Journal of Biophysical Chemistry
Vol.1 No.1(2010), Article ID:1879,14 pages DOI:10.4236/jbpc.2010.11003
Molecular dynamics simulation analyses of viral fusion peptides in membranes prone to phase transition: effects on membrane curvature, phase behavior and lipid-water interface destabilization
Department of Laboratory Medicine, School of Medical Technology, Teikyo University, Tokyo, Japan; *Corresponding Author:
kazunet@med.teikyo-u.ac.jp
Received 8 January 2010; revised 3 February 2010; accepted 18 February 2010.
Keywords: molecular simulation; fusion peptide; stalk formation; lipid mixing; hemifusion
ABSTRACT
To gain insight into the atomistic details of membrane fusion induced by fusogenic peptides, molecular dynamic simulations of synthetic peptides, derived from viral fusion proteins, contained in lipid bilayers were performed. A 20 amino acid peptide from the N-terminus of the influenza HA fusion peptide (WT20) assumed the oblique orientation at the interface between water and the membrane made up of dipalmitoylphosphatidylcholine (DPPC)/palmitic acid (PA), as reported previously for different membranes. Simulations of WT20 embedded in bilayer membranes made up of dioleoylphosphatidylethanolamine (DOPE) and DPPC/PA showed a positive curvature-inducing effect, whereas WT20 showed a negative curvature- inducing effect on a DPPC bilayer. In phase reconstitution analyses starting from a random mixture of DPPC, PA and water molecules, WT20 weakly stabilized an inverted hexagonal phase. In the latter analyses WT20 preferentially assumed a transmembrane orientation as opposed to the interfacial orientation, regardless of the phase to which the system settled (lamellar vs. inverted hexagonal). In another set of analyses using systems containing a water layer between the apposed DPPC/PA (and DOPE) monolayers, the behavior of WT20 during the formation of an intermembrane connection (or stalk) was examined. Comparison among the mutants supports a view that the oblique orientation of WT20 facilitates the perturbation of the lipid-water interface and the stalk formation. Taken together, these results imply that the influenza HA fusion peptide can have substantial effects on the membrane curvature and can assume a wide range of orientation/position in membranes depending on the local environment of the lipid/water system. Its movability and oblique orientation appear to be associated with its ability to perturb membrane/water interfaces.
1. INTRODUCTION
Fusion of viral and plasma or endosomal membranes is mediated by envelope glycoproteins, known as fusion proteins. For many viruses, fusion requires major structural modification of the fusion protein. Such modification has been established, for example, for influenza virus hemagglutinin (HA) and HIV gp41 proteins [1-6]. The ectodomain of HA has been shown to consist of six polypeptides forming a trimer of HA1-HA2 complexes. The N-terminus of HA2, critical for HA fusion activity, has a highly conserved, hydrophobic sequence, referred to as the fusion domain or fusion peptide (FP) [7,8]. In response to low pH in the endosome, HA changes conformation and projects its fusion peptide towards the target cell membrane [2,9].
Sequence and mutational analyses have identified and characterized FPs of fusion proteins for a number of viruses. Many findings support the direct involvement of FP, which is typically located at the N-terminus, in fusion between viral and cellular membranes (e.g. [7,10-15]). Studies of the full-length fusion protein and in vitro assay of fusion peptide activities provided important clues on protein-induced membrane fusion events. In addition, synthetic FPs have been shown to exhibit membrane fusion and lytic activities [16-26].
The FP of the Newcastle Disease Virus was shown to adopt an oblique angle in a DPPC monolayer [27]. Since then, the tilted orientation has been reported for FPs of several fusion proteins involving SIV gp32 [28], HIV-1 gp41 [29] and HA2 [30]. More recently, Tamm and coworkers, using NMR and EPR, showed that the FP of HA (or HAFP) forms a shallow-angle inverted V structure [31,32]. Their subsequent studies of the mutants G1V and W14A support the view that the inverted V shape and/or oblique orientation plays an important role in the fusogenicity of the peptide [33,34]. The N-terminus FP and membrane proximal external region (MPER) of HIV-1 gp41 also exhibit lipid destabilizing and fusion activity against liposomes [25,26,28,35,36]. Several other peptides from proteins involved in lipid metabolism or from neurotoxic proteins have also been shown to adopt a tilted orientation (for review see [27,37]). Charloteaux et al. [37] have proposed that the tilted orientation may be responsible for the lipid destabilizing activity of FPs. For HAFP, although experimental findings vary in several aspects, there is general consensus that the significant α-helical content and oblique orientation of HAFP in the membrane are important for fusogenicity [30,38,39].
Recent computational molecular simulations have been extended to studies of phase transitions of lipids, providing opportunities to study how the formation of nonlamellar phases is affected by lipid composition and peptides [40]. Marrink and Tieleman carried out atomistic molecular dynamics (MD) simulations of monoolein in an inverted cubic phase of the diamond type [41]. Importantly, a dipalmitoylphosphatidylcholine (DPPC)/palmitic acid (PA)/water 1:2:20 system exhibits membrane fusion and transformation from a lamellar to the inverted hexagonal phase in a manner similar to experimental systems [42]. Coarse-grained (CG) simulations of DOPE and DOPE/DOPC (dioleoylphosphatidylcholine) bilayers showed that spontaneous formation of stalks between the lamellar bilayers is triggered either by increasing the temperature or by reducing the hydration level [43]. It has also been shown that a transmembrane peptide induces lipid sorting as well as the lamellar to inverted phase transition in CG simulations [44].
Given the progress in molecular dynamics simulations, one may ask whether membrane fusion induced by FPs can be studied with MD simulations. Several simulation studies of FPs with explicit treatment of the water/lipid membrane have been carried out [45-48]. They showed that the oblique and kinked structure of FPs is stable for at least 5-18 ns in DMPC (dimyristoyl-phosphatidylcholine) bilayer [47] and in POPC (palmitoyloleoylphosphatidylcholine) bilayers [46,48]. A recent study on oligomerization of HAFP, which utilized an implicit treatment of water, has shown that many different configurations (peptide/peptide and peptide/membrane) have similar energy levels [49]. How such structural features of FPs are associated with its fusogenicity is important. However, MD simulation analysis of membrane fusion is generally in its infancy. Hence, using the DPPC/PA and DOPE systems that have been shown to reproduce the phase behavior reported for the corresponding experimental system [42,50,51], we performed the following sets of simulations (~3 ms in total) to examine dynamics of HAFP and other peptides.
First, the equilibrium position/orientation in the bilayer membranes of HAFP was analyzed (in the “equilibrium analysis” section). Second, using relatively large patches of membranes, the effect of HAFP on the membrane curvature was examined (“curvature analysis”). Third, using the phase reconstitution system made up of DPPC/PA/water, the effect of HAFP on the phase behavior was analyzed (“phase analyses”). Finally, in the Discussion section, we show some non-equilibrium MD simulation results to examine the behavior of HAFP and MPER during the formation of an intermembrane connection (“stalk propensity analyses”). Overall results are consistent with recent experimental and theoretical findings, suggesting the potential usefulness of the atomistic simulation approach for studying peptide-induced fusion. However, given the previous simulation studies of HAFP with the DMPC and POPC membranes [46-48], we here attempted to examine the peptide dynamics in lipid structures involving non-lamellar ones. Therefore, the simulation conditions are not physiological and corresponding experiments involving the peptides have not been done. Since this approach cannot stand alone, experimental and structural studies that solidify the hypothesis are necessary.
2. SIMULATION DETAILS
The GROMACS 3.3.1 program was used for MD simulations [52]. For DPPC, the parameters by Berger were used [53]. For palmitic acid (PA), the protonated state was used as in Knecht et al. [42]. The PA carboxyl group was parameterized based on the glutamic acid from the GROMOS-87, whereas for the remaining acyl chain, the Berger parameter was used [53]. For DOPE, the parameter by Berger et al. [53] with modification by de Vries [50] was used. For the HAFP, X31 sequence, referred to as WT20, was used as in [32]. For the MPER of HIV-1, the segment of amino acids 662-683 was used based on Sun et al. [54]. The sequence of the peptides analyzed are:
WT20:GLFGAIAGFIENGWEGMIDG
W14A:GLFGAIAGFIENGAEGMIDG
G1V:VLFGAIAGFIENGWEGMIDG
WT13:GLFGAIAGFIENG
WT8:GLFGAIAG
MPER: ELDKWASLWNWFNITNWLWYIK
Note that W14A and G1V are identical with WT20 except for the altered residue shown with an underline. For the peptides and their mutants, the GROMOS-96 parameter set was used. For water, the simple-point charge (SPC) model [55] was used as in Knecht et al. [42]. For all simulations, the bond lengths of lipids and water molecules were restrained using the LINCS and SETTLE respectively [56,57]. To account for the long-range electrostatic interactions, the particle-mesh Ewald (PME) algorithm [58] was used with the realspace cutoff at 9Å and the maximal grid size of 0.12Å. Berendsen coupling was used for temperature control [59]. The pressure was controlled by the Berendsen barostat at 1 atm coupling all directions of the box to 1 bar allowing the box to deform.
As stated in the Introduction, this study consists of the equilibrium analysis, the curvature analysis, the phase analysis and the stalk propensity analysis. For all DPPC/PA membranes, the initial coordinates were created by randomly placing the DPPC and PA in a manner shown in [42]. The DOPE bilayer was prepared as described in [50]. Our preliminary analyses reproduced the phase behavior of the DPPC/PA/water system reported in [42]. 1:20 DOPE/water did not show a HII transition at 353 K within 10 ns. For peptides, coordinates for the initial structures were based on NMR analyses [31,33,34,54]. For WT20, G1V and W14A, model 8 (of PDB code 1IBN), model 2 (of 1XOP) and model 17 (of 2DCI) were used respectively, based on the root mean square deviation (RMSD) of the backbone heavy atoms from the other models. Likewise, for the MPER the model 3 of 2PV6 was used [54]. For the HAFP, the N-terminus of the peptides was protonated as in Zhou et al. [60], whereas the C-terminus was capped with NH2. The Nand C-termini of MPER were acetylated and amidated, respectively. The sidechains of Glu11 and Glu15 of the HAFP were protonated except for set 16 based on the study by Zhou et al. [60]. For ionizable residues of the MPER, the standard protonation state at pH 7 was used.
For the equilibrium analysis (Figure 1), a membrane composed of DPPC/PA/water 1:2:40 was pre-equilibrated, then the peptides were embedded in a position/orienta-tion similar to those in [31,33,34,54]. A 100 ps run was carried out with positional restraints on all peptide atoms, followed by a 10 ns run at 323 K with restraints only on the dihedral angles (Figure 1 and see below).
For the curvature analysis (Figure 3), three different bilayers were used: 1) 192 DPPC, 384 PA and 15909 water molecules, 2) 270 DOPE and 10324 water molecules, and 3) 240 DPPC and 11065 water molecules. The approximate size of the simulation box (in x, y, z dimensions) was for: 1) 4.5 × 23.3 × 9.4 nm3, 2) 4.5 × 18 × 8.5 nm3, and 3) 4.5 × 20 × 8.0 nm3. Three WT20 peptides were placed at the lipid/water interface and 20 ns simulations were carried out. The curvature was measured using the distance d, which is the distance between the
 (a)
(a)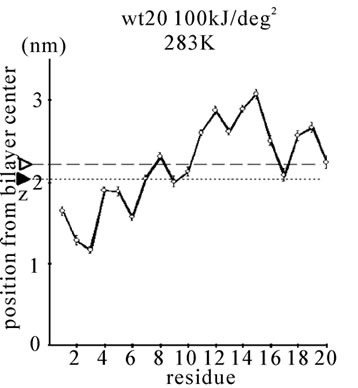 (b)
(b) (c)
(c)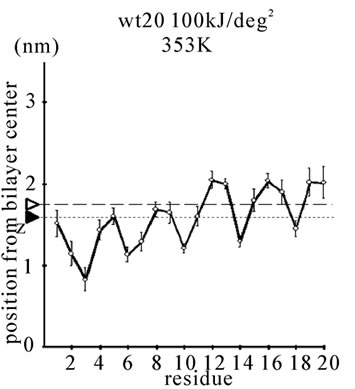 (d)
(d)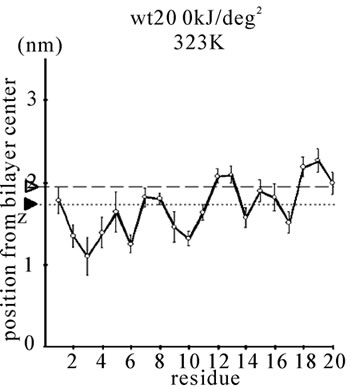 (e)
(e) (f)
(f)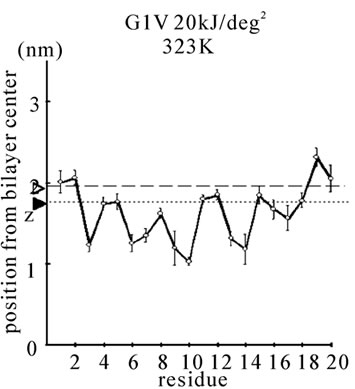 (g)
(g)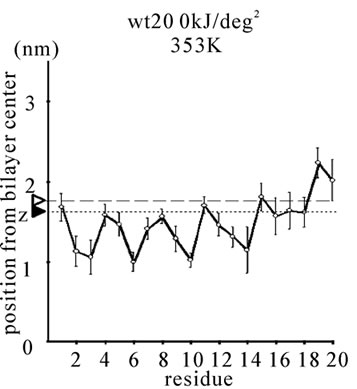 (h)
(h)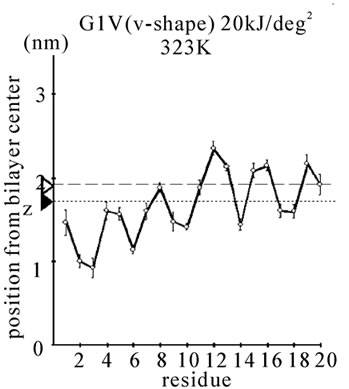 (i)
(i) (j)
(j)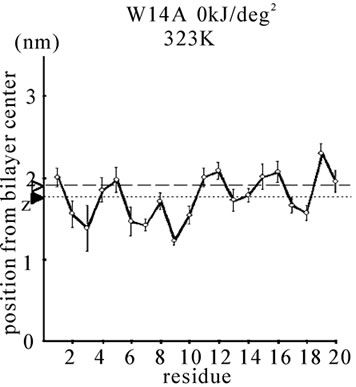 (k)
(k)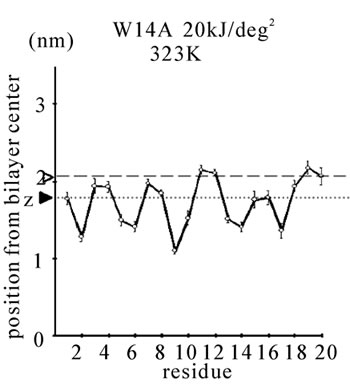 (l)
(l)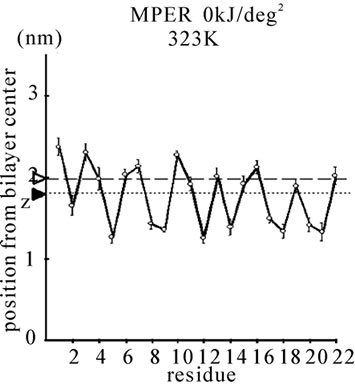 (m)
(m)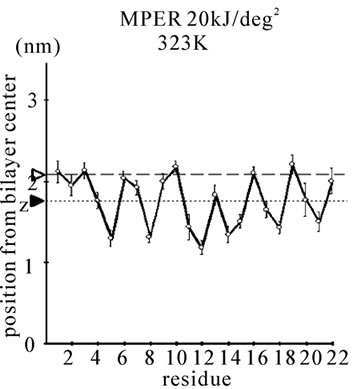 (n)
(n)
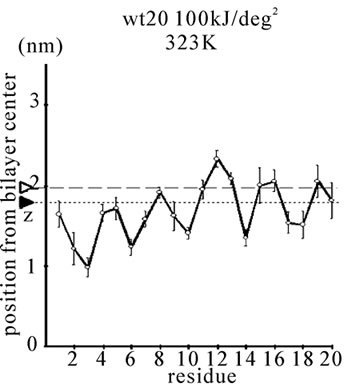
Figure 1. The average z-positions of peptide sidechains in the equilibrium analysis. Shown are the COMs (the center of mass) of side chains and standard deviation (error bars) obtained from the period between t = 5 and 10 ns. (a-c) DPPC/PA/
water/WT20 1:2:40:1 system with the dihedral restraints 100 kJ/(deg)2 at 283 K (a), 323 K (b) and 353 K (c). (d, e) Results of the same system as in (a-c) but with using 20 kJ/(deg)2 restraints at 323 K (d) and 353 K (e). (f, g) Results of the same system as (a-c) but without dihedral restraints at 323 K (f) and 353 K (g). The average positions of the phosphorus atoms in lipid molecules are shown by arrowheads. (h) G1V data at 323 K with 20 kJ/(deg)2 restraints. (i) G1V data at 323 K without restraints. (j) G1V data at 323 K restrained to the 1IBN (pH 5 NMR-structure of WT20) with 20 kJ/(deg)2 dihedral restraints. (k) W14A at 323 K with 20 kJ/(deg)2 restraints. (l) W14A at 323K without restraints. (m) DPPC/PA/water/MPER 1:2:40:1 system with the 20 kJ/(deg)2 restraints, at 323 K. (n) Same as (m), but without the restraints. In each figure, an open triangle represents the average position of the COM of phosphorus atoms of DPPC, whereas a closed triangle represents that of carbonyl oxygen atoms of DPPC.
bilayer midplane and the COM of the group of the lipid molecules whose x-coordinate of the COM is located within the range covered by the three peptides as shown by the cartoon in the Figure 3.
For the phase analysis (Figure 4), the procedure was based on [42]. A system composed of 32 DPPC, 64 PA, 640 SPC water molecules with or without WT20 was used. 30 ns simulations were performed starting from the initial structures generated randomly using the Genbox module of the Gromacs suite.
For the stalk propensity analysis (Figure 5), the initial coordinates of lipids and peptides were sampled from the last 0.5 ns of the equilibrium analysis runs (at 323 K), whereas the hydration level was reduced to e.g., DPPC/PA/water 1:2:20. (The structure at 353 K was not used because the reduction of the hydration level from 1:2:40, if carried out at 353 K, appears to be too strong a perturbation.) The amount of water between the monolayers was adjusted without moving the peptide or lipid. To reduce the bias further, we sampled different frames from the equilibrium analysis. Care was taken to vary the coordinates and initial velocities among the runs. In this study, a stalk was defined as follows. First, a connection of lipid molecules between the lipid monolayers was found which existed for > 1 ns. Second, this connection was judged to be a stalk when it could be regarded as a cylinder that contained some of the atoms of three or more lipid molecules and 10 or less water molecules, within any 3Å segment of the cylinder. For the two-peptide stalk propensity analysis, the lipid/peptide coordinates at 323 K were aligned such that the peptides contained in the proximal monolayers apposed to each other and oriented in three different relative configurations (parallel, cross and anti-parallel). For example, for the cross configuration, two peptides form a ‘cross’ when viewed from the top, whereas, for the anti-parallel configuration, the N-terminus of one peptide and the C-terminus of the other overlap when viewed from top. For all the configurations, the xand y-coordinates of the center of mass of the two peptides were identical.
One feature of this study is that many simulations utilized restraints on the f and y dihedral angles of the peptide backbone, using the initial structures (i.e., the models from 1IBN and 2PV6) as the reference structures. For the restraints, a coefficient of 100 kJ/(deg)2 (strong restraint) and 20 kJ/(deg)2 (weak restraint) were tried. With the weak restraints at the high temperature (353 K), the helicity of WT20 was preserved, but the kink-like structure was less prominent (Figure 1 and data not shown). However, to minimize the effect of the artificial restraints, we utilized the weak restraints in many of the simulations. As the Results section shows, when the dihedral restraints were removed, structural variation was large in the pre-runs and in our preliminary analyses; the stalk-forming ability of the peptide was low compared to the peptide with the restraints. This led us to apply restraints in many of the simulations. The effects of the restraints must be critically assessed in future studies.
In this study, the z-axis is perpendicular to bilayer. For the secondary structure analysis, the DSSP program was used based on [61]. Calculations were carried out on 15 AMD 2.2-GHz processors. All molecular images were made with VMD [62].
3. RESULTS
3.1. Equilibrium Analysis
First, the equilibrium position and structure of the peptides were analyzed. The positions of the COM of the amino acid sidechains in DPPC/PA/water/WT20 1:2:40:1 simulations were analyzed (Figures 1(a)-1(g)). The composition was chosen because the phase behavior (i.e., lamellar vs. inverted hexagonal) has been studied by both experiments [51] and simulations [42] under similar conditions. The temperature was varied widely (from 283 K to 353 K) because of the potential usefulness of the system for peptide dynamics analyses with lipids in a non-lamellar phase. For Gly residues the position of the Cα atom is shown in Figure 1. Under this water-rich condition, no intermembrane connection was formed during 10 ns. Note that for Figures 1(a)-1(c), the 100 kJ/(deg)2 restraints on the peptide backbone dihedral angles were used (see Simulation Details). At 323 K, WT20 resided in a slightly deeper part of the membrane than at 283 K, most likely because the area per lipid molecule increased and the headgroup layer was able to accommodate the peptide better at 323 K (Figures 1(a) and 1(b)). (In the pilot analyses, the area of the membrane(128 DPPC and 256 PA molecules) was 52.3, 67.3 and 78.4 nm2 at 283, 323 and 353 K respectively). At 323 K, the position and orientation of WT20 were largely similar to those of previous simulations [46-48]. At 353 K, the structure was similar to that at 323 K, with the exception that the lipid acyl chains were opened wider, making the bilayer thinner than at 323 K (Figure 1(c)).
As previous simulation studies have described [46-48], the overall structure was V-like in shape, but the angle of V was larger than the pH 5 NMR-structure (PDB code 1IBN) by Han et al. [31].
When the restraints were weakened to 20 kJ/(deg)2, the general orientations were similar to those found with 100 kJ/(deg)2 restraints (Figures 1(d) and 1(e)). The α-helix content was also similar to that with 100 kJ/(deg)2 restraints (Figures 2(a) and 2(b)). At 353 K, α-helical content was 63.5 ± 10.3% for 100 kJ/(deg)2 and 68.1 ± 8.1% for 20 kJ/(deg)2 for the last 5 ns (Figures 2(a) and 2(b)), similar to ~66.8% for the pH 5 structure (1IBN).
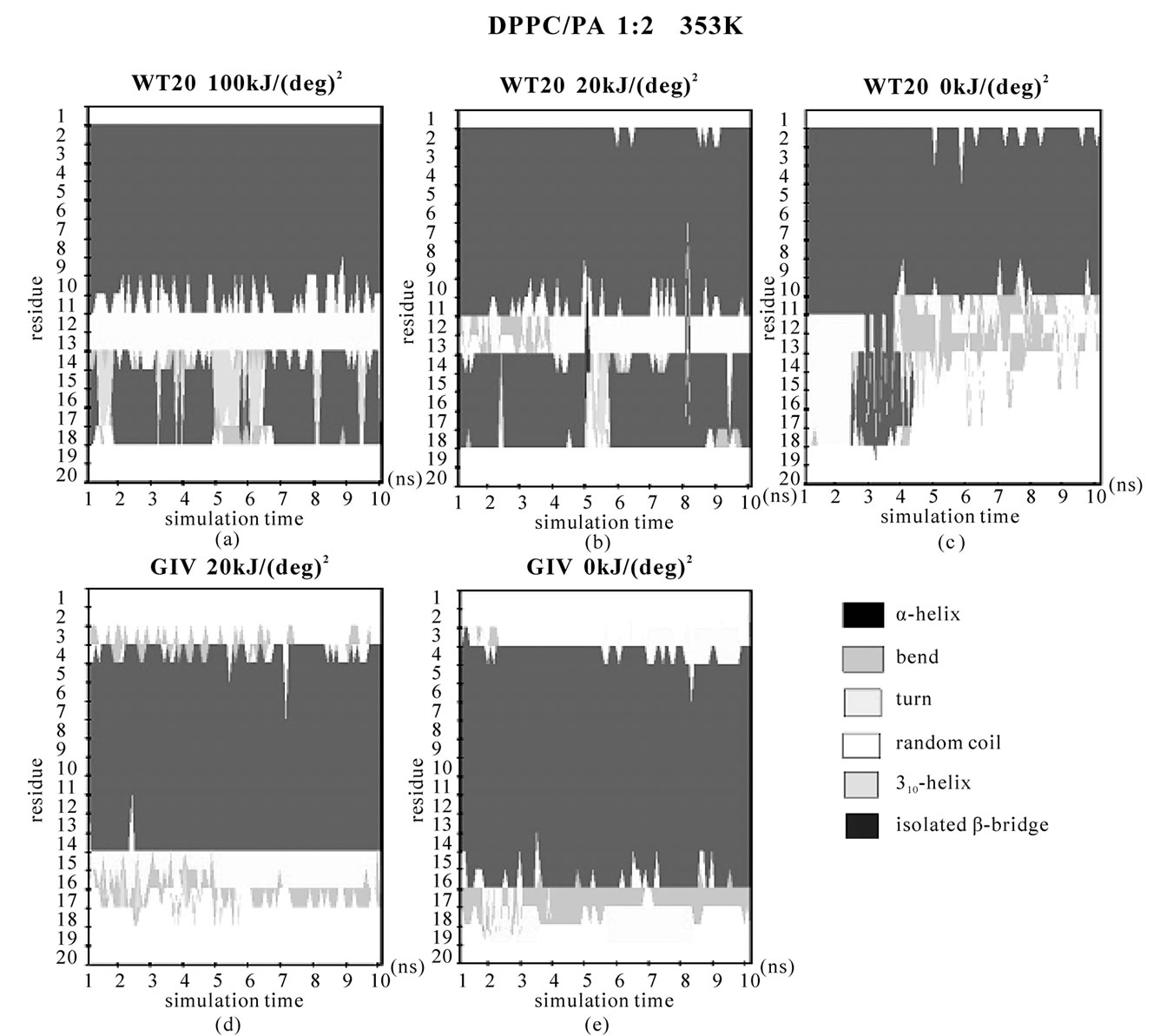
Figure 2. Secondary structure analysis of the peptide backbone based on DSSP [54]. (a, b, c) WT20 with DPPC/PA/water 1:2:40 system at 353 K. (a) Simulation with 100 kJ/(deg)2 dihedral restraints. (b) Simulation with 20 kJ/(deg)2 dihedral restraints. (c) Simulation with no dihedral restraints; (d, e) G1V with DPPC/PA/water 1:2:40 system at 353 K. (d) With 20 kJ/(deg)2 restraints. (e) With no dihedral restrains.
However, when no dihedral restraints were used at 353 K, the orientation was flatter and the helical content decreased to ~45% (Figure 2(c) and data not shown). For runs with dihedral restraints of 100 kJ/(deg)2, 20 kJ/(deg)2 and no restraints, the RMSDs (the average of all Cα atoms) for the last 5 ns of the 353 K pre-run were 1.9 Å, 3.4 Å and 5.2 Å, respectively, with respect to the pH5 NMR-structure (1IBN). (The RMSDs compared with the structure at t = 8 ns were 0.7 Å and 1.8 Å and 2.0 Å.)
Taken together, without the restraints, WT20 exhibits substantial deformation at 353 K from the pH 5 NMRstructure. Removal of the dihedral restraints resulted in a substantial change in the overall structure and an increase in α-helical content also in the case with G1V and W14A mutants (Figures 1(h), 1(i), 1(k), 1(l) and Figures 2(d), 2(e).). Of note, for G1V and W14A, the restraints based on the reported structures (PDB code 1XOP and 2DCI, respectively) were used. Considering the potential importance of structure, simulations were performed mainly with dihedral restraints of 20 kJ/(deg)2 in the following, whereas some simulations were carried out without the restraints.
3.2. Curvature Analysis
The second line of the analyses examined the effect of WT20 on the curvature of bilayer membranes. As Figure 3 illustrates, three WT20 molecules were embedded in the headgroup layer of each side of bilayers and 20 ns simulations were carried out at 313 K without the dihedral restraints. Intriguingly, the presence of the three WT20 molecules led to a slightly positive curvature of a DPPC/PA 1:2 bilayer, with the d value (Figure 3(a)) being ~0.53 nm (set-L1, Figure 3(b)). When three WT20 peptides were placed such that they aligned with the x-axis, the curvature occurred over the x-direction (set-L1). When the WT20 peptides were aligned with the y-axis, very weak positive curvature was observed over the x-direction (set-L2, Figure 3(c)). The positive curvature-inducing effect on a DOPE bilayer was also observed when WT20 peptides were aligned with the x-axis and also when aligned with y-axis (set-L3 and L4 in Figures 3(d) and 3(e), respectively). However, the effect on a DPPC bilayer was unstable when the WT20 molecules were aligned with the x-axis, as suggested by the large s.d. of d value (as shown in Figure 3, set-L5), whereas a negative curvature was induced when the WT20 molecules were aligned with the y-axis (set-L6 in Figure 3). Overall, the effect on the curvature is dependent on the lipid composition and the peptide orientation. It is currently unclear why the variance was large in the case of DPPC membrane with peptides aligned with the x-axis (set-L5). As a positive control, a DPPC bilayer containing lysophosphatidylcholine (with palmitic acid as the acyl chain) and phosphatidic acid induced curvature in 5 ns simulations of the DPPC/PA bilayer (data not shown), and therefore at least for our setting, 20 ns simulations were likely to be sufficiently long to examine the peptide effects on membrane curvature.
3.3. Phase Analysis
The effect of WT20 on ‘phase behavior’ of the DPPC/ PA/ water system was analyzed in the manner described in [42]. In this method, simulations starting from random structures/positions of molecules were performed, allowing spontaneous reconstitution of a lamellar or inverted hexagonal phase (Figure 4). As set-R1 of Figure 4 shows, 60% of the runs (n = 20) performed at 333 K without WT20 formed an inverted hexagonal phase, while 35% of the runs formed a lamellar (bilayer) phase consistent with previous results [42]. For the simulations performed at 333 K with a WT20 peptide (set-R2), a slight increase in the propensity for the hexagonal phase was observed; 75% of runs (n = 20) formed an inverted hexagonal phase.
The effect of WT20 was more pronounced in the simulations performed at 313 K (set-R3 and -R4) than in simulations performed at 333 K; the presence of WT20 increased the propensity of hexagonal phase formation from 35% (set-R3) to 65% (set-R4). These simulations were performed with the 20 kJ/(deg)2 dihedral restraints on WT20 to the 1IBN structure. When the dihedral restraints were removed and 313 K simulations performed, the results were largely similar (set-R5) with 60% were judged to be an inverted hexagonal phase. Overall, although the effect appeared to be subtle, the presence of WT20 caused a small shift toward the hexagonal phase. One notable feature of the results was that the WT20 tended to reside in the hydrophobic part of the bilayer or hexagonal structures, protruding into the lipid acyl chains, with the Nand C-termini interacting with distinct water columns (or layers) (Figures 4(d) and 4(e)). In fact, such position/orientation was observed for all the simulations that assumed an inverted hexagonal phase and for 13 out of the 14 simulations that assumed a lamellar phase. Of course, many more simulations have to be carried out to draw any conclusion regarding the equilibrium position because the amount of water seems to be a critical factor governing the equilibrium position of the peptide. Nonetheless, these features are interesting in that such versatility of WT20 positioning may be relevant to its function in later stages of membrane fusion, such as hemifusion formation and rupture of hemifusion diaphragm.
4. DISCUSSION
In the above, the results of the equilibrium analysis, the curvature analysis and the phase analysis of WT20 were shown. In the equilibrium analysis, the oblique orientation of WT20 was largely similar to that reported
 (a)
(a)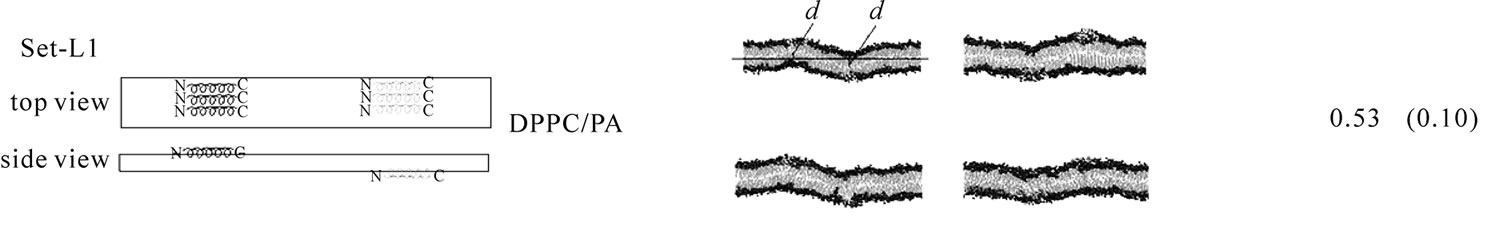 (b)
(b) (c)
(c) (d)
(d) (e)
(e) (f)
(f) (g)
(g)
Figure 3. The effect of WT20 on the membrane curvature. (a) Scheme for peptide embedding and curvature measurement. Three WT20 molecules were embedded in the direction along the x-axis or y-axis and 20 ns simulations carried out. The curvature assessment is based on the distance d shown in the cartoon. (b-g) The average d value and representative snapshots at about 20 ns are shown for each set. If the curvature was positive, the value of the average of d becomes positive. Note that two d values per simulation were obtained and in total eight d values were obtained when four simulations were performed. Yellow trace shows the peptide backbone. Red and blue spheres show lipid oxygen and nitrogen atoms. Cyan lines show lipid acyl chains. Water molecules are hidden for clarity. (b) set-L1. Four simulations with a DPPC/PA 1:2 bilayer. WT20 molecules were oriented along the x-axis. The average of d = 0.53 nm with s.d. = 0.10. (c) set-L2. Same as (b) but the WT20 molecules are oriented along the y-axis. Two simulations were performed. The average of d = 0.41 nm. (s.d. = 0.24). (d) set-L3. DOPE membrane with the WT20 peptides oriented along the x-axis. Two simulations were performed. The average of d = 0.27 nm (s.d. = 0.07). (e) set-L4. Same as (d) but the WT20 peptides are oriented along the y-axis. Two simulations were performed. The average of d = 0.42 nm (s.d. = 0.13). (f) set-L5 DPPC membrane with the WT20 peptides oriented along the x-axis. Three simulations were performed. The average of d = –0.21 nm (s.d. = 0.46). (g) set-L5 DPPC membrane with the WT20 peptides oriented along the y-axis. Three simulations were performed. The average of d = –0.85 nm (s.d. = 0.25).
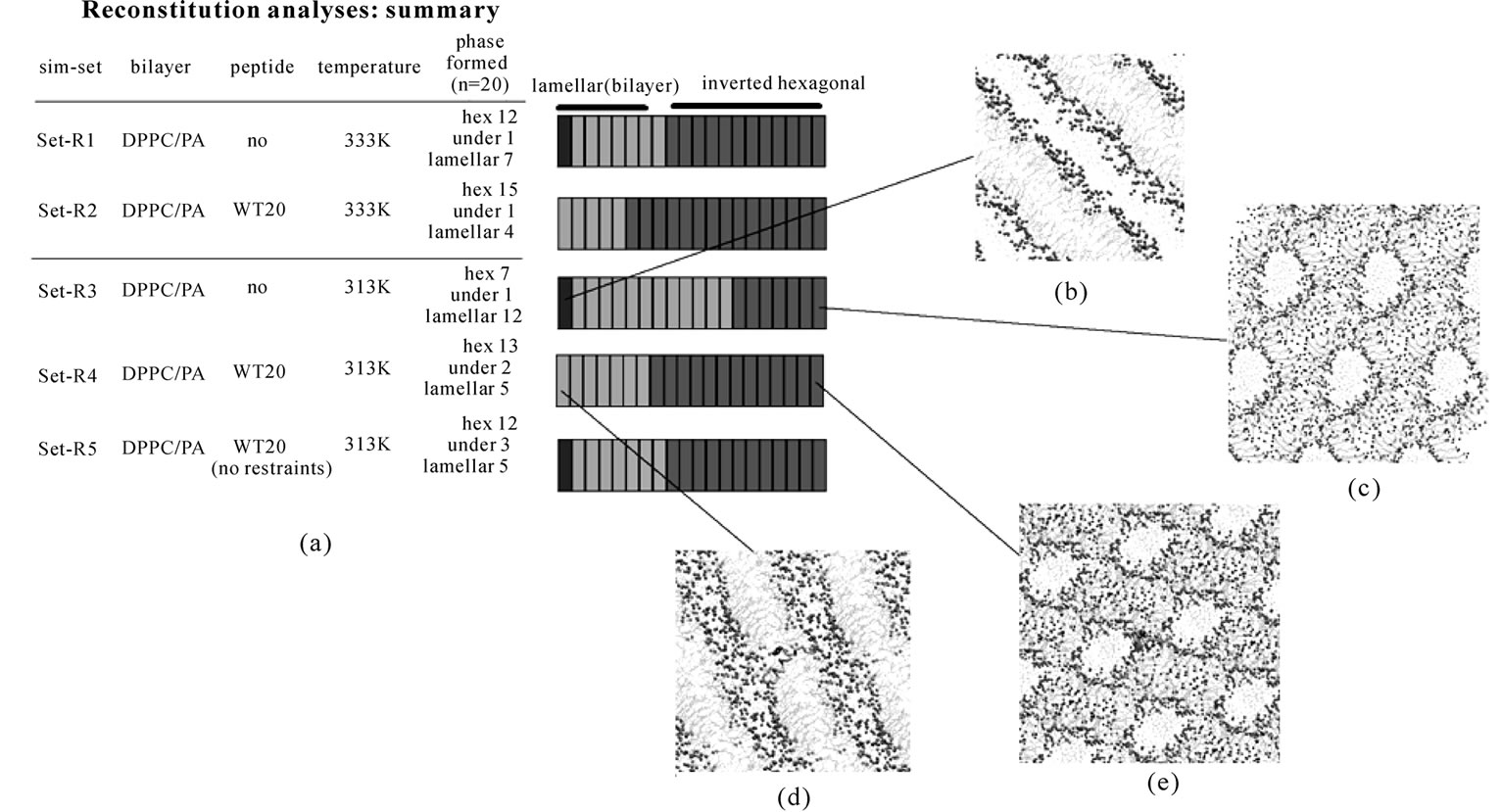
Figure 4. Analyses based on spontaneous formation of the bilayer or the inverted hexagonal phase. (a) Summary of five sets of simulations (set-R1 to R5): (set-R1) DPPC/PA/water, 333 K; (set-R2) DPPC/PA/water/WT20, 333 K; (set-R3) DPPC/PA/water, 313 K; (set-R4) DPPC/PA/water/WT20, 313 K; (set-R5) DPPC/PA/water/WT20 (no restraints), 313 K. 32 DPPC, 64 PA and 640 water molecules were used. For each set, twenty simulations were performed starting from different random structures. Note that for set-R2 and R4, the dihedral angles of WT20 were restrained (20 kJ/(deg)2) to the 1IBN as the reference structure. Shown in the right column is a summary of the phases obtained in the simulations. For each set, the final phase of separate 40 ns simulations is judged to be one of an inverted hexagonal ('hex'), a lamellar phase or an undefined (‘undef’) phase as in [42]. A structure mainly consisting of a bilayer is judged as a lamellar phase although this phase is likely to correspond to a gel phase in [42]. A lamellar phase is indicated in red (pink denotes a defect containing lamellar phase), an inverted hexagonal phase is depicted in green. Gray denotes an undefined phase. (b-e) Representative images of the phase formed: a lamellar phase formed in a set-R3 simulation run (b), a hexagonal phase in a set-R3 run (c), a lamellar phase in a set-R4 run (d), a hexagonal phase in set-R4 run (e). In (d, e) the WT20 peptide backbone is represented by an orange trace and the N-terminus as a yellow sphere. The membrane representation is similar to that described in the legend of Figure 3.
previously, despite of our use of the DPPC/PA membrane. At the high temperature, the orientation was less oblique than at the low temperature, yet the introduction of the dihedral restraints restored the oblique angle. This finding may have technical implications for future simulation studies of peptide-induced membrane fusion.
Does WT20 alter the membrane curvature? Experiments using dipalmitoleoylphosphatidylethanolamine (DiDOPE) by Epand and Epand [63] have shown that the wildtype peptide lowers the temperature of the phase transition from bilayer to hexagonal at pH 5 while it raises the temperature at pH 7.4. When added to phosphatidylethanolamine (PE), fusion peptides of some viral fusion proteins induce QII (inverted cubic) phases that do not form spontaneously without the addition of the peptides [64,65]. These and other findings suggest that HAFP lowers the activation energy for steps in the fusion process, rather than stabilizing a particular structure such as the hexagonal phase. Although the difference in lipid composition makes it difficult to compare the experimental findings with our results, our findings suggest a possibility that MD simulations can complement experimental phase analyses in the presence of peptides. In our analyses, the WT20 effect leading to the slightly positive curvature in DPPC/PA membrane (Figures 3(b) and 3(c)) may appear to be inconsistent with the result of the reconstitution analyses (Figure 4) in which the presence of WT20 led to an increased propensity of an inverted hexagonal phase. However, the peptide protruded into the hydrophobic part of the hexagonal structure with the Nand C-termini interacting with distinct water columns (Figures 4(d) and 4(e)). Therefore, the phase behavior of the DPPC/PA membrane may be difficult to interpret simply based on the peptide effect on the curvature. The HA peptide may assume distinct orientations and functions depending on the lipid membrane composition and physical properties.
The transmembrane orientation of WT20 observed in the phase analysis may be relevant to the later steps of liposome fusion (e.g., the formation of hemifusion and the hemifusion diaphragm rupture), which deserve future analyses. The previous studies using EPR and NMR have unambiguously shown that WT20 resides in the outer leaflet of the bilayer membranes [31]. Therefore after binding to the membrane, WT20 is most likely stay in the outer leaflet with the tilted orientation. On the other hand, it has been proposed that, after the stalk formation WT20 assumes a more vertical and/or transmembrane orientation, interacts with transmembrane domain of the fusion protein and destabilizes the stalk and/or the hemifusion diaphragm [4,5]. In this context, more analyses using the reconstitution of various lipids and peptides may give clue to peptide functions in membrane fusion.
4.1. Stalk Propensity Analysis
HAFP and many other peptides derived from virus fusion proteins exhibit lipid destabilizing and fusion activity against liposomes. So, one could be tempted to ask whether the fusogenicity of such peptides in vitro experiments can be studied in silico. For the DPPC/PA system, the stalk formation is the critical (rate-limiting) step in the transition from a lamellar to an inverted hexagonal phase [42]. For this system, stalk occurrence can easily be observed just by raising the temperature or reducing the hydration level. So, we initially attempted to analyze the peptide behavior during the very early phase of stalk formation. In this attempt, we unexpectedly found that WT20 can facilitate the stalk formation in the system. Therefore, we carried out several nonequilibrium analyses, in which one or two peptide(s) were contained in two apposed lipid monolayers. Figures 5(a) and 5(b) illustrate the setting of the oneand two-peptide simulations, respectively. It may be envisaged that this set of analyses mimics the situation in which dehydration drives two liposomes containing peptides to collide with each other. The initial structures were carefully set up as commented in the Simulation Details section. Table 1 summarizes the results of onepeptide simulations (mainly using the DPPC/PA/water/WT20 1:2:20:1 system). The stalk propensity score represents the proportion of simulations in which a stalk was formed (Table1).
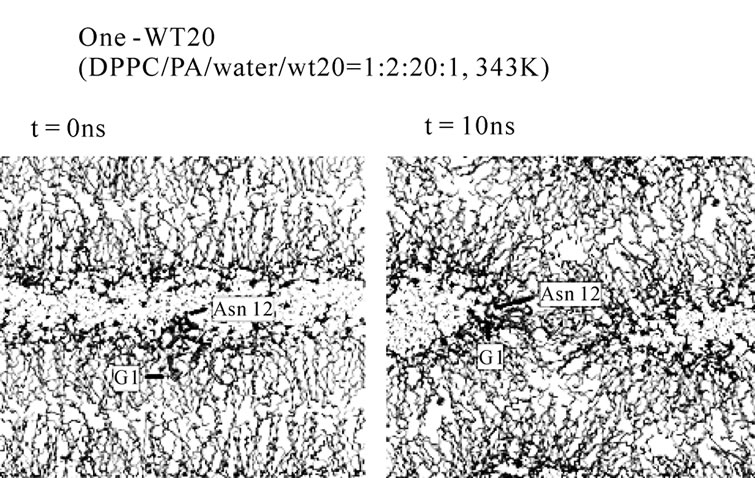
For all five simulations in set 1, a stalk was formed promptly at t = ~1-5 ns (Table 1, set 1). By contrast, the stalk propensity was weak (0.3) for the system without WT20 (set 2). When the hydration level was increased, the stalk propensity decreased (set 3 and set 4), consistent with the results reported [42]. To reduce the rate of stalk formation from that in the control (i.e., set 2) simulation, we reduced the temperature, instead of increasing the hydration level. At 343 K, the stalk propensity was 0.9 for the 1:2:20:1 system (set 6), whereas it was 0.2 without WT20 (set 5). Figure 5(a) shows snapshots from a representative simulation of set 6. In many stalkforming events, the polar residues belonging to the kinked region (i.e., Glu11, Asn12 and Glu15) associated
 (a)
(a)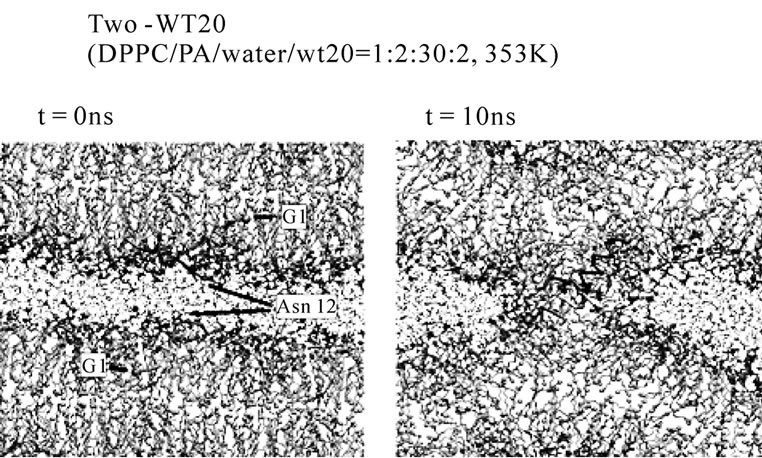 (b)
(b)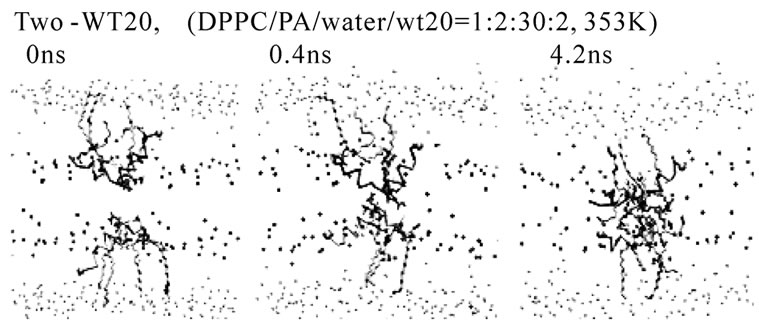 (c)
(c)
Figure 5. Stalk formation in MD simulations. Snapshots at indicated time points of representative simulations are shown. (a) DPPC/PA/water/WT20 1:2:20:1 system at 343 K (set 6). (b) DPPC/PA/water/WT20 1:2:30:2, 353 K. (c) Lipid molecule movements of a representative stalk-forming event of a 1:2:30:2 simulation. For (a, b), the thick orange trace (also the yellow trace in (b)) shows the peptide backbone. The yellow sphere shows the Cα atom of the G1 residue. Purple licorice indicates the sidechain of Asn12. Cyan lines show lipid acyl chains. The terminal C atoms of lipid acyl chains are represented as cyan balls. Red and blue spheres show lipid oxygen and nitrogen atoms. Black lines indicate water molecules. For (c), the representation scheme is similar to the one used in (a, b), but several lipid molecules are highlighted by thick licorice representation, whereas water and most of lipid acyl chains are hidden. Red licorice shows the sidechain of Glu11.
with the headgroups of lipid molecules of the apposed membrane. Our analyses showed that, once formed, a
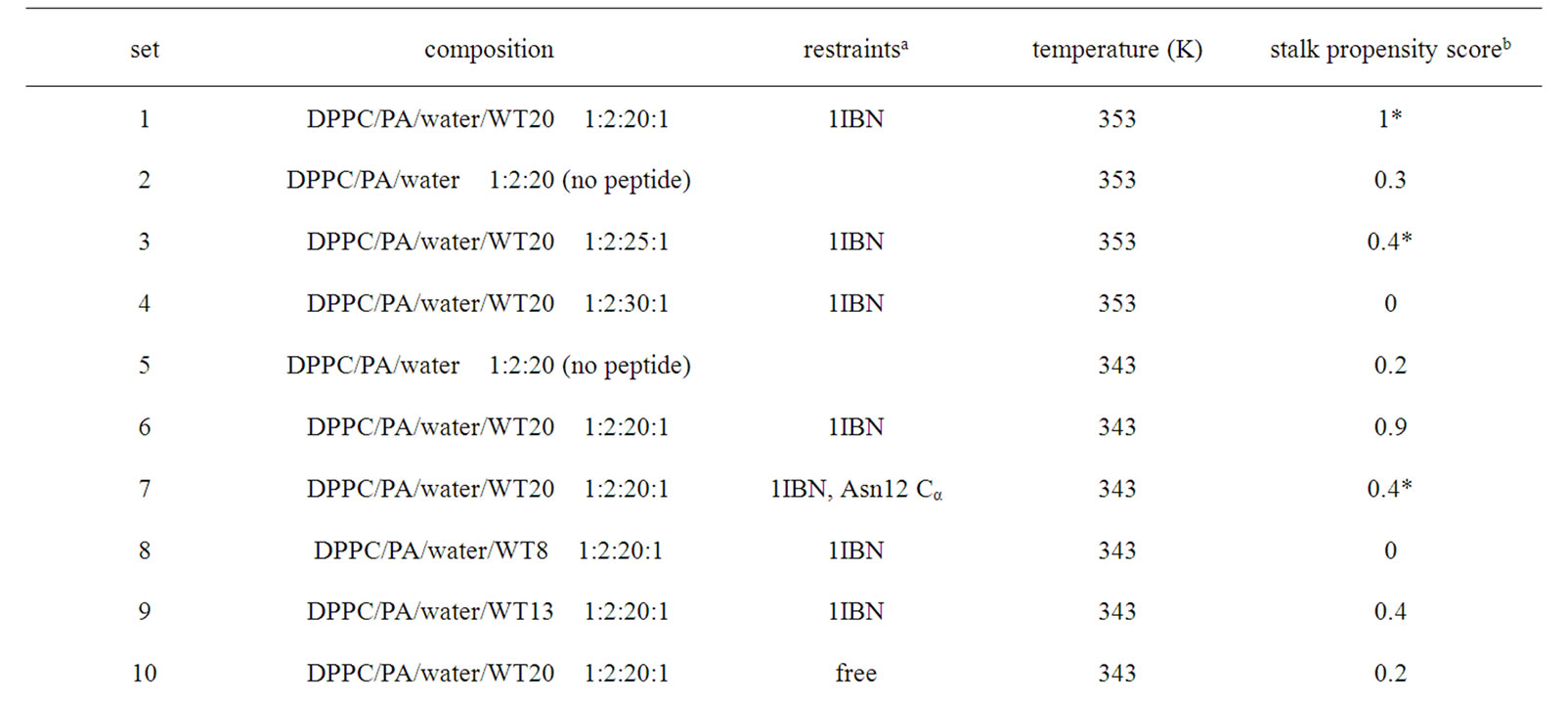

Table 1. Stalk propensity analysis of one peptide molecule.
stalk consisting of 3-4 lipid molecules tends to grow thicker consisting of > 8 molecules. Stalk formation was typically accompanied by a subtle upward movement of WT20. When the z-position of Asn12 Cα in WT20 was restrained relative to the z-position of the COM of lipid phosphorus atoms of the same (i.e., cis) monolayer, the stalk propensity was weak (set 7). Similar analyses were carried out for the MPER. Figures 1(m) and 1(n) show the equilibrium position of the MPER at 353 K. For both cases with and without the restraints, the helical structure was stable and the orientation was largely parallel to the membrane surface. The stalk propensity of MPER was small (set 13).
The system containing one peptide in each of the apposed monolayers was also examined (i.e., two-peptide analysis). Despite the relatively high hydration level was used (DPPC/PA/water/WT20 = 1:2:30:2, the same as set 4), the stalk propensity was > 0.8 regardless of the configuration. Typically, the stalk formation was preceded by self-association of WT20 (Figures 5(b) and 5(c)). When the z-position of the Asn12 Cα of both WT20 molecules was restrained, no stalk formation occurred. Compared with the one-WT20 simulations, stalk formation was much quicker in the two-WT20 simulations. The association between the two WT20 peptides was typically mediated by ~2-3 hydrogen bonds formed by sidechains of residues Glu11, Asn12 and Glu15 of both peptides (data not shown). When simulations were performed on WT8 (i.e., a segment of eight residues at the N-terminus of WT20), no stalk propensity was found (set 8 and data not shown for the two-peptide system), consistent with the experimental finding [66]. The stalk propensity of WT13 was weak (set 9. The propensity was 0.3 for the 1:2:30:2 system). A stalk-inducing effect was also observed with two WT20 molecules in the DOPE membrane at 353 K, although the effect appears weak (set 14, 15). In several simulations, prior to the stalk formation, WT20 moved underneath the DOPE molecules. As WT20 moving to a shallower position, DOPE molecules also moved into the water layer (details not shown). Importantly, for both oneand two-peptide analysis, most of the observed stalks occurred in the proximity of the peptide. Our inspection showed that the lead-off lipid molecule (i.e., the first lipid molecule that moved out of the bilayer and stayed in the stalk for > 100 ps) was frequently associated with Phe3, Phe9 and Glu15.
The following findings implicate the structure and orientation of the peptide for its ability to perturb the membrane-water interface. For most of the stalk propensity analysis, we introduced 20 kJ/(deg)2 restraints on the peptide dihedral angles based on the 1IBN structure. When the restraints were removed, structural variation was large (Figures 1(f) and 1(g) and data not shown) and WT20 showed weak stalk propensity (set 10. The stalk propensity was 0.3 for the 1:2:30:2 system). Further insights may be obtained from mutants G1V and W14A. Gly1, the residue at the N-terminus, is critical for functions of the HA protein [7,33,67]. In the equilibrium analysis, G1V adopted a linear or slightly U-shaped structure (e.g., Figures 1(h) and 1(i)). With the 20 kJ/(deg)2 dihedral restraints at its reported (linear) structure (PDB code: 1XOP), the stalk propensity of G1V was small (set 11. The stalk propensity was 0.3 for the 1:2:30:2 system). When the backbone dihedrals of WT20 were restrained to those of the NMR-determined G1V structure (i.e., linear), WT20 adopted a linear structure and the stalk-inducing activity became very weak, the propensity being 0.1 for the 1:2:30:2 system. Strikingly, when the dihedrals of G1V were restrained to those of the 1IBN (i.e., v-shape), the stalk propensity was high (0.9 for 1:2:30:2). When W14A was examined in the one-peptide system, the propensity was weak (set 12). When two W14A mole cules in anti-parallel and parallel configurations were examined in the 1:2:30:2 system (dihedral restraints set to the reported W14A structure, 2DCI), there was no significant stalk-inducing effect, the propensity being 0.1.
Overall, these results are consistent with the experimental results [33] which argue the importance of the V-shaped structure and/or the oblique orientation for the stalk-inducing effect of the peptides. Although it is difficult to draw conclusion based on our MD simulations, our simulations showed that WT20 in the inverted V-shape has more opportunities for interaction with lipids or the peptide in the apposed monolayer membrane than G1V and W14A. The shape appears to enable the exposure of the kinked region to the water layer. However, due to the unphysiological nature of our system, this issue requires many more analyses.
4.2. Technical Limitations of Peptide-Induced Membrane Fusion Assay in Silico
While our stalk propensity results were consistent with experiments, it should be borne in mind that the membranes rich in palmitic acid or DOPE are unphysiological. It should also be stressed that, while we started simulations from lamellar structures, the membrane we used have ‘per se’ a propensity to form non-bilayer structures at the temperature based on the analysis by Knecht et al. [42]. While such non-equilibrium simulations may provide some information on kinetical effect of the peptides, more analyses close to equilibrium conditions are necessary to draw more reliable conclusions. The use of the high temperature allowed us to obtain some statistics, but as a trade-off, restraints were introduced on the peptide backbone dihedral angles. Thus, most of the analyses are dependent on the “reference” structure, instead of the equilibrium structure for the force field and conditions used. It should also be pointed out that the limited size of the simulation system can cause artifacts. The effects of the artificial setting have to be carefully examined through more rigorous future experimental and theoretical studies. Future computational approaches may include coarse-grained (CG) simulations and Brownian dynamics simulations. Recent CG-based studies of membrane fusion involve Kasson et al. [68]. Analyses using these methods may elucidate more robust features about the behavior of the fusion peptides.
The amount of water was critical in this study, emphasizing the importance of the dehydration for the membrane fusion. Future directions may involve oligomerization of peptides (not limited to the N-terminal fusion peptides) because the oligomerization may help the dehydration. β-sheet aggregation of HAFP may recruit HA trimers into a fusion site [66]. HAFP may also interact with the transmembrane (TM) domain or the other peptide segments contained in the external region [33]. The presence of other segments of HA protein may increase the chance for peptide oligomerization further [69]. Recent MD simulation analyses of oligomer configurations have shown that the HAFP oligomer can assume a variety of configurations (of peptide/peptide and peptide/membrane) [49]. The oligomerized peptides may have several roles ranging from stalk formation, to hemifusion, and to full fusion [e.g., 70]. Future computational efforts may well be focused on oligomerization of transmembrane peptides as well as fusion peptides (e.g., Kim et al. [71]).
To summarize, the dynamics of the HAFP within lipid membranes was studied. When the HAFP was located at the DPPC/PA membrane-water interface, it assumed an oblique orientation as previously reported. The HAFP exhibited a significant effect on the membrane curvature. The effect was dependent on the lipid composition of the membranes: i.e., a positive curvature for the DPPC/PA membrane and the DOPE membrane whereas a negative curvature for the DPPC membrane. In the phase reconstitution analysis starting from the random DPPC/PA system, the HAFP exhibited a weak stabilization of an inverted hexagonal phase, which is suggestive of enhancement of the negative curvature. The distinct effects (i.e., positive vs. negative curvature) observed in the two DPPC/PA systems is likely to be related to the result that in the latter system the HAFP assumed a transmembrane orientation more frequently than an interfacial orientation. It can be envisaged that HAFP induces different types of curvature, depending on lipid composition and on the location and orientation within membrane. HAFP also increased the rate of stalk-formation in our nonequilibrium MD simulations in which the spontaneous stalk formation is slow without peptides. However, there are clearly many difficulties if one tries to interpret rigorously the non-equilibrium simulation results. Many more efforts to bring the system closer to a physiological system and to deal with longer time scales are necessary.
5. ACKNOWLEDGEMENTS
The authors thank the anonymous reviewers for their helpful suggestions. This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
REFERENCES
- Wilson, I.A., Skehel, J.J. and Wiley, D.C. (1981) Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature, 289(5796), 366-373.
- Carr, C.M. and Kim, P.S. (1993) A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell, 73(4), 823-832.
- Skehel, J.J. and Wiley, D.C. (2000) Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annual Review Biochemistry, 69, 531-569.
- Tamm, L.K., Crane, J. and Kiessling, V. (2003) Membrane fusion: A structural perspective on the interplay of lipids and proteins. Current Opinion in Structural Biology, 13, 453-466.
- Earp, L.J., Delos, S.E., Park, H.E. and White, J.M. (2005) The many mechanisms of viral membrane fusion proteins. Current Topics in Microbiology and Immunology, 285, 25-66.
- Chan, D.C., Fass, D., Berger, J.M. and Kim, P.S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell, 89(2), 263-273.
- Gething, M.J., Doms, R.W., York, D. and White, J. (1986) Studies on the mechanism of membrane fusion: Sitespecific mutagenesis of the hemagglutinin of influenza virus. The Journal of Cell Biology, 102(1), 11-23.
- Cross, K.J., Wharton, S.A., Skehel, J.J., Wiley, D.C. and Steinhauer, D.A. (2001) Studies on influenza haemagglutinin fusion peptide mutants generated by reverse genetics. EMBO Journal, 20, 4432-4442.
- Chen, J., Skehel, J.J. and Wiley, D.C. (1999) Nand C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proceedings of the National Academy of Sciences USA, 96, 8967-8972.
- Gallaher, W.R. (1987) Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell, 50(3), 327-328.
- Durell, S.R., Martin, I., Ruysschaert, J.M., Shai, Y. and Blumenthal, R. (1997) What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion. Molecular Membrane Biology, 14(3), 97- 112.
- Bosch, M.L., Earl, P.L., Fargnoli, K., Picciafuoco, S., Giombini, F., Wong-Staal, F. and Franchini, G. (1989) Identification of the fusion peptide of primate immunodeficiency viruses. Science, 244(4905), 694-697.
- Dürrer, P., Galli, C., Hoenke, S., Corti, C., Glück, R., Vorherr, T. and Brunner, J. (1996) H+-induced membrane insertion of influenza virus hemagglutinin involves the HA2 amino-terminal fusion peptide but not the coiled coil region. The Journal of Biological Chemistry, 271(23), 13417-13421.
- Harter, C., James, P., Bächi, T., Semenza, G. and Brunner, J. (1989) Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the “fusion peptide”. The Journal of Biological Chemistry, 264(11), 6459-6464.
- Stegmann, T., Delfino, J.M., Richards, F.M. and Helenius, A. (1991) The HA2 subunit of influenza hemagglutinin inserts into the target membrane prior to fusion. The Journal of Biological Chemistry, 266, 18404-18410.
- Lear, J.D. and DeGrado, W.F. (1987) Membrane binding and conformational properties of peptides representing the NH2 terminus of influenza HA-2. The Journal of Biological Chemistry, 262(14), 6500-6505.
- Murata, M., Takahashi, S., Kagiwada, S., Suzuki, A. and Ohnishi, S. (1992) pH-dependent membrane fusion and vesiculation of phospholipid large unilamellar vesicles induced by amphiphilic anionic and cationic peptides. Biochemistry, 31(7), 1986-1992.
- Wharton, S.A., Martin, S.R., Ruigrok, R.W., Skehel, J.J. and Wiley, D.C. (1988) Membrane fusion by peptide analogues of influenza virus haemagglutinin. Journal of General Virology, 69, 1847-1857.
- Burger, K.N., Wharton, S.A., Demel, R.A. and Verkleij, A.J. (1991) The interaction of synthetic analogs of the N-terminal fusion sequence of influenza virus with a lipid monolayer. Comparison of fusion-active and fusion-defective analogs. Biochimica et Biophysica Acta, 1065(2), 121-129.
- Soltesz, S.A. and Hammer, D.A. (1997) Lysis of large unilamellar vesicles induced by analogs of the fusion peptide of influenza virus hemagglutinin. Journal of Colloid and Interface Science, 186(2), 399-409.
- Epand, R.F., Macosko, J.C., Russel, C.J., Shin, Y.-K. and Epand, R.M. (1999) The ectodomain of HA2 of influenza virus promotes rapid pH dependent membrane fusion. Journal of Molecular Biology, 286(2), 489-503.
- Tamm, L.K. and Han, X. (2000) Viral fusion peptides: A tool set to disrupt and connect biological membranes. Bioscience Reports, 20(6), 501-518.
- Zhelev, D.V., Stoicheva, N., Scherrer, P. and Needham, D. (2001) Interaction of synthetic HA2 influenza fusion peptide analog with model membranes. Biophysical Journal, 81(1), 285-304.
- Nieva, J.L. and Agirre, A. (2003) Are fusion peptides a good model to study viral cell fusion? Biochimica et Biophysica Acta, 1614(1), 104-115.
- Rafalski, M., Lear, J.D. and DeGrado, W.F. (1990) Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry, 29(34), 7917-7922.
- Nieva, J.L., Nir, S., Muga, A., Goni, F.M. and Wilschut, J. (1994) Interaction of the HIV-1 fusion peptide with phospholipid vesicles: Different structural requirements for fusion and leakage. Biochemistry, 33(11), 3201-3209.
- Brasseur, R., Lorge, P., Goormaghtigh, E., Ruysschaert, J.M., Espion, D. and Burny, A. (1998) The mode of insertion of the paramyxovirus F1 N-terminus into lipid matrix, an initial step in host cell/virus fusion. Virus Genes, 1(4), 325-332.
- Martin, I., Dubois, M.C., Defrise-Quertain, F., Saermark, T., Burny, A., Brasseur, R. and Ruysschaert, J.M. (1994) Correlation between fusogenicity of synthetic modified peptides corresponding to the NH2-terminal extremity of simian immunodeficiency virus gp32 and their mode of insertion into the lipid bilayer: An infrared spectroscopy study. The Journal of Virology, 68, 1139-1148.
- Martin, I., Schaal, H., Scheid, A. and Ruysschaert, J.M. (1996) Lipid membrane fusion induced by the human immunodeficiency virus type 1 gp41 N-terminal extremity is determined by its orientation in the lipid bilayer. The Journal of Virology, 70, 298-304.
- Lüneberg, J., Martin, I., Nüssler, F., Ruysschaert, J.M. and Herrmann, A. (1995) Structure and topology of the influenza virus fusion peptide in lipid bilayers. The Journal of Biological Chemistry, 270(46), 27606-27614.
- Han, X., Bushweller, J.H., Cafiso, D.S. and Tamm, L.K. (2001) Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nature Structural Biology, 8(8), 715-720.
- Han, X. and Tamm, L.K. (2000) A host-guest system to study structure-function relationships of membrane fusion peptides. Proceedings of the National Academy of Sciences USA, 97(24), 13097-13102.
- Li, Y., Han, X., Lai, A.L., Bushweller, J.H., Cafiso, D.S. and Tamm, L.K. (2005) Membrane structures of the hemifusion-inducing fusion peptide mutant G1S and the fusion-blocking mutant G1V of influenza virus hemagglutinin suggest a mechanism for pore opening in membrane fusion. The Journal of Virology, 79(18), 12065- 12076.
- Lai, A.L., Park, H., White, J.M. and Tamm, L.K. (2006) Fusion peptide of influenza hemagglutinin requires a fixed angle boomerang structure for activity. The Journal of Biological Chemistry, 281(9), 5760-5770.
- Sáez-Cirión, A., Nir, S., Lorizate, M., Agirre, A., Cruz, A., Pérez-Gil, J. and Nieva, J.L. (2002) Sphingomyelin and cholesterol promote HIV-1 gp41 pretransmembrane sequence surface aggregation and membrane restructuring. The Journal of Biological Chemistry, 277(24), 21776- 21785.
- Lorizate, M., Huarte, N., Sáez-Cirión, A. and Nieva, J.L. (2008) Interfacial pre-transmembrane domains in viral proteins promoting membrane fusion and fission. Biochimica et Biophysica Acta, 1778(7-8), 1624-1639.
- Charloteaux, B., Lorin, A., Brasseur, R. and Lins, L. (2009) The “Tilted Peptide Theory” links membrane insertion properties and fusogenicity of viral fusion peptides. Protein and Peptide Letters, 2009, 16(7), 718-725.
- Efremov, R.G., Nolde, D.E., Volynsky, P.E., Chernyavsky, A.A., Dubovskii, P.V. and Arseniev, A.S. (1999) Factors important for fusogenic activity of peptides: molecular modeling study of analogs of fusion peptide of influenza virus hemagglutinin. FEBS Letters, 462(1-2), 205-210.
- Gray, C., Tatulian, S.A., Wharton, S.A. and Tamm, L.K. (1996) Effect of the N-terminal glycine on the secondary structure, orientation, and interaction of the influenza hemagglutinin fusion peptide with lipid bilayers. Biophysical Journal, 70(5), 2275-2286.
- Marrink, S.-J., de Vries, A.H. and Tieleman, D.P. (2009) Lipids on the move: Simulation of membrane pores, domains, stalks and curves. Biochimica et Biophysica Acta, 1788(1), 149-168.
- Marrink, S.-J. and Tieleman, D.P. (2001) Molecular dynamics simulation of a lipid diamond cubic phase. Journal of the American Chemical Society, 123(49), 12383- 12391.
- Knecht, V., Mark, A.E. and Marrink, S.-J. (2006) Phase behaviour of a phospholipid/fatty acid/water mixture studied in atomic detail. Journal of the American Chemical Society, 128(6), 2030-2034.
- Marrink, S.-J. and Mark, A.E. (2004) Molecular view of hexagonal phase formation in phospholipid membranes. Biophysical Journal, 87(6), 3894-3900.
- Nielsen, S.O., Lopez, C.F., Ivanov, I., Moore, P.B., Shelley, J.C. and Klein, M.L. (2004) Transmembrane peptide-induced lipid sorting and mechanism of Lalpha-to-inverted phase transition using coarse-grain molecular dynamics. Biophysical Journal, 87(4), 2107-2115.
- Kamath, S. and Wong, T.C. (2002) Membrane structure of the Human Immunodeficiency Virus gp41 fusion domain by molecular dynamics simulation. Biophysical Journal, 83(1), 135-143.
- Vaccaro, L., Cross, K.J., Kleinjung, J., Straus, S.K., Thomas, D.J., Wharton, S.A., Skehel, J.J. and Fraternali, F. (2005) Plasticity of influenza haemagglutinin fusion peptides and their interaction with lipid bilayers. Biophysical Journal, 88(1), 25-36.
- Huang, Q., Chen, C.L. and Herrmann, A. (2004) Bilayer conformation of fusion peptide of influenza virus hemagglutinin: A molecular dynamics simulation study. Biophysical Journal, 87(1), 14-22.
- Lagüe, P., Roux, B. and Pastor, R.W. (2005) Molecular dynamics simulations of the influenza hemagglutinin fusion peptide in micelles and bilayers: Conformational analysis of peptide and lipids. Journal of Molecular Biology, 354(5), 1129-1141.
- Sammalkorpi, M. and Lazaridis, T. (2007) Configuration of influenza hemagglutinin fusion peptide monomers and oligomers in membranes. Biochimica et Biophysica Acta, 1768(1), 30-38.
- de Vries, A.H., Mark, A.E. and Marrink, S.J. (2004) The binary mixing behavior of phospholipids in a bilayer: A molecular dynamics study. The Journal of Physical Chemistry B, 108(7), 2454-2463.
- Seddon, J.M., Templer, R.H., Warrender, N.A., Huang, Z., Cevc, G. and Marsh, D. (1997) Phosphatidylcholine–fatty acid membranes: Effects of headgroup hydration on the phase behaviour and structural parameters of the gel and inverse hexagonal (HII) phases. Biochimica et Biophysica Acta, 1327, 131-147.
- Lindahl, E., Hess, B. and van der Spoel, D. (2001) GROMACS 3.0: A package for molecular simulation and trajectory analysis. Journal of Molecular Modeling, 7(8), 306-317.
- Berger, O., Edholm, O. and Jähnig, F. (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophysical Journal, 72(5), 2002-2013.
- Sun, Z.Y., Oh, K.J., Kim, M., Yu, J., Brusic, V., Song, L., Qiao, Z., Wang, J.H., Wagner, G. and Reinherz, E.L. (2008) HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity, 28(1), 52-63.
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F. and Hermans, J. (1981) Intermolecular forces, interaction models for water in relation to protein hydration. D. Reidel Publishing, Dordrecht, The Netherlands.
- Hess, B., Bekker, H., Berendsen, H.J.C. and Fraaije, J.G.E.M. (1997) LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463-1472.
- Miyamoto, S. and Kollman, P.A. (1992) SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry, 13(8), 952-962.
- Darden, T., York, D. and Pedersen, L. (1993) Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. Journal of Chemical Physics, 98(12), 10089- 10092.
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., DiNola, A. and Haak, J.R. (1984) Molecular dynamics with coupling to an external bath. Journal of Chemical Physics, 81(8), 3684-3690.
- Zhou, Z., Macosko, J.C., Hughes, D.W., Sayer, B.G., Hawes, J. and Epand, R.M. (2000) 15N NMR study of the ionization properties of the influenza virus fusion peptide in zwitterionic phospholipid dispersions. Biophysics Journal, 78(5), 2418-2425.
- Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: Pattern recongnition of hydrogen-bond and geometrical features. Biopolymers, 2, 2577-2637.
- Humphery, W., Dalke, A. and Schulten, K. (1996) VMD--visual molecular dynamics. Journal of Molecular Graphics, 14, 33-38.
- Epand, R.M. and Epand, R.F. (1994) Relationship between the infectivity of influenza virus and the ability of its fusion peptide to perturb bilayers. Biochemical and Biophysical Research Communications, 202(3), 1420-1425.
- Colotto, A. and Epand, R.M. (1997) Structural study of the relationship between the rate of membrane fusion and the ability of the fusion peptide of influenza virus to perturb bilayers. Biochemistry, 36(25), 7644-7651.
- Siegel, D.P. and Epand, R.M. (2000) Effect of influenza hemagglutinin fusion peptide on lamellar/inverted phase transitions in dipalmitoleoylphosphatidylethanolamine: implications for membrane fusion mechanisms. Biochimica et Biophysica Acta, 1468(1-2), 87-98.
- Han, X. and Tamm, L.K. (2000) pH-dependent selfassociation of influenza hemagglutinin fusion peptides in lipid bilayers. Journal of Molecular Biology, 304(5), 953-965.
- Qiao, H., Armstrong, R.T., Melikyan, G.B., Cohen, F.S. and White, J.M. (1999) A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Molecular Biology Cell, 10(8), 2759-2769.
- Kasson, P.M., Kelley, N.W., Singhal, N., Vrljic, M., Brunger, A.T. and Pande, V.S. (2006) Ensemble molecular dynamics yields submillisecond kinetics and intermediates of membrane fusion. Proceedings of the National Academy Sciences USA, 103(32), 11916-11921.
- Peisajovich, S.G. and Shai, Y. (2003) Viral fusion proteins: multiple regions contribute to membrane fusion. Biochimica et Biophysica Acta, 1614(1), 122-129.
- Lau, W.L., Ege, D.S., Lear, J.D., Hammer, D.A. and DeGrado, W.F. (2004) Oligomerization of fusogenic peptides promotes membrane fusion by enhancing membrane destabilization. Biophysics Journal, 86(1), 272- 284.
- Kim, J.H., Hartley, T.L., Curran, A.R. and Engelman, D.M. (2009) Molecular dynamics studies of the transmembrane domain of gp41 from HIV-1. Biochimica et Biophysica Acta, 1788(9), 1804-1812.