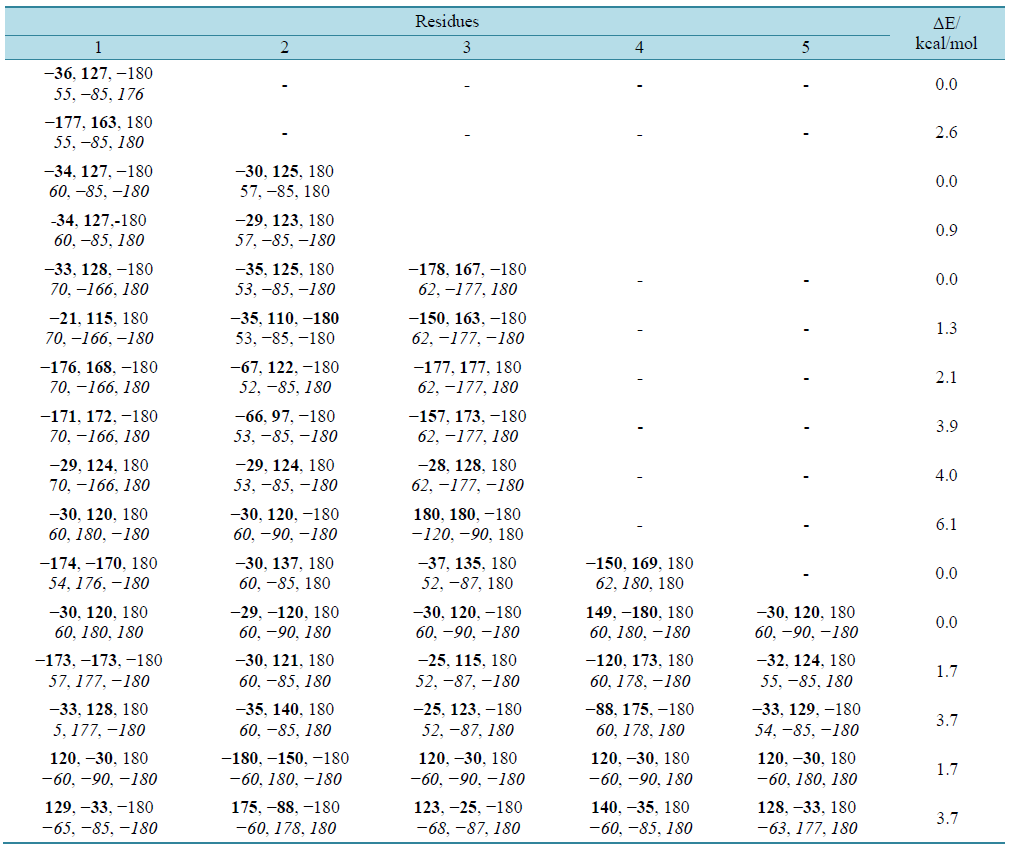
The results in table 1 clearly depict that the conformational flexibility of glutamine is very limited and has preference for two set of ϕ, ѱ values of ~−30, 130 or extended state. Likewise the side chain adopts χ values are found to fall in two categories, i.e., ~60, −90, 180 degree or 60, 180, and 180. A molecular view of the model dipeptide Ac-Gln-NHMe in the most stable state shown in figure 1 is stabilized by carbonyl-carbonyl interactions [31] with do…c = 2.4 Å [32] . In addition, the hydrogen bond between the NH moiety of the backbone and carbonyl group of the side chain with χ values of the side chain in the neighborhood of 60, −90, 180 (g, −90, t) also contributes to the stability. The model tetra-peptide, Ac-(Gln)3-NHMe also adopt the same set of ϕ and ѱ values i.e., approximately −30, 125 stabilized by carbonyl-carbonyl interactions (backbone-backbone and side chain-side chain) and CH-O interactions involving carbonyl oxygen of the first residue with γ-methylene group of the side chain of first and β, γ methylene group of the third residue (figure 2). The γ-methylene group of the first residues is also involved in CH-O interactions with carbonyl oxygen of the second residue. The side chain of the first and third residue with χ values ~60, 180, 180 (g, t, t) is involved in hydrogen bond formations through their amide linkages. Interestingly, the side chain of the second residue with χ values of ~ 60, −90, and 180 is involved in hydrogen bond formation through its carbonyl oxygen with NH moiety of the same residue giving rise to a seven member ring. It is due to these interactions poly Q has the tendency to adopt poly-L-proline type II structure. A molecular view of the peptide depicting all the interactions is shown in figure 2.
The conformational results for polyglutamine peptides with chain length 4 to 7 summarized in table 1 clearly indicate that with increasing chain length, the conformational behavior of polyglutamine peptides changes and not more than three consecutive residues adopt the same set of ϕ, ψ, ω and χ values. The ϕ, ψ values −35, 120 are intercepted by ϕ, ψ value corresponding to extended state, with no regular pattern and the conformational behaviour is very restricted. The states with ϕ and ψ values corresponding to the regular secondary structures (i.e. helices and beta sheets) are found to lay at high energy i.e., do not adopt regular secondary structure like helices and beta sheets. In the most stable states of all the peptides the χ values for the side chain of all the residues (except the terminal ones) fall in two categories i.e., either of the type g, t, t or of the type g, −90, t. Side chains of amino acid residues with χ values of g, t, t type are involved in inter hydrogen bond formation, whereas side chains of amino acid residues with χ values of g, −90, t form are involved in hydrogen bond formation between NH of the amide linkage (side chain) and backbone carbonyl oxygen of the previous residues. The latter interaction restricts both the ϕ, ψ value, partially similar to polyproline. A molecular view of the peptide Ac-Gln7-NHMe depicting the various interactions i.e., carbonyl-carbonyl interactions between backbone-backbone carbonyl groups and between backbone carbonyl & side chain carbonyl is shown in figure 3. It is due to the interactions between backbone amide linkage and side chain amide, the conformational behaviour is very
 (a)
(a)  (b)
(b)
Table 1. Conformational results for glutamine peptides in terms of ϕ, ψ, ω and χ values in degrees of the type Ac-(Gln)n- NHMe with (a) n = 1 to 5, (b) n = 6 & 7.
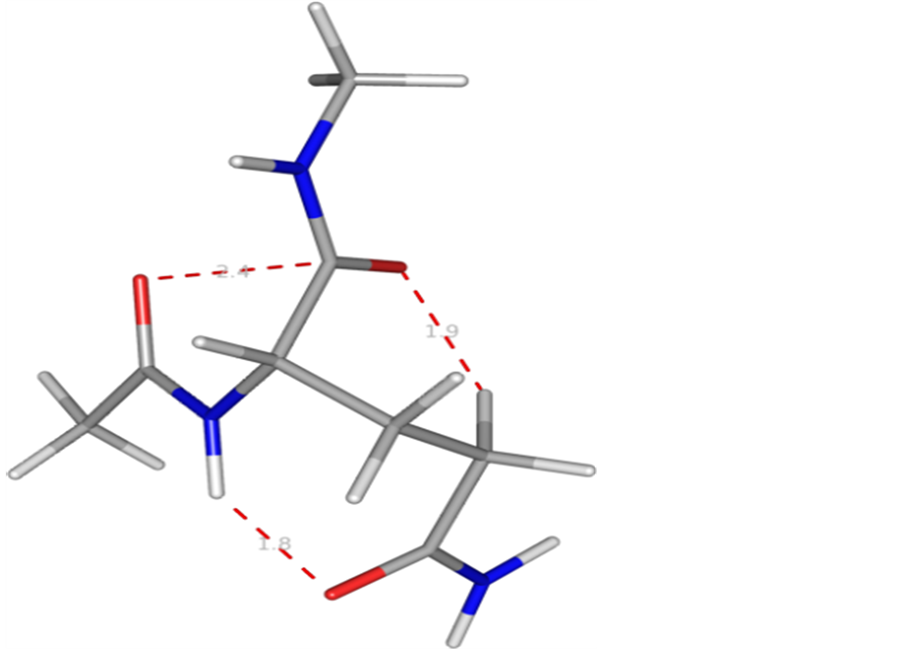
Figure 1. A molecular view of the Ac-Gln-NHMe with Φ = −36, Ψ = 127˚ depicting the carbonyl-carbonyl interaction, CH-O interaction and hydrogen bond formation between amino group and carbonyl oxygen of the side chain of the same residue.
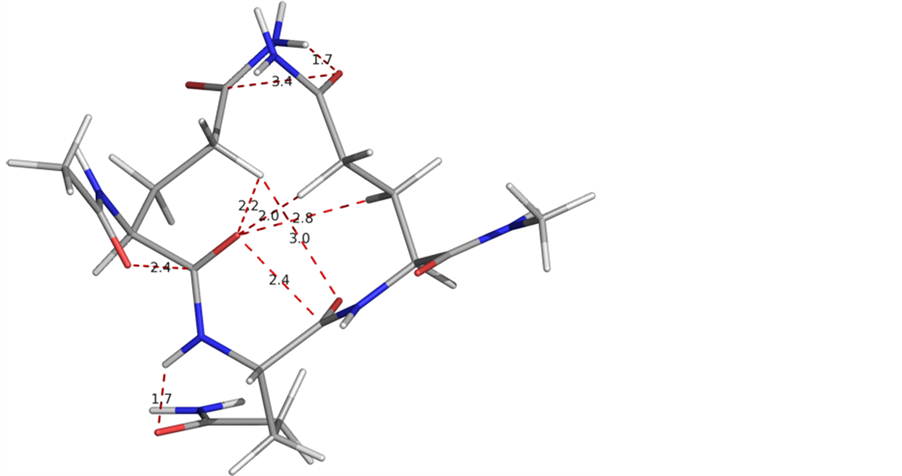
Figure 2. A graphical display of the molecule Ac-Gln3-NHMe displaying the backbone-backbone and side chain-side chain carbonyl interactions and CH-O interactions and hydrogen bonding.
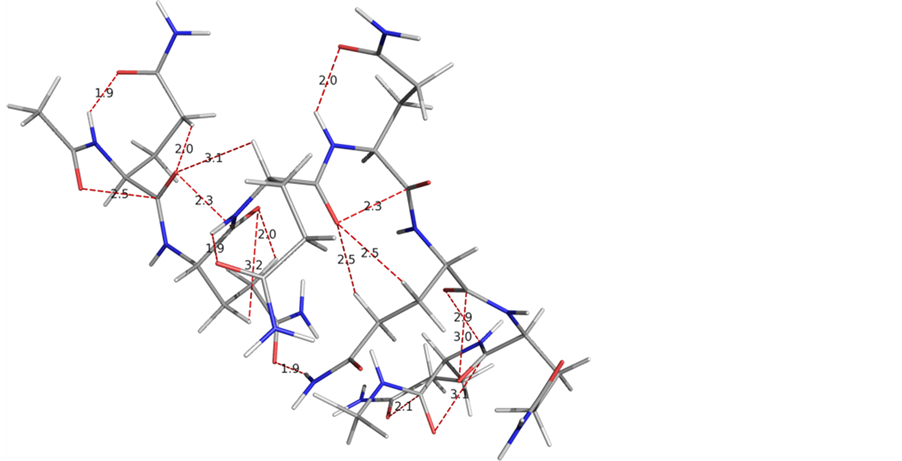
Figure 3. A graphical view of the peptide Ac-Gln7-NHMe in the most stable state showing the different interactions including backbone and side chain carbonyl-carbonyl.
restricted, and the peptide adopts compact structure. This may imply that the solubility of the poly Q may increase in hydrophobic environments.
4. Simulations
Preparation of the Starting Geometry for Simulations
The quantum mechanical results in table 1 clearly indicate that though the ϕ and ψ values do fall in the allowed region of the Ramachandran map, yet polyglutamine peptides do not have the tendency to be populated in single conformational state. The quantum mechanical calculations are restricted to small molecules, to prepare starting geometries for simulations of glutamine peptide of chain length more than seven, four starting geometries with ϕ, ψ values corresponding to different regions in Ramachandran map were considered, i.e., β-strand, inverse collagen, right handed helix and collagen type. These states were first optimised by Deep-View (SPDBV) [33] at dielectric constant of 80 and the optimised structures thus obtained are named as I, II, III, and IV, respectively. The ϕ, ѱ values thus obtained for Q16 given in table 2 and it is obvious from the results that most of the residues with starting geometry with ϕ and ψ values, 30, −130 (inverse collagen type) acquire ϕ and ψ values in the right handed helical region. The ϕ and ψ values in all the starting geometries undergo change with minimum changes for the right handed helical structure. Even, there is a change in the ω values and this has been explained in terms of carbonyl-carbonyl interactions and hydrogen bond formation between carbonyl group of the backbone and NH2 group of the side chain.
5. Simulations in Water
The SPDBV optimized geometries served as starting geometries for simulation studies under NVT conditions (i.e., corresponds to thermodynamic equilibrium). The 10 ns simulation results for poly Q6 are given in table 3 reveal that the ϕ, ѱ values of the starting geometry II and IV have not opened completely as they still have the residues involved in the hydrogen bond formation with ϕ, ѱ values in the helical region. Therefore, these structures were further simulated for additional 10 ns. After 20 ns simulations, the ϕ and ѱ value for the starting geometries I, II and IV were found to be in the 2nd quadrant of Ramachandran map. The helical structure takes longer time for opening i.e. structure III has not opened even for simulation up to 20 ns. These results form the basis for simulation studies of larger polyglutamine peptides with starting geometry I. It is due to the interaction of water molecule with the carbonyl groups and NH moieties, the ϕ, ψ, ω and χ values undergo changes and
Table 2. Optimized geometries obtained for simulation studies for Q16 with different conformational states with ϕ, ψ, ω and χ’s values of −130, 130 (I); 30, −130 (II); −57, −47 (III) and −30, 130 (IV) by SPDBV at ε 80.
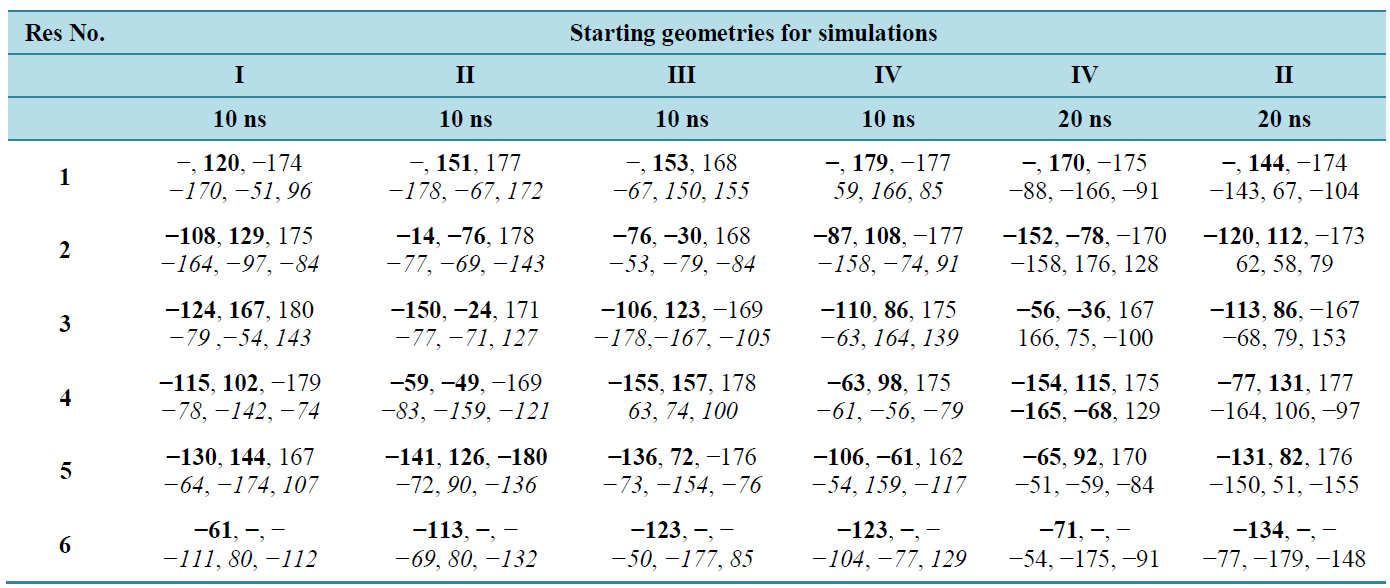
Table 3. Simulation results under NVT conditions for poly Q6 with different starting geometries.
φ, ψ, ω, & χ’ s values in bold, normal and italics respectively.
some of the amide/peptide bonds are available for interactions. This may imply that short chain poly Q peptides are soluble in water. A fact is consistent with the experimental observations [5] . The structure is stabilized by interaction of water molecules with the carbonyl groups and NH moieties both of the side chains as well as of the backbone, carbonyl-carbonyl interactions and hydrogen bonding between the side chains amide linkages. It is due to these interactions deviation in omega and planarity in the side chain amides ware observed. A molecular view of Q6 with ϕ, ψ values corresponding to β-strand after simulation studies with starting geometry-I with water molecule within 3Å of the peptide surface depicting the various stabilizing interactions is shown in figure 4.
Likewise, the simulations were carried out for polyglutamine Q10 and Q16. In simulation studies (table 4) also like the quantum mechanical results, the ϕ and ψ values for not more than three consecutive residues were found to be the same. The molecular view of Q10 and Q16 after simulations (with starting geometry-I) with water molecule within 3Å of the peptide surface are shown in figure 5. Poly Q10 adopts curved structure, whe
Table 4. Conformational results for Q16 after simulations for 10 ns with different starting geometries.
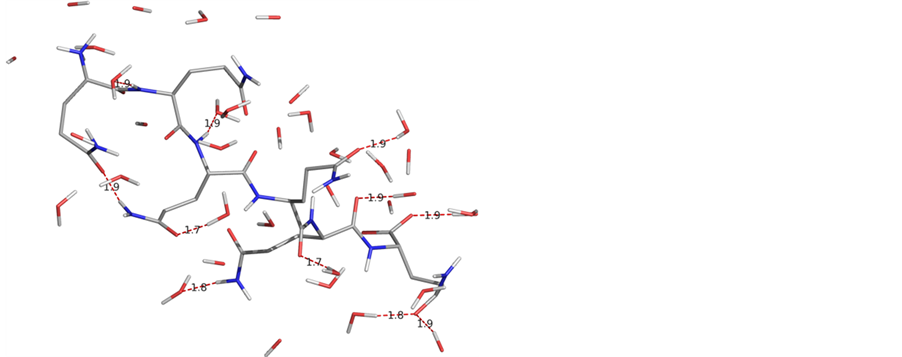
Figure 4. A molecular view of the peptide Q6 after simulations with starting geometry I with water molecules within 3 Å of the peptide surface adopts a beta strand like structure.
Figure 5. A graphical views of Q10 and Q16 after 10ns simulations with water molecules within 3 Å of the peptide surface clearly depict that the conformational behaviour is purely chain length dependent.
reas, in poly Q16 structure has the bends at the terminals positions. Both these structures are different from the structure of poly Q6, i.e., the conformational behavior of the glutamines peptides is chain length dependent. The ϕ and ψ values for Q16 as apparent from the table do not correspond to β-strand. These structures are stabilized by carbonyl-carbonyl interactions between 1) the backbones carbonyls 2) the backbone & side chain carbonyls 3) hydrogen bond formation involving backbone and side chain amide linkages. The network of these interactions is more in Q16. These interactions between the hydrophilic moieties eventually leads to decrease in hydrophilic character of glutamine residues and this observation is in consistent with the experimental finding that solubility of the poly Q decreases with increasing chain length.
6. Conclusion
The quantum mechanically and simulation results predict that the conformational behavior of polyglutamine is dictated by the interactions between the peptide bond of the side chain amide linkages. The conformational behavior of poly Q is found to be chain length dependant. The quantum mechanically results predict the conformational behavior of polyglutamine is found to resemble poly-L-proline type II. The polyglutamine structures are stabilized by various interactions like carbonyl-carbonyl, CH-O and hydrogen bonds. This study will form the basis to arrive at the structures of large poly Q peptides and hence their aggregation behavior.
References
- Fleischer, B. (1918) Uber myotonische Dystrophie mit Katarakt. Albrecht von Graefes Archiv für Ophthalmologie, 96, 91-133. http://dx.doi.org/10.1007/BF02018704
- Christopher, E.P., Kerrie, N.E. and John, D.C. (2005) Repeat Instability: Mechanisms of Dynamic Mutations. Nature Reviews Genetics, 6, 729-742. http://dx.doi.org/10.1038/nrg1689
- Williams, A.J. and Paulson, H.L. (2008) Polyglutamine Neurodegeneration: Protein Misfolding Revisited. Trends in Neurosciences, 31, 521-528. http://dx.doi.org/10.1016/j.tins.2008.07.004
- Poirier, M.A., Jiang, H. and Ross, C.A. (2005) A Structure-Based Analysis of Huntingtin Mutant Polyglutamine Aggregation and Toxicity: Evidence for a Compact Beta-Sheet Structure. Human Molecular Genetics, 14, 765-774. http://dx.doi.org/10.1093/hmg/ddi071
- Chen, S. and Wtezel, R. (2001) Solubilization and Disaggregation of Polyglutamine Peptides. Protein Science, 10, 887-891. http://dx.doi.org/10.1110/ps.42301
- Papaleo, E. and Invernizzi, G. (2011) Conformational Diseases: Structural Studies of Aggregation of Polyglutamine Proteins. Current Computer-Aided Drug Design, 7, 23-43. http://dx.doi.org/10.2174/157340911793743574
- Poirier, M.A., Li, H., Macosko, J., Cai, S., Amzel, M. and Ross, C.A. (2002) Huntingtin Spheroids and Protofibrils as Precursors in Polyglutamine Fibrilization. The Journal of Biological Chemistry, 277, 41032-41037. http://dx.doi.org/10.1074/jbc.M205809200
- Perutz, M.F., Johnson, T., Suzuki, M. and Finch, J.T. (1994) Glutamine Repeats as Polar Zippers: Their Possible Role in Inherited Neurodegenerative Diseases. Proceedings of the National Academy of Sciences of the United States of America, 91, 5355-5358. http://dx.doi.org/10.1073/pnas.91.12.5355
- Kim, M.W., Chelliah, Y., Kim, S.W., Otwinowski, Z. and Bezprozvanny, I. (2009) Secondary Structure of Huntingtin Amino-Terminal Region. Structure, 17, 1205-1212. http://dx.doi.org/10.1016/j.str.2009.08.002
- Poirier, M.A., Jiang, H. and Ross, C.A. (2005) A Structure-Based Analysis of Huntingtin Mutant Polyglutamine Aggregation and Toxicity: Evidence for a Compact Beta-Sheet Structure. Human Molecular Genetics, 14, 765-774. http://dx.doi.org/10.1093/hmg/ddi071
- Albà, M.M. and Guigó, R. (2004) Comparative Analysis of Amino Acid Repeats in Rodents and Humans. Genome Research, 14, 549-554. http://dx.doi.org/10.1101/gr.1925704
- Verdaguer, N., Urpi, L., Fita, I. and Subirana, J.A. (1988) Molecular Structure of L-Lysyl-L-Alanyl-L-Alanine: A Tripeptide Found in Histone H1. Biopolymers, 27, 1887-1896. http://dx.doi.org/10.1002/bip.360271203
- Nandel, F.S. and Jaswal, R. (2007) New Type of Helix and 27 Ribbon Structure Formations in PolyΔLeu Peptides: Construction of a Single-Handed Template. Biomacromolecules, 8, 3093-3101. http://dx.doi.org/10.1021/bm700504h
- Nandel, F.S. and Saini, A. (2011) Peptoids with Aliphatic Side Chains as Helical Structures without Hydrogen Bonds and Collagen/Inverse-Collagen Type Structures. Journal of Biophysical Chemistry, 2, 37-48. http://dx.doi.org/10.4236/jbpc.2011.21006
- Pullman, B. and Pullman, A. (1974) Molecular Orbital Calculations on the Conformations of the Amino Acid Residues of Proteins. Advances in Protein Chemistry, 28, 347-526. http://dx.doi.org/10.1016/S0065-3233(08)60233-8
- Weiner, S.J., Kollman, P.A., Nguyen, D.T. and Case, D.A. (1986) An All Atom Force Field for Simulations of Proteins and Nucleic Acids. Journal of Computational Chemistry, 7, 230-252. http://dx.doi.org/10.1002/jcc.540070216
- Mohle, K. and Hoffman, H.J. (1998) Secondary Structure Formation in N-Substituted Peptides. Journal of Peptide Research, 51, 19-28. http://dx.doi.org/10.1111/j.1399-3011.1998.tb00412.x
- Aduzbei, A.A. and Sternberg, J.E. (1993) Left-Handed Polyproline II Helices Commonly Occur in Globular Proteins. Journal of Molecular Biology, 229, 472-493. http://dx.doi.org/10.1006/jmbi.1993.1047
- Liermann, J.C., Opatz, T., Kolshorn, H., Hof, L.A.C. and Anke, H. (2009) Omphalotins E-I, Five Oxidatively Modified Nematicidal Cyclopeptides from Omphalotus olearius. European Journal of Organic Chemistry, 2009, 1256-1262.
- Fusetani, N. and Matsunaga, S. (1993) Bioactive Sponge Peptides. Chemical Reviews, 93, 1793-1806. http://dx.doi.org/10.1021/cr00021a007
- Van der, S.D., Lindahl, E., Hess, B., van Buuren, A.R., Apol, E., Meulenhoff, P.J., Tieleman, D.P., Sijbers, A.L.T.M., Feenstra, K.A., van Drunen, R. and Berendsen, H.J.C. (2005) Gromacs User Manual, Version 4.0.
- van Gunsteren, W.F., Billeter, S.R., Eising, A.A., Hünenberger, P.H., Krüger, P., Mark, A.E., Scott, W.R.P. and Tironi, I.G. (1996) Biomolecular Simulation: The GROMOS96 Manual & User Guide. Hochschulverlag AG an der ETH Zürich, Z ̈urich.
- Berendsen, H.J.C., Postma, J.P.M., DiNola, A. and Haak, J.R. (1984) Molecular Dynamics with Coupling to an External Bath. Journal of Chemical Physics, 81, 3684-3690. http://dx.doi.org/10.1063/1.448118
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F. and Hermans, J. (1981) Interaction Models for Water in Relation to Protein Hydration. In: Intermolecular Forces, D. Reidel Publishing Company, 331-342. http://dx.doi.org/10.1007/978-94-015-7658-1_21
- Sun, T.G., Liu, M., Chen, W.Z. and Wang, C.X. (2010) Molecular Dynamics Simulation of the Transmembrane Subunit of BtuCD in the Lipid Bilayer. Science China Life Sciences, 53, 620-630. http://dx.doi.org/10.1007/s11427-010-0103-7
- Hockney, R.W. and Eastwood, J.W. (1981) Computer Simulation Using Particles. McGraw-Hill, New York.
- Nose, S. (1984) A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. Journal of Chemical Physics, 81, 511-519. http://dx.doi.org/10.1063/1.447334
- Hess, B., Bekker, H., Berendsen, H.J.C. and Fraaije, J.G.E.M. (1997) LINCS: A Linear Constraint Solver for Molecular Simulations. Journal of Computational Chemistry, 18, 1463-1472. http://dx.doi.org/10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
- Essmann, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H. and Pedersen, L.G. (1995) A Smooth Particle Mesh Ewald Method. Journal of Chemical Physics, 103, 8577-8592. http://dx.doi.org/10.1063/1.470117
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F. and Hermans, J. (1981) Interaction Models for Water in Relation to Protein Hydration. In: Pullman, B., Ed., Intermolecular Forces, Springer, Berlin, 331-342. http://dx.doi.org/10.1007/978-94-015-7658-1_21
- Allen, F.H., Baalham, C.A., Lommerse, J.P.M. and Raithb, P.R. (1998) Carbonyl-Carbonyl Interactions Can Be Competitive with Hydrogen Bonds. Acta Crystallographica Section B, 54, 320-329. http://dx.doi.org/10.1107/S0108768198001463
- Deane, C.M., Allen, F.H., Taylor, R. and Blundell, T.L. (1999) Carbonyl-Carbonyl Interactions Stabilize the Partially Allowed Ramachandran Conformations of Asparagines and Aspartic Acid. Protein Engineering Design and Selection, 12, 1025-1028. http://dx.doi.org/10.1093/protein/12.12.1025
- Guex, N. and Peitsch, M.C. (1997) SWISS-MODEL and the Swiss-Pdb Viewer: An Environment for Comparative Protein Modelling. Electrophoresis, 18, 2714-2723. http://dx.doi.org/10.1002/elps.1150181505
NOTES

*Corresponding author.