Green and Sustainable Chemistry
Vol.1 No.4(2011), Article ID:8501,4 pages DOI:10.4236/gsc.2011.14021
Halogen Exchange in Near-Critical Water
1Department of Chemistry, University of Louisiana at Monroe, Monroe, USA
2Department of Chemistry, Grambling State University, Grambling, USA
E-mail: *junk@ulm.edu
Received June 20, 2011; revised August 12, 2011; accepted August 25, 2011
Keywords: Hydrothermal, Halogen Exchange
Abstract
Bromoaromatics are ubiquitous in chemistry, and their manufacture is often wasteful. Halogen exchange under hydrothermal conditions constitutes a viable alternative for their synthesis in some cases. The preparation of 1,2-dibromobenzene and 1-bromo-2-chlorobenzene from 1,2-dichlorobenzene, by treatment with hydrobromic acid in hydrothermal media at temperatures ranging from 240˚C to 320˚C was investigated as a viable alternative to de novo synthesis. The effects of temperature, exchange duration and the presence of Fe3+ salts on product yields are discussed. Yields for both targeted haloarenes of up to 37% and 48%, respectively, were achieved, with very limited formation of 1,3- and 1,4-dihalobenzene isomers. A mechanism for halogen exchange was proposed.
1. Introduction
Superheated and supercritical aqueous reaction media are capable of supporting novel reactions, which are difficult to implement in conventional solvent systems. While attempting to implement isotope exchange in near-critical deuterated media [1], the authors observed concurrent halogen exchange for halobenzenes. Thus, treatment of chlorobenzene with aqueous hydrobromic acid at nearcritical conditions resulted in Cl-Br exchange. A further study of such exchange reactions seemed warranted, aimed at minimizing the need for the de novo manufacture of bromobenzenes by utilizing environmentally hazardous chlorobenzenes obtained as industrial waste products.
The chosen test substrate, 1,2-dichlorobenzene (DCB), is a byproduct in the manufacture of chlorobenzene and finds rather limited application [2]. In contrast, the demand for brominated aromatics is increasing in light of the emergence of palladium catalyzed cross-coupling reactions [3]. The target compounds, 1,2-dibromobenzene (DBB) and 1-bromo-2-chlorobenzene (BCB), commonly are prepared by nitration, reduction to the corresponding anilines, and Sandmeyer displacement, an expensive and wasteful approach [4]. Consequently, DBB retails at approximately fifty times the cost of DCB [5].
In contrast to halogen exchange in non-aromatic positions, few examples exist for the efficient synthesis of haloaromatics by transhalogenation. The conversion of 1,3,5-trichlorobenzene to chlorofluorobenzenes in polar aprotic solvents has been demonstrated [6], but the reported yields are modest. Displacement of iodine by chlorine is observed under relatively mild conditions [7], apparently driven by the relative stabilities of the respective carbon-halogen bonds. However, such displacements are of limited practical interest.
In this study, the authors report the yields of DBB and BCB as a function of time and temperature, as well as the effects of iron salts, a common industrial contaminant. Mechanistic insights gained from product distributions were complemented by ab-initio DFT calculations, in an effort to gain mechanistic insight into this reaction.
2. Experimental
Reagent grade dihalobenzenes, 48% aqueous hydrobromic acid, sodium bromide, ortho-xylene and 1,2,4,5- tetramethylbenzene were supplied by Sigma-Aldrich Chemical Company. Methyl tert-butylether (MTBE) was furnished by Acros Organics. All reagents were used as received.
2.1. Exchange Conditions
Experiments were carried out with 0.20 g of DCB unless stated otherwise. Aqueous 48% w/w hydrobromic acid (2.50 g) served as source of bromide, corresponding to a molar Br/Cl ratio of approx. 5.5 during exchange. Reactants were flame sealed into Pyrex tubes of 8 mm internal diameter and approx. 7 mL internal volume. Partial pressure compensation was achieved by placing the tubes in 60 mL Hastelloy C-22 autoclaves that were half filled with water to approximate the internal pressure of the tubes and prevent rupture. Autoclaves were then placed in a preheated muffle furnace equipped with electronic temperature control and timer, and heated to within ±2˚C of the nominal temperature. After cooling, the sealed tubes were broken open and ortho-xylene and 1,2,4,5- tetramethylbenzene added as standards. Products were extracted with reagent grade methyl-tert-butyl ether (MTBE). All data presented here reflect duplicate runs unless stated otherwise.
2.2. Sample Analyses
Analyte concentrations were determined by gas chromatography using a Varian CP 3800 gas chromatograph equipped with flame ionization detector, split-splitless injector, and a 25 m × 0.53 mm × 1.0 µm OV-17 column. The initial hold was 60˚C for 3 min, followed by a 10˚C /min ramp and a final hold at 250˚C for 10 min. The internal standard selected for bromobenzene, chlorobenzene and dichlorobenzene isomers was ortho-xylene, that for all possible bromochlorobenzene and dibromobenzene isomers was 1,2,4,5-tetramethylbenzene. Concentrations were determined based on five point, forced zero-intercept calibrations that included all possible dichloro-, bromochloroand dibromobenzene isomers. All calibrations and sample dilutions were carried out using MTBE as solvent. Concentrations are expressed as mol% relative to DCB initially present (set to 100%). Within the reported temperature regime, no evidence was found for significant mineralization or the formation of semivotatiles other than those included in this study. Consequently, the mol% sum of all aromatics present during and after the reported exchange experiments was found to be essentially invariant.
2.3. Exchange Modeling
Molecular modeling results of competing mechanisms leading to nucleophilic aromatic substitution were reported previously [8], but clearly do not fully apply to the highly acidic conditions employed here. Modeling of plausible reaction intermediates was carried out using Gaussian 03 Rev. E.01 [9]. The initial structures were built using GaussView 5.09. Density functional theory (DFT) at the B3LYP/6-31G(d,p) level was used throughout [10,11]. Three mechanisms were considered, as shown in Figures 1-3, and stabilities for the candidate intermediates B and C determined by ab initio calculation as stated above. These were subsequently compared to benzyne intermediates such as A (in Figure 1), for which stabilities in the order of ΔG ≈ 38 kcal/mol with respect to the organic precursor molecules are generally accepted [12]. It should be cautioned that these values neglect solvation effects; however, all three intermediates being considered are non-polar, making this comparison reasonable.
3. Results
3.1. Exchange Temperature
As indicated in Figure 4, halogen exchange remains relatively slow below 280˚C, even after prolonged exchange times. At and above 320˚C, a sharply increased
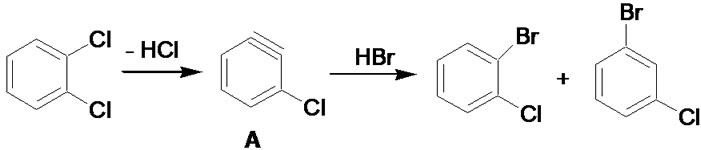
Figure 1. Halogen exchange via benzyne intermediate A.
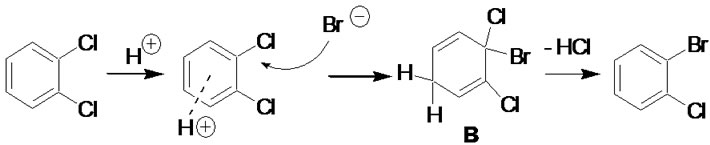
Figure 2. Halogen exchange via 1,4-cyclohexadiene intermediate B.
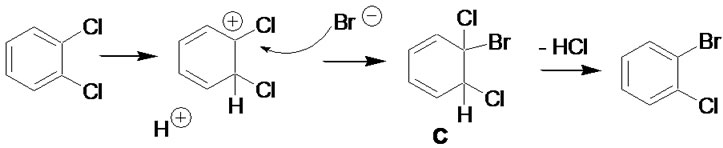
Figure 3. Halogen exchange via 1,2-cyclohexadiene intermediate C.
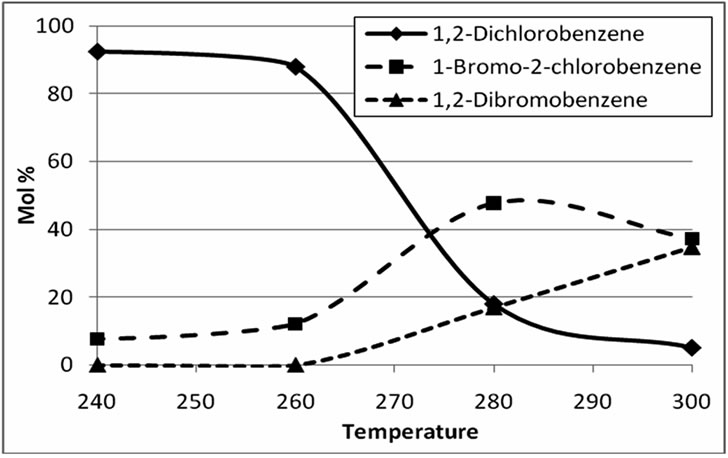
Figure 4. Concentrations of 1,2-dichlorobenzene, 1-bromo- 2-chlorobenzene, and 1,2-dibromobenzene as a function of exchange temperatures between 240˚C and 300˚C. Exchange time, 4 h.
incidence of tube failures (rupture of Pyrex tube inside autoclave) along with significant dehalogenation to monohalobenzenes was observed. In practical terms, it is obviously desirable to keep the reaction temperature as low as possible to minimize the difficulties associated with working under hot and highly acidic conditions, and to minimize exchange times. Thus, exchange temperatures of 280˚C - 320˚C were selected for subsequent work.
Gas chromatographic analyses indicated that the formation of semi-volatile products other than monoand dihalobenzenes (e.g. unidentified chlorinated phenols, aryl ethers) was limited to less than approx. 5 mol% of the total benzene derivatives present within the chosen temperature range. This was complemented by the analysis of one representative sample using micellar electrokinetic capillary chromatography (MEKC) [13], which failed to find significant quantities of polar, nonvolatile products. Consequently, product distributions in this study are represented as relative mol% of the cumulative molar quantity of halobenzenes present, which remained essentially constant under the conditions chosen. Table 1 exemplifies that, in addition to unreacted DCB, BCB and DBB were the dominant constituents of the reaction mixtures.
3.2. Exchange Times
The composition of reaction mixtures as a function of time, in terms of the dominant halobenzenes, is shown in Figure 5. It appears that after exchange times of approx. 3 h, the system approaches a state of equilibrium. The most notable dehalogenation product observed was bromobenzene, but its formation remained fairly limited. The yield of BCB peaked after approx. 1 h at 48%, that of DBB after 3 h at 37%.
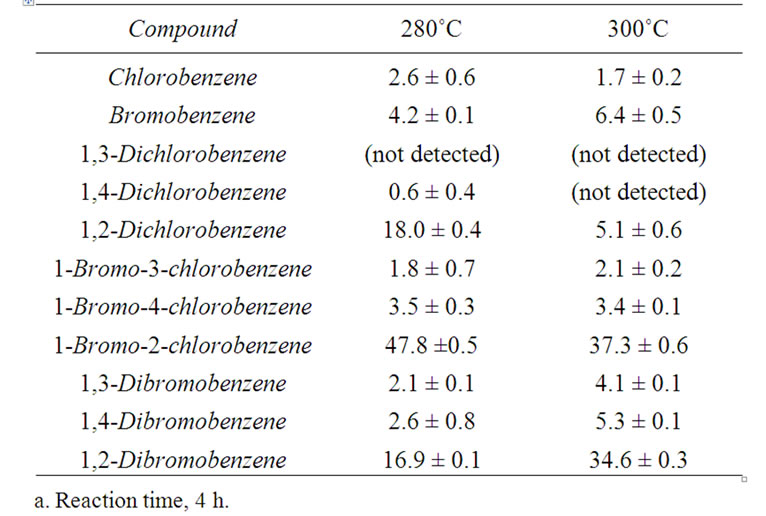
Table 1. Relative mol% of halobenzenes at 280˚C and 300˚C, respectivelya.
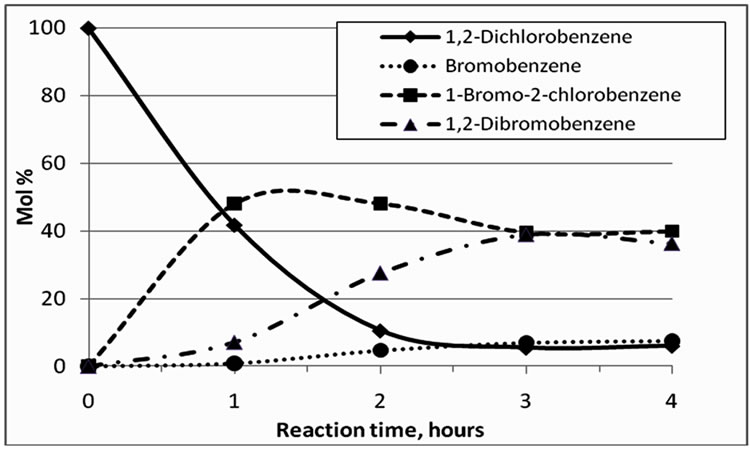
Figure 5. Mol% composition of dominant halobenzenes, 0-4 h at 280˚C.
3.3. Exchange in the Presence of Salts
When aqueous hydrobromic acid was replaced by an equimolar concentration of aqueous sodium bromide, no formation of BCB or DBB was observed and >98% of the starting material was recovered. Addition of FeBr3 at moderate to high concentrations during exchange reduced the molar percentage of DBB in the resulting product mixture while increasing those of both chlorobenzene and unreacted DCB (Table 2). Its effect on other exchange products, such as BCB, was less pronounced.
4. Conclusions
Superheated aqueous media are well suited for halogen exchange, as exemplified by the conversion of DCB to chemically more reactive BCB and DBB. Such exchange protocols promise to reduce the need for the de novo synthesis of bromobenzene derivatives, an often wasteful process, by utilizing byproducts of chlorobenzene manufacture and water as the primary reaction medium. The presence of iron bromide was found to significantly lower the yield of DBB.
The limited formation of isomerized products is noteworthy. Hydrolysis of halobenzenes to phenols in superheated aqueous phase is well known to proceed via benzyne intermediates [14]. Halogen exchange based on a similar mechanism would lead to extensive isomerization as
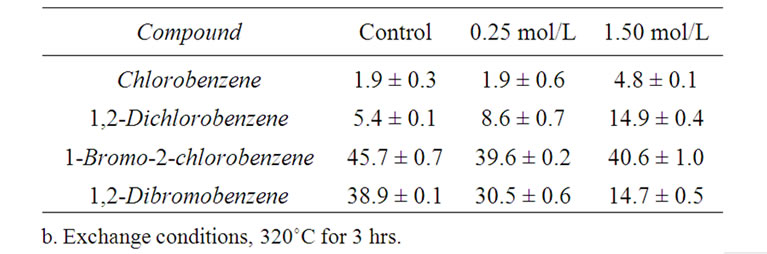
Table 2. Effect of FeBr3 on 1-bromo-2-chlorobenzene and 1, 2-dibromobenzene yieldsb.
a result of HBr addition in random orientation to benzyne intermediates (Figure 1), as well as the competitive formation of phenolic products by addition of water. The resulting compound mixtures would be difficult to separate, limiting the utility of this method. Observed product distributions discount this and are in agreement with computational results, favoring intermediate B in Figure 2 (33.3 kcal/mol) over intermediate C in Figure 3 (34.9 kcal/mol) or benzyne intermediates. The observed need for highly acidic media further supports an additionelimination sequence initiated by arene protonation. It is noteworthy that, in contrast to virtually all other halogen exchange reactions published to date, stronger carbonchlorine bonds are replaced by weaker carbon-bromine bonds, pointing to a halogen concentration gradient rather than relative bond stabilities as the driving force for exchange.
5. Acknowledgments
We are indebted to the late Dr. W. James Catallo for his assistance with the MEKC analysis and advice during the early stages of this work. T. Junk gratefully acknowledges funding from the Petroleum Research Fund (Grant #34409-B4) for the purchase of high pressure autoclaves.
6. References
[1] T. Junk and W. J. Catallo, “Preparative Supercritical Deuterium Exchange in Arenes and Heteroarenes,” Tetrahedron Letters, Vol. 37, No. 20, 1996, pp. 3445-3448. doi:10.1016/0040-4039(96)00615-6
[2] M. Rossberg, W. Lendle, G. Pfleiderer, A. Tögel, E.-L. Dreher, E. Langer, H. Jaerts, P. Kleinschmidt, H. Strack, R. Cook, U. Beck, K.-A. Lipper, T. R. Torkelson, E. Löser and K. K. Beutel, “Chlorinated Hydrocarbons,” Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2006. doi:10.1002/14356007.a06_233. pub2
[3] J. F. Hartwig, “Organotransition Metal Chemistry. From Bonding to Catalysis,” University Science Books, New York, 2010.
[4] R. Huisgen and J. Sauer, “Nucleophile Aromatische Substitution über Arine,” Angewandte Chemie, Vol. 72, No. 2, 1960, pp. 91-108. doi:10.1002/ange.19600720302
[5] “Chemical Sources USA,” Annual Edition, Chemical Sources International, Inc., Clemson, 1824.
[6] R. G. Pews and J. A. Gall, “Aromatic Fluorine Chemistry. Part 4. Preparation of 2,6-Difluoroaniline,” Journal of Fluorine Chemistry, Vol. 52, No. 3, 1991, pp. 307-316. doi:10.1016/S0022-1139(00)80345-0
[7] J. H. Clark, C. W. Jones, C. V. Duke and J. M. Miller, “Halogen Exchange Reactions of Aryl Halides Using Supported Copper (I),” Journal of Chemical Research Synopses, Vol. 8, 1989, pp. 1-238.
[8] M. N. Glukhovtsev, R. D. Bach and S. Laitner, “SingleStep and Multistep Mechanisms of Aromatic Nucleophilic Substitution of Halobenzenes and Halonitrobenzenes with Halide Anions: Ab Initio Computational Study,” Journal of Organic Chemistry, Vol. 62, No. 12, 1997, pp.4036-4046. doi:10.1021/jo962096e
[9] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M.Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople. Gaussian, Inc., Wallingford CT, 2004.
[10] A. D. Becke, “Density-Functional Thermochemistry. III. The Role of Exact Exchange,” Journal of Chemical Physics, Vol. 98, No. 7, 1993, pp. 5648-5652. doi:10.1063/1.464913
[11] C. Lee, W. Yang and R. G. Parr, “LYP Gradient-Corrected Functional,” Physical Review B, Vol. 37, No. 2, 1988, pp. 785-789. doi:10.1103/PhysRevB.37.785
[12] R. Lindh, A. Bernhardsson and M. Schütz, “Benzyne Thermochemistry: A Benchmark Ab Initio Study,” Journal of Physical Chemistry A, Vol. 103, No. 48, 1999, pp. 9913-9920. doi:10.1021/jp991919b
[13] S. Terabe, K. Otsuka, A. Tsuchiya and T. Ando, “Electrokinetic Separations with Micellar Solutions and OpenTubular Capillaries,” Analytical Chemistry, Vol. 56, No. 1, 1984, pp. 111-113. doi:10.1021/ac00265a031
[14] A. T. Bottini and J. D. Roberts, “Mechanism for Liquid Phase Hydrolyses of Chlorobenzenes and Halotoluenes,” Journal of the American Chemical Society, Vol. 79, 1957, pp. 1458-1462. doi:10.1021/ja01563a050