Journal of Diabetes Mellitus
Vol. 3 No. 2 (2013) , Article ID: 31331 , 10 pages DOI:10.4236/jdm.2013.32009
Role of sphingosine kinases and sphingosine 1-phosphate in mediating adipogenesis
![]()
Department of Pediatrics, School of Medicine and Biomedical Sciences, University at Buffalo, New York, USA; ldm@buffalo.edu
Copyright © 2013 Lucy D. Mastrandrea. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 13 March 2013; revised 15 April 2013; accepted 23 April 2013
Keywords: Adipocyte; Adipogenesis; Obesity; Sphingosine Kinase; 3T3-L1 Cells; Sphingosine 1-Phosphate; Sphingosine 1-Phosphate Receptor
ABSTRACT
Background: Recent development of obesity involves promotion of preadipocyte differrentiation. This study investigated the role that sphingosine kinases (SPHK) and ceramide-derived sphingosine 1-phosphate (S1P) play in adipocyte terminal differentiation. Materials and Methods: The mouse 3T3-L1 cell line was used as a model for adipogenesis. Cells were harvested at specific time points after initation of differentiation, and SPHK activity was measured. 3T3-L1 cells were treated with S1P and expression of early adipogenesis transcription markers was measured by real time PCR. The expression of S1P-receptors (S1PRs) during differentiation was measured. Results: SPHK activity is induced when 3T3-L1 cells are treated with insulin, dexamethasone, and isobutylmethylxanthine to induce differentiation. SPHK1 is active in preadipocytes and early in the differentiation process. Both SPHK1 and SPHK2 isozymes contribute to activity in differentiated adipocytes. Inhibition of SPHK1 attenuates adipocyte differentiation; however, extracellular S1P does not rescue the effect of SPHK1 inhibition on adipogenesis. Although treatment of preadipocytes with S1P induced message expression of the early adipogenesis transcription factor CC AAT/ binding protein-alpha, continued treatment did not fully support the development of differentiated adipocytes. Sphingosine 1-phosphate receptors (S1PRs) are expressed in preadipocytes and message expression declines markedly during adipocyte differentiation. Conclusion: These results demonstrate that the contribution of SPHK and S1P to adipogenesis is mediated primarily through biphasic activation of SPHK1 and 2 with extracellular S1P and S1PRs playing little role during preadipocyte differentiation.
1. INTRODUCTION
Adipogenesis involves the activation of a complex series of transcriptional pathways which induce the expression of proteins required for the development of lipid-laden adipocytes [1]. Differentiation is initiated when preadipocytes undergo growth arrest followed by mitotic clonal expansion, and then entry into the terminal differentiation pathway. Two transcription factors which play a predominant role in adipogenesis are CCAAT/ enhancer-binding protein-α (CEBP-α) and peroxisome proliferator-activated receptor-γ (PPAR-γ) [2]. The mouse 3T3-L1 cell line serves as a model for adipogenesis and is used as a system to identify additional signaling molecules that contribute to the terminal differentiation process.
Sphingosine 1-phosphate (S1P) is a novel bioactive phospholipid which has been demonstrated to participate in a wide array of cellular processes. S1P serves dual roles both as an intracellular signaling molecule and as an extracellular ligand for G protein-coupled S1P recaptors (S1PR1-5) [3]. These cell surface receptors are ubiquitously, yet differentially, expressed in tissues, and they mediate diverse signal transduction pathways eliciting responses specific to the cell type [4,5]. S1P acting through plasma membrane S1PRs has been demonstrated to play a role in cell differentiation, modulating angiogenesis [6], promoting embryonic stem cell transition to cardiomyocytes [7], and inducing development of mast cells from hematopoietic precursors [8]. Intracellular S1P regulates calcium release [9] and serves as a second messenger promoting cell proliferation in opposition to apoptosis [10]. Finally, intranuclear S1P has been demonstrated to alter histone acetylation, a major epigenetic factor in chromatin remodeling and gene expression [11].
Sphingosine kinases (SPHK) are responsible for S1P biosynthesis from sphingosine [12]. Two SPHK isoforms have been identified [12-14] . SPHK1 has been described primarily in growth-promoting and anti-apoptotic activities, while SPHK2 is reported to be involved in apoptosis induction and inhibition of DNA synthesis/cell growth [11,15,16] . Knockout studies in mice suggest that SPHK1 and SPHK2 have redundant functions since deletion of either isozyme alone does not have a specific phenotype, whereas deletion of both genes results in defects in neurogenesis and vascular development with embryonic lethality [17]. Both isoforms are expressed in adipocytes and at least one isoform (SPHK1) participates in adipogenesis [18].
Differentiated rat adipocytes express S1PR1-3 and S1PR5 [19]. G-protein coupled signal transduction pathways attributed to the receptors include inhibition of adenylyl cyclase (S1PR1), activation of phospholipase C (S1PR3), and upregulation of mitogen activated protein kinase and c-Jun N-terminal kinase activities (S1PR2-5) (reviewed in [20]). Extracellular application of S1P to rat adipocytes elicits increases in intracellular calcium and inositol-1,4,5-trisphosphate (IP3) and cAMP generation that is dependent on S1PR activation. In addition, S1P inhibits insulin-induced leptin production and activates lipolysis in a cAMP-dependent manner [19]. The specific S1PRs which mediate these activities have not been delineated. While it has been reported that intracellular S1P levels increase during adipocyte differentiation, and knockdown of SPHK1 partially blocks accumulation of lipid within 3T3-L1 cells, the mechanism by which S1P participates in adipocyte differentiation is not clear [18]. In this study, the 3T3-L1 cell model is used to study the participation of S1P in adipocyte differentiation. While SPHK enzyme activity increases robustly early in the process of adipocyte differentiation, expression of three S1PRs decreases, and treatment of preadipocytes with S1P only partially supports adipogenesis. The results of this study indicate that participation of S1P in adipocyte differentiation is biphasic, with intracellular S1P generation necessary and sufficient for adipogenesis.
2. MATERIALS AND METHODS
2.1. Materials
S1P and sphingosine were from Biomol Research Laboratory Inc. (Plymouth Meeting, PA). [32γ]Adenosine triphosphate (ATP) (3000 Ci/mmol) and [32P]-orthophosphate (8500 - 9120 Ci/mmol) were from Perkin Elmer (Boston, MA). Fetal bovine serum (FBS) was from PAA Laboratories (Bedford, MA), and fetal calf serum (FCS) from ATCC (Manassas, VA). Culture media and molecular boilogy reagents were from Invitrogen (Grand Island, NY). SPHK1 and SPHK2 antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). SPHK-I2 was from Cayman Chemicals (Ann Arbor, MI). Other reagents were from Sigma Chemical Co. (St. Louis, MO).
2.2. Cell Culture
Mouse 3T3-L1 cells were obtained from ATCC (Manassas, VA). Fibroblasts were maintained in DMEM containing 4.5 g/L D-glucose, 1.5 g/L NaHCO3, 110 mg/L sodium pyruvate, 100 U/L penicillin, 0.1 mg/L streptomycin, and 10% FCS (growth media) at 37˚C, 5% CO2 environment. When cells reached confluence, media was changed to include 10% FBS instead of FCS for 24 hours. Differentiation was induced (Day 0) by addition of media supplemented with 10% FBS, isobutylmethylxanthine (IBMX), dexamethasone, and insulin (0.1 mM, 1 µM, and 1 µg/ml). After 72 hours, induction media was removed and replaced with media containing 10% FBS and 1 µg/ml insulin. Media was changed every 2 - 3 days. To assess whether S1P could support adipocyte differentiation, 3T3-L1 preadipocyte cultures were maintained in DMEM + 10% FBS lacking IBMX, dexamethasone, and insulin with or without S1P (1 µM). Media was changed as above with re-addition of S1P at each media change. Cells were harvested as indicated.
2.3. In Vitro Assay of Sphingosine Kinase Activity
Cells (10 × 30 mm dish) were harvested at times indicated in the text. Harvested cells were lysed by freeze-thawing in SPHK buffer [21]. Lysates were centrifuged at 15,000 × g for 20 min at 4˚C, and the protein concentration of the supernatant fraction was determined. SPHK activity assays included (unless otherwise indicated): 40 - 80 mg protein/reaction, 50 mM sphingosine complexed with FAF-BSA (4 mg/ml suspension), and [32P]-ATP (2.5 mCi, 1 mM) containing MgCl2 (10 mM) [12]. Reaction linearity was determined in preliminary assays. Biochemical characterization of SPHK isozyme activities was determined in the presence of 400 mM potassium chloride (KCl) or 1% Triton X-100 (TX-100), as described previously [13]. Labeled S1P was extracted [21] and separated by thin-layer chromatography (TLC) on heat-activated silica gel G plates using a solvent system containing 1-butanol/methanol/acetic acid/water (80/ 20/10/20, v/v). [32P]S1P was quantitated and SPHK activity was expressed as picomoles of S1P formed/min/ mg of total protein [22].
2.4. Immunoblot Analysis
Cells were harvested in total protein lysis buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM EDTA, pH 7.4, 1 mM PMSF, 10 µg/ml aprotinin, and 1% TX-100. Equal amounts of whole-cell lysate were resolved by SDS-PAGE (12.5% gel) and transferred to PVDF membrane. Membranes were blocked using Trisbuffered saline with Tween 20 (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, and 0.05% Tween (TBST) containing 5% milk for 1 hour, and then incubated 2 hours at room temperature with the indicated SPHK antibodies (1:200 dilution). After washing three times with TBST, the membranes were probed with donkey α-goat horseradish peroxidase-conjugated secondary antibody to detect bands via WestPico chemiluminescence reagent (Pierce Biotechnology, Rockford, IL).
2.5. Oil Red O Staining
3T3-L1 cells were differentiated in the presence of SPHK-I2 inhibitor (0.5 - 10 µM) with or without S1P (1 or 10 µM). Inhibitor and S1P were re-added to the cell cultures at each media change. Adipocytes were washed with PBS and then fixed with 3.8% formaldehyde for 1 hour. Cells were then washed and stained with Oil Red O (3 mg/ml) and isopropyl alcohol in water (60/40, v/v) for 30 minutes. To assess the degree of differentiation, stained cells were then washed 4x with water, and Oil Red O extracted with isopropyl alcohol. The resultant staining was quantified by measuring OD500nm. Un-differentiated 3T3-L1 preadipocytes cultured in DMEM + 10% FBS for 15 days were stained and served as background control.
2.6. mRNA Quantitation
3T3-L1 cells were treated with S1P (1 µM) in the presence or absence of differentiation media. Levels of CEBP-α, adipocyte complement-related protein of 30 kDa (Acrp30) and glucose transporter type 4 (GLUT4) mRNA were determined by real-time QPCR [23]. Adipocyte RNA was extracted following treatment, as described previously [24]. QPCR was carried out using using 1 µg of total RNA with primers to CEBP-α: sense, 5’-TGGACAAGAACAGCAACGAG-3’; antisense, 5’- CCTTGACCAAGGAGCTCTCA-3’ (225 bp), Acrp-30: sense, 5’-GTTGCAAGCTCTCCTGTTCC-3’; antisense, 5’TCTCCAGGAGTGCCATCTCT-3’ (192 bp), and GLUT-4: sense, 5’TTCCTTCTATTTGCCGTCCTC-3’; antisense, 5’TGGCCCTAAGTATTCAAGTTCTG-3’ (168 bp). To measure S1PR mRNA during adipogenesis, QPCR was carried out using 1 μg of RNA harvested from cells at indicated time with primers to S1PR1: sense, 5’-AAGTCTCTGGCCTTGCTGAA-3’; antisense, 5’-GATGATGGGGTTGGTACCTG-3’ (183 bp), S1PR2: sense, 5’-CCTCATCACCACCATCCTCT-3’; antisense, 5’-CCTCATCACCACCATCCTCT-3’ (227 bp), S1PR3: sense, 5’- TCTCAGGGCATGTCACTCTG-3’; antisense, 5’-CAGCTTTTGTCACTGCCGTA-3’ (163 bp). QPCR was carried out with the iQ SYBR Green Supermix. Reaction conditions were: initial denaturation at 95˚C for 3 min. followed by 50 cycles of 95˚C for 10 sec and 61˚C for 30 seconds. Specificity of the reaction was verified by melt curve analysis. In each reaction, gene expression was normalized to 18S rRNA level and calculated using the 2−ΔΔCT method. Expression of each adipocyte differentiation gene at day 0 (undifferentiated) served as the control.
2.7. Statistical Analysis
Values are means ± SE. Significant differences between treatment groups were determine by Student’s t test (paired) or one-way ANOVA with post hoc analysis using the Student-Newman-Keuls multiple comparison test. For data expressed as percent of basal or fold increase in expression, two-tailed, one-sample hypothesis test was used. QPCR expression data for adipogenesis genes was log transformed to address variability in absolute fold expression between experiments. Values of P ≤ 0.05 were accepted as significant. Data analysis was performed using Statmost 32 (Dataxiom Software, CA).
3. RESULTS
3.1. SPHK Activity Increases during Adipogenesis
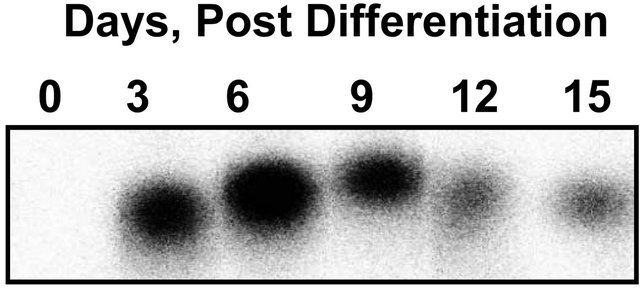
3T3-L1 cells were differentiated and cytosolic SPHK activity was measured at various time points after initiation of differentiation. SPHK activity was detectable in undifferentiated cells and increased early in the differentiation process, with maximal activity at day 6 post-differentiation (Figure 1(a)). The level of SPHK activity declined in a time-dependent manner after day 6; however, in fully differentiated adipocytes (day 15) SPHK activity remained elevated by approximately 2-fold compared to undifferentiated cells (Figure 1(b)).
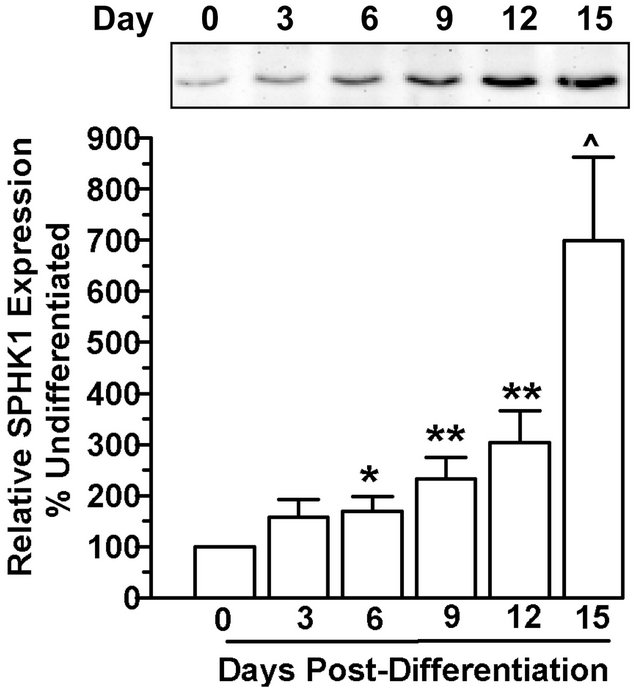
SPHK activity did not correlate directly with SPHK isoform protein levels. For SPHK1 protein, although there were modest but significant increases in protein expression as early as day 6 post-differentiation, there were continued increases in protein levels on days 9 and 12, and maximal expression identified by day 15 with an approximately 7-fold change in SPHK1 compared to undifferentiated cells (Figure 2(a)). In contrast, SPHK2 protein increased maximally at day 6 post-differentiation and remained stable throughout the differentiation process (Figure 2(b)).
3.2. SPHK Isozyme Characterization during Adipocyte Differentiation
The contribution of isozymes SPHK1 and SPHK2 to
 (a)
(a)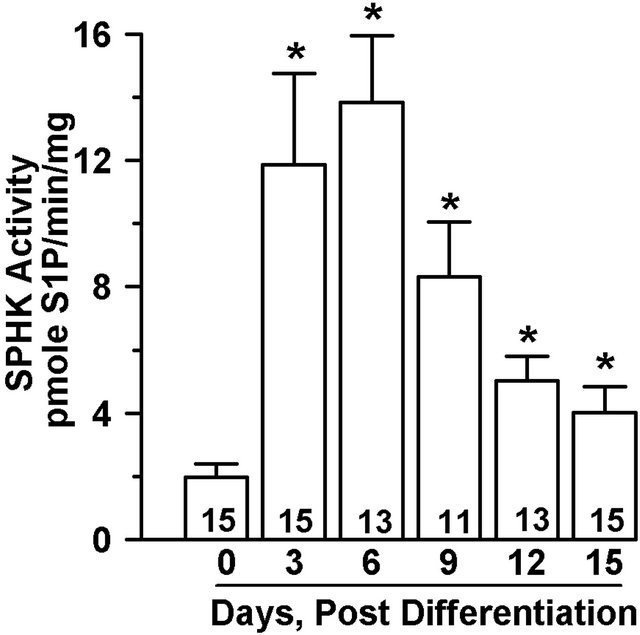 (b)
(b)
Figure 1. SPHK activity is induced during adipoctye differentiation. 3T3-L1 cells 2 days postconfluence were induced to differentiate for three days, followed by culture in media containing 1 µg/ml insulin for up to 15 days. Undifferentiated (Day 0) or differentiated cells were harvested as indicated. Cell cytosolic fractions (60 µg) were analyzed for SPHK activity in the presence of sphingosine (50 µM)/FAF-BSA (4 mg/ml) complex. (a) Representative phosphoimage of S1P produced, extracted, and isolated by thin-layer chromatography. (b) SPHK activity is expressed as pmole S1P/mg/min and values are mean ± SE for the number of experiments shown at the base of each bar. *p < 0.01 vs. day 0 for all samples. P values determined by one-way ANOVA and multiple comparison test.
total SPHK activity in differentiating adipocytes was determined by comparison of their activities in the presence of high salt (KCl) and detergent (TX-100) [13]. SPHK1 activity is inhibited by high salt and activated by TX-100, whereas SPHK2 behaves in a reciprocal manner. SPHK activity in preadipocytes was low and was activated by TX-100 indicating that SPHK1 activity predominates in preadipocytes (Figure 3). During the early phase of preadipocyte differentiation, total SPHK activity increases by approximately 4-fold on day 3 (Figures 1 and 3). However, neither KCl nor TX-100 treatment resulted in complete inhibition of SPHK activity (Figure 3), suggesting that both isozymes contribute to S1P production during adipogenesis. On the other hand, a significant 1.9-fold activation by TX-100 compared to the
 (a)
(a)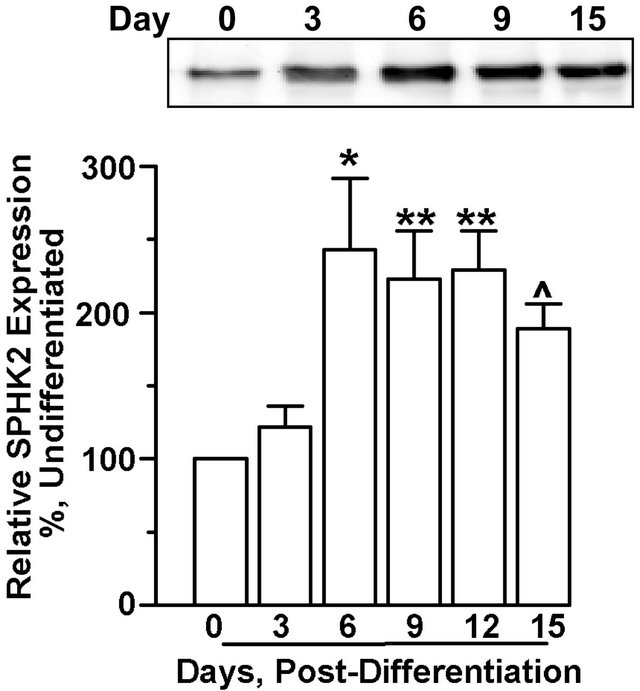 (b)
(b)
Figure 2. SPHK protein level expression does not correlate with activity during adipocyte differentiation. 3T3-L1 cells were induced to differentiate and cells were harvested on indicated days post-differentiation. Total protein extract (40 µg) was subjected to immunoblot analysis with either (a) SPHK1 or (b) SPHK2 specific antibodies. Representative immunoblots are shown. Quantitative analysis of the Western blots for 8-10 individual experiments showed that (a) SPHK1 levels and (b) SPHK2 levels increased following differentiation. *p < 0.05; **p < 0.01; ^p < 0.005 vs. day 0 (preadipocytes) as determined by one-way ANOVA and multiple comparison test.
total activity on day 3 suggests that SPHK1 predominates at this stage. SPHK activity in fully differentiated adipocytes (day 15) was neither significantly inhibited nor activated in response to treatment with KCl or TX- 100.
To further address the contribution that SPHK plays in adipocyte differentiation, 3T3-L1 cells were treated with
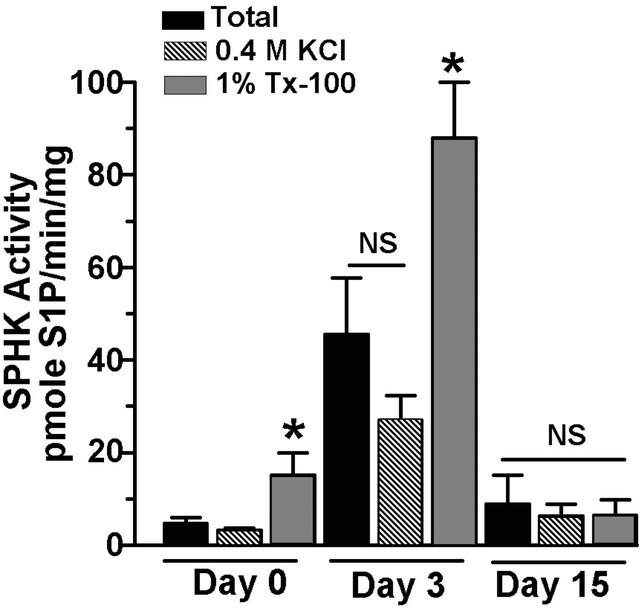
Figure 3. SPHK1 and SPHK2 isozymes are active during adipocyte differentiation. 3T3-L1 cells were induced to differentiate and cells were harvested on day 0, 3, and 15 post-differentiation. SPHK activity was determined in cytosolic fraction (60 µg) in the absence or presence of 0.4 M KCl and 1% Triton X-100 (TX-100), as indicated. Values are means ± SE for 7 independent experiments. P values were determined by one-way ANOVA with multiple comparison test. *p < 0.05 vs. corresponding day total SPHK activity.
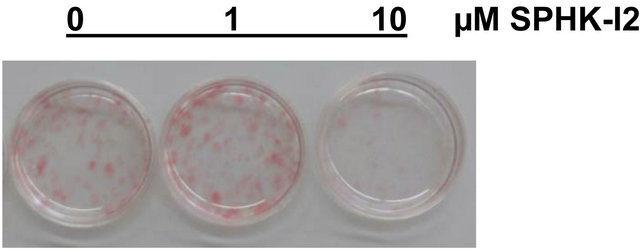
the SPHK1 specific inhibitor SPHK-I2 from the onset of differentiation. SPHK-I2 (10 µM) partially inhibited adipocyte differentiation as measured by the accumulation of lipid measured by Oil Red O staining on day 15 (Figure 4).
3.3. Effect of Extracellular S1P and SPHK1 Inhibition on Adipocyte Differentiation
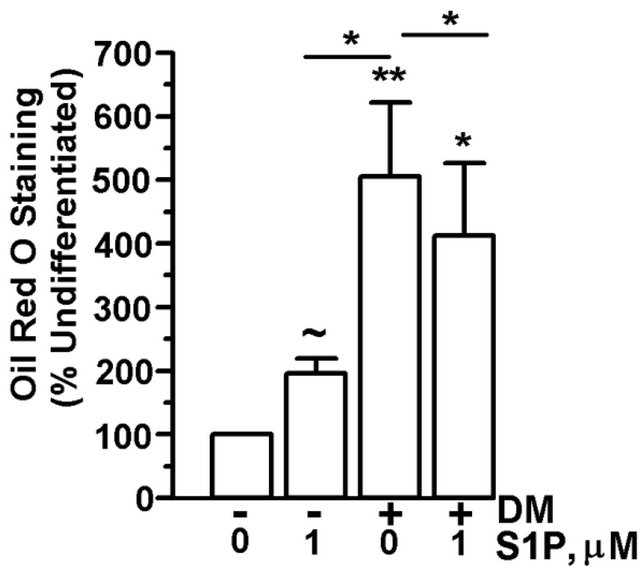
In order to determine whether extracellular S1P affected adipogenesis or could rescue the partial block of adipocyte differentiation by biochemical inhibition of SPHK1, 3T3-L1 cells were differentiated for 15 days in the presence of SPHK-I2 (10 µM) with or without exogenous S1P. While undifferentiated preadipocytes had a low level of Oil Red O staining, treatment with S1P did induce an approximately 2-fold increase in lipid accumulation (Figure 5(a)). In the presence of differentiation media, cells had a 5-fold increase in Oil Red O staining vs. undifferentiated cells. However, the addition of S1P with differentiation media to cells significantly reduced Oil Red O staining by about 25% vs. cells treated with differentiation media only (Figure 5(a)). Co-culture of differentiating adipocytes with SPHK-I2 significantly inhibited lipid accumulation in the cells vs. cells treated with differentiation media only (Figure 5(b)). Treatment of cells with S1P (1 µM or 10 µM) did not compensate for the inhibition of SPHK1 and S1P biosynthesis by SPHK-I2, and lipid levels remained significantly lower than differentiated control cells (Figure 5(b)).
 (a)
(a)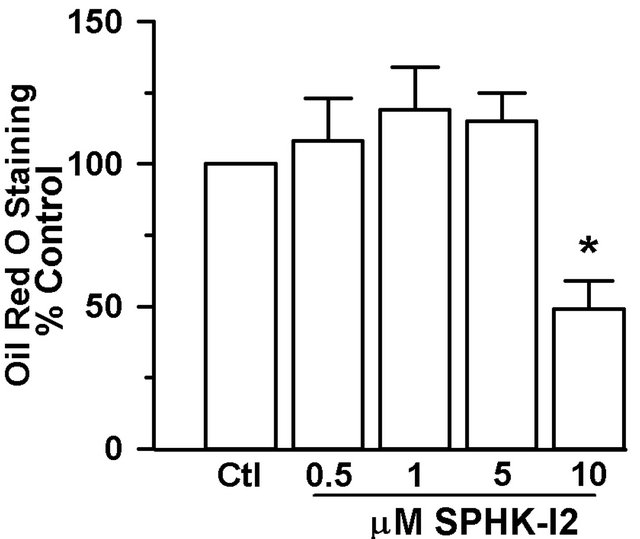 (b)
(b)
Figure 4. Pharmacologic inhibition of SPHK1 blocks lipid accumulation in adipocytes. 3T3- L1 cells were induced to differentiate in the presence of DMSO (vehicle control) or SPHK1 specific inhibitor SPHK-I2 at concentrations 0.5 - 10 µM. Media was changed every 2 - 3 days, and inhibitor was included throughout the entire differentiation period. On day 15, cells were subjected to Oil Red O staining. (a) Representative photograph of stained adipocytes differentiated in the presence or absence of SPHK-I2. (b) The degree of Oil Red O staining was expressed as a percent of the A500nm compared to the vehicle-treated cells. Values are means ± SE for 3 independent experiments. P value was determined by two-tailed, one-sample hypothesis test. *p < 0.05 vs. vehicletreated adipocytes.
3.4. Extracellular S1P Effects on Early Adipocyte Differentiation Markers
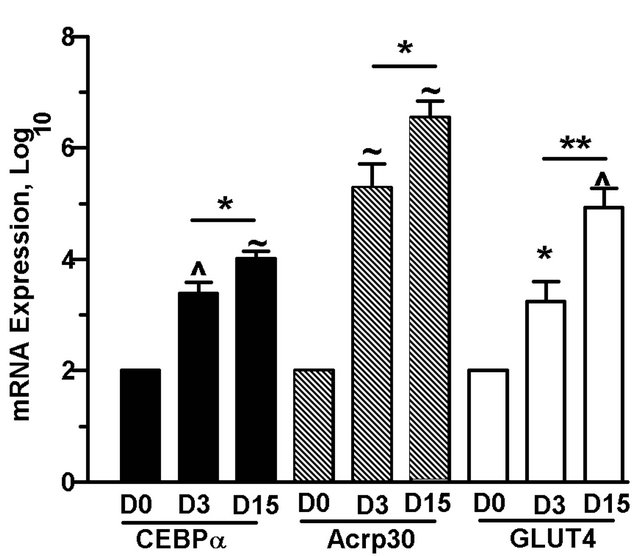
Since treatment of preadipocytes with S1P resulted in significant accumulation of lipid compared to untreated preadipocytes (Figure 5(a)), S1P might serve as an early differentiation stimulus. Expression of CEBPα mRNA, an early indicator of commitment to adipogenesis [25], as well as expression of downstream adipocyte specific genes, Acrp30 and GLUT4, increased significantly in a time-dependent manner from days 3 to15 in 3T3-L1 cells cultured in differentiation media (Figure 6(a)). To address whether early adipogenesis activators are expressed in response to extracellular S1P, preadipocytes were treated with S1P (1 µM) in the absence of differenttiation media for up to 15 days. Cells treated with S1P demonstrated an increase in CEBPα message expression by day 15 of the differentiation process compared to un-
 (a)
(a)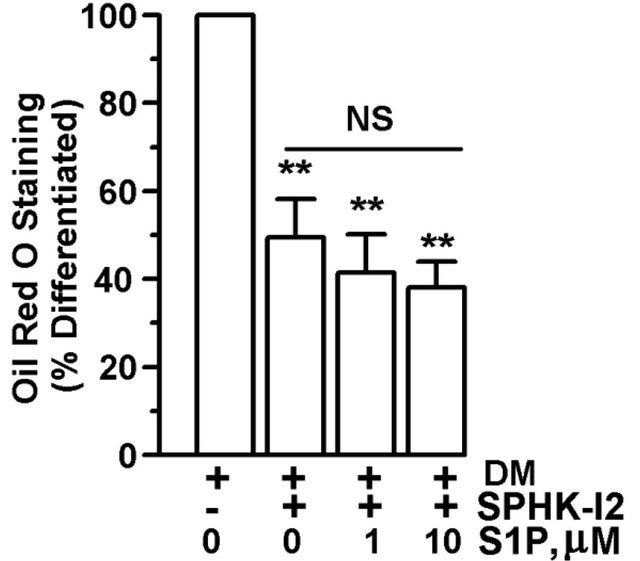 (b)
(b)
Figure 5. S1P does not fully support adipocyte differentiation. 3T3-L1 cells were induced to differentiate in the presence of DMSO (vehicle control), SPHK-I2 (10 µM), with (+) or without (−) the addition of S1P (1 or 10 µM). Control cells were treated with DMEM + 10% FBS lacking differentiation cocktail. Media was changed every 2 - 3 days, and cells were retreated with inhibitor and S1P throughout the entire experiment. Cells were subjected to Oil Red O staining. (a) Oil Red O staining expressed as a percent of A500nm compared to undifferentiated cells cultured in DMEM + 10% FBS. Values are means ± SE for 6 independent experiments. (b) Oil Red O staining expressed as a percent of A500nm compared to differentiated cells not treated with either S1P or SPHK-I2. Values are means ± SE for 4 independent experiments. P value was determined by two-tailed, one-sample hypothesis test. *p < 0.05, **p < 0.025, ~p < 0.01.
treated cells (Figure 6(b)). While Acrp30 and GLUT4 mRNA levels did increase (Acrp30, 437 fold ± 357 fold; p < 0.025; GLUT4, 293 fold ± 187 fold; p < 0.0.08) in day 15 cells treated with S1P compared to undifferentiated (day 0) cells, the increase was not statistically significant compared to cells cultured for 15 days in the absence of S1P (data not shown).
 (a)
(a)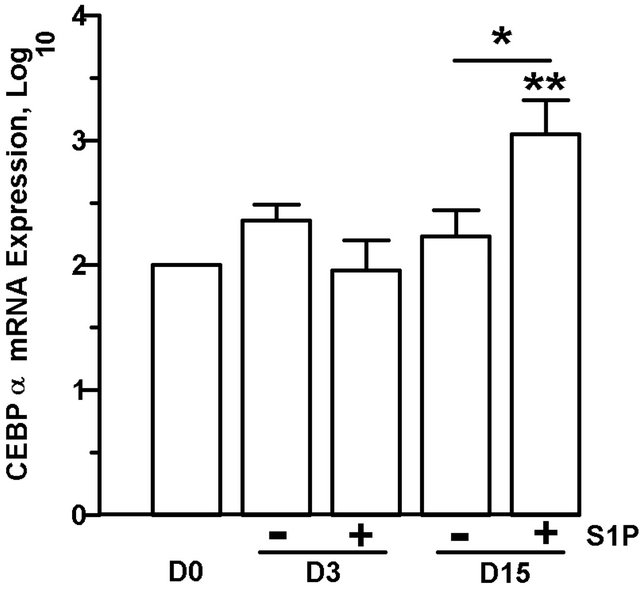 (b)
(b)
Figure 6. Prolonged culture of 3T3-L1 cells with S1P increases CEBPα mRNA expression. 3T3-L1 were induced to differentiate or were cultured in DMEM + 10% FBS with (+) or without (−) the addition of S1P (1 µM). Cells were harvested on days (D) 0, 3 or 15, and mRNA expression of CEBPα, Acrp30, and GLUT 4 relative to 18S rRNA were determined by qPCR. (a) mRNA expression in differentiated 3T3-L1 cells. (b) CEBPα m RNA expression in 3T3-L1 cells treated with S1P in the absence of differentiation media. Fold increase values over day 0 cells were log transformed and means ± SE were calculated for 4 - 7 experiments. P values were determined by twotailed, one-sample hypothesis test (vs. D0) or two-tailed, two-sample hypothesis test (D3 vs. D15). *p ≤ 0.05, **p ≤ 0.025, ^p ≤ 0.005, ~p ≤ 0.0005.
3.5. S1PR Expression during Adipocyte Differentiation
S1PRs are expressed in both primary rat preadipocytes and adipocytes [19], however, the expression pattern of S1PRs in the 3T3-L1 model has not been examined. One explanation for the observation that extracellular S1P does not fully support adipogenesis is that expression of S1PRs in the undifferentiated state is low, thereby limiting S1P-mediated signaling events. Gene expression for three S1PRs—S1PR1, S1PR3, and S1PR2—was measured during the differentiation process by real-time PCR. For all three receptors, mRNA levels were highest in the preadipocytes and declined significantly in a time-dependent manner during the 15 days of differentiation (Figure 7). Message expression during differentiation declined by 50% or more at day 3 for all S1PRs analyzed. By day 15, all three S1PR mRNA levels were markedly reduced, with S1PR1 mRNA levels as low as 12 ± 1.4% of preadipocyte levels.
4. DISCUSSION
S1P acts both as an extracellular receptor ligand and intracellular signaling molecule in multiple cell types. A physiologic role for S1P has been demonstrated in mature adipocytes where it signals through S1P-receptors to stimulate lipolysis and modulate leptin secretion [19]. In addition, SPHK isoforms play a role in adipocyte differentiation [18]. The present study demonstrates that SPHK activity increases rapidly and remains elevated in 3T3-L1 cells undergoing terminal differentiation. Total SPHK activity in differentiating adipocytes increased by 6-fold at day 3 and 2-fold at day 15 compared to undifferentiated cells. However, the rapid increase in SPHK activity did not correlate directly with accumulation of SPHK isoform expression. The peak protein levels occurred later than peak enzyme activities and were evident at day 15 for SPHK1 and at day 6 for SPHK2. The early increase in SPHK activity confirms the observations of Hashimoto et al. [18] in which intracellular S1P levels increased approximately 3-fold by day 3 post-differentiation, preceding increases in SPHK message and protein. These results indicate that activation of endogenous enzyme is a more important contributor than new en-
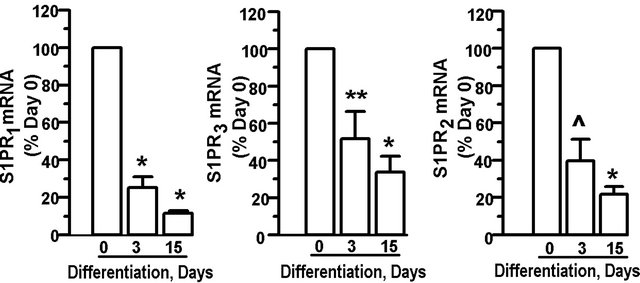
Figure 7. S1P receptor expression declines during adipocyte differentiation: 3T3-L1 cells were induced to differentiate. Cells were harvested at day 0 (preadipocytes), and day 3 and day 15 after initiation of differentiation. Adipocyte levels of S1PR mRNA relative to 18S rRNA for S1PR1, S1PR3, and S1PR2, were determined. Data is expressed as percent of day 0 expression and values are means ± SE for 7 independent experiments. P value was determined by two-tailed, one-sample hypothesis test. ^p < 0.025, *p < 0.001, **p < 0.0025.
zyme biosynthesis in the early/acute induction of SPHK during adipocyte differentiation. The question of what mediates the activation of SPHK during adipogenesis points to a likely involvement of cAMP in the process. Intracellular cAMP activates SPHK in osteoblast cells [26]. One of the earliest regulators of adipocyte differenttiation are the CEBPs [25]. The expression of these transcriptional activators is at least partially mediated by cAMP [27], which increases in preadipocytes treated with differentiation cocktail containing the phosphordiesterase inhibitor IBMX. Treatment of rat adipocytes with S1P (30 µM) also increases cAMP production via S1PR mediated signaling [19], and in the present study, extracellular S1P partially supported adipocyte differentiation. Another regulatory pathway involves protein kinase C (PKC), since phorbol esters induce phosphorylation of SPHK1 [28] through activation of PKC and extracellular signal-regulated kinase 1/2 (ERK)-mediated phosphorylation [29]. Transient activation of the ERK pathway promotes the early stages of adipocyte differentiation [30]. Thus, both cAMP and the ERK pathway may contribute to the increase in SPHK activity seen with initiation of adipogenesis.
While activation of endogenous enzyme likely represents the mechanism by which S1P levels rise early in the adipocyte differentiation process, SPHK message and protein accumulate during the differentiation process as well [18]. SPHK activity is regulated both acutely and chronically. Agents which have been demonstrated induce acute activation of SPHK include cytokines, glucose, phorbol esters, and G-protein coupled receptor ligands [22,31-33] . Chronic regulation of SPHK at the level of both transcription and translation has also been demonstrated. Transcriptional regulation of human SPHK1 by phorbol 12-myristate 13-acetate (PMA) in the human leukemia cell line MEG-O1 maps to the 5’ promoter region just upstream of the first exon [34]. Promoter mapping identified sequence containing a candidate activator protein-2 (AP-2) site that is essential for PMA regulation of hSPHK1. AP-2 is induced by both PMA and cAMP participating in the activation of PMAand cAMP-inducible genes [35]. In dopaminergic neuronal cells, neurotrophic factors increase SPHK1 transcription through CEBPs [36]. Initiation of adipocyte differentiation is dependent on accumulation of cAMP; thus, the activtion of SPHK during adipogenesis at the transcriptional level may be mediated by a similar transcriptional regulatory element. In addition, computer analysis using the TFSEARCH program (http://www.cbrc.fp/research/db/TFSEARCH.html) reveals a putative CEBPα transcription factor binding site upstream of the murine SPHK1 transcription start site that may be involved in the chronic regulation of SPHK1 message and protein during differentiation.
Several SPHKs have been reported for species including human, rat, mouse, yeast, and plants [14]. The contribution of individual SPHK isoforms to cellular function appears to be at least partially redundant as evidenced by the fact that specific phenotypes are demonstrated only when both SPHK isoforms are deleted [17]. In the present study, Western blot analysis of cytosolic lysates demonstrated that both SPHK1 and SPHK2 protein are induced in differentiating adipocytes. SPHK2 increased maximally early in the differentiation process, whereas SPHK1 increased more gradually with a maximum by 15 days. Such differences may be due to translation rates of the different genes, or to half-lives of the proteins themselves. We have previously demonstrated in insulinoma cells that the both SPHK isoforms are very stable, suggesting that upregulation of translation is the predominant mechanism for enzyme accumulation during adipocyte differentiation [33]. Moreover, since neither KCl nor TX-100 completely inhibited SPHK activity, both isozymes contribute to the total SPHK activity in preadipocytes and during adipocyte differentiation. SPHK activity was significantly activated by TX-100, which has been demonstrated to stimulate SPHK1 activity in cellular lysates. These results suggest that both SPHK1 and SPHK2 are expressed and active during adipocyte differentiation, with SPHK1 contributing more significantly to overall activity early in the differentiation process. The data also show that SPHK activity is inactivated or desensitized at the terminal stage of differentiation, even though SPHK1 and 2 protein levels are significantly elevated.
The observation that SPHK1 isozyme contributes significantly to the overall activity induced early in the differentiation process is confirmed by treatment with a specific pharmacologic SPHK1 inhibitor, SPHK-I2, which resulted in a partial inhibition of adipogenesis, mirroring inhibition of adipogenesis via genetic down-regulation of SPHK1 activity [18]. In human cancer cell lines overexpressing SPHK1, SPHK-I2 inhibits proliferation at lower concentrations than used in this study (IC50 1 - 4.6 μM) [37]. However, the finding that differentiation was not completely inhibited by a SPHK1 inhibitor supports previous evidence that SPHK2 can partially compensate for SPHK1 activity during development and differentiation [17]. Alternatively, while extracellular S1P did not restore adipogenesis in the face of SPHK1 inhibition by SPHK-I2, S1P in this compartment may initiate adipogenesis.
The process of adipocyte differentiation is characterized by growth arrest, synchronous mitotic clonal expansion, followed by terminal differentiation [1]. Stimulation of adipogenesis leads to increases in intracellular S1P production, correlating with SPHK activation (Figure 1). However, it is not clear whether S1P participates in adipocyte differentiation via intracellular or extracellular signaling events. Extracellular S1P-mediated signal transduction occurs through the family of S1PRs which participate in cell growth, differentiation, and protection from apoptosis. Treatment of preadipocytes with micromolar concentrations of S1P leads to cAMP generation [19], an essential step in the promotion of adipogenesis. In this study, we tested whether extracellular S1P was sufficient to support adipogenesis. Although treatment of preadipocytes with S1P did result in a two-fold increase in lipid accumulation, overall, the degree of differentiation was significantly less than that with standard differentiation media. In fact, co-treatment of preadipocytes with DM and S1P attenuated lipid accumulation. In addition, while culture of preadipoctyes with S1P alone resulted in accumulation of CEBPα mRNA, the degree (~30% stimulation) and time-course of activation (as late as day 15) were significantly lower and later than that seen with differentiation media. S1P did promote adipogenesis as evidenced by Oil Red O staining, although the magnitude of the response was about half that of differentiation media alone. The failure of extracellular S1P to fully support adipogenesis may be in part explained by S1PR expression. Although expression of S1PR1-3 mRNA is actually highest in undifferentiated cells, the mRNA levels of each receptor precipitously decline throughout the course of differentiation. If receptor protein parallels the mRNA expression levels, then S1P signal transduction could be severely impacted as differentiation proceeds. Alternatively, since S1P has growth-promoting effects, treatment of preadipocytes with S1P may interfere with the ability of the cells to undergo growth arrest, which is essential for commitment to adipocyte differentiation.
In summary, the evidence strongly suggests that intracellular SPHK activity and S1P are important contributors to adipogenesis. SPHK is active in differentiating adipocytes, with enzyme activation preceding changes in protein levels. Both SPHK1 and 2 are expressed, and enzyme activity is modulated during initiation of differentiation. Moreover, pharmacologic inhibition of SPHK1 negatively impacts adipogenesis. However, the evidence argues against a major role for extracellular S1P in modulating adipogenesis. Treatment of preadipocytes with extracellular S1P induced a modest accumulation of lipid that was not additive with and even reduced the effectiveness of differentiation media. Moreover, the sphingolipid did not rescue adipogenesis from the inhibition of SPHK1 by SPHK-I2. Rapid declines in S1PR message during differentiation may explain why S1P does not fully support adipogenesis. Alternatively, the loss of these receptors may represent a biologic transition from a rapidly dividing cell line to a growth arrested lineage capable of differentiating.
5. ACKNOWLEDGEMENTS
The author has no conflicts of interest to disclose. This work was supported by the Louis Sklarow Memorial Fund. LDM thanks Alice DelToro, M.A. for her technical support and Dr. Suzanne Laychock for her thoughtful criticisms of this manuscript.
REFERENCES
- Otto, T.C. and Lane, M.D. (2005) Adipose development: From stem cell to adipocyte. Critical Reviews in Biochemistry and Molecular Biology, 40, 229-242. doi:10.1080/10409230591008189
- Feve, B. (2005) Adipogenesis: Cellular and molecular aspects. Best Practice & Research Clinical Endocrinology & Metabolism, 19, 483-499. doi:10.1016/j.beem.2005.07.007
- Alvarez, S.E., Milstien, S. and Spiegel, S. (2007) Autocrine and paracrine roles of sphingosine-1-phosphate. Trends in Endocrinology & Metabolism, 18, 300-307. doi:10.1016/j.tem.2007.07.005
- Spiegel, S. and Milstien, S. (2000) Sphingosine-1-phosphate: Signaling inside and out. FEBS Letters, 476, 55- 57. doi:10.1016/S0014-5793(00)01670-7
- Spiegel, S. and Milstien, S. (2002) Sphingosine 1-phosphate, a key cell signaling molecule. The Journal of Biological Chemistry, 277, 25851-25854. doi:10.1074/jbc.R200007200
- Visentin, B., Vekich, J.A., Sibbald, B.J., Cavalli, A.L., Moreno, K.M., Matteo, R.G., et al. (2006) Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angio-genesis in multiple tumor lineages. Cancer Cell, 9, 225- 238. doi:10.1016/j.ccr.2006.02.023
- Sachinidis, A., Gissel, C., Nierhoff, D., Hippler-Altenberg, R., Sauer, H., Wartenberg, M., et al. (2003) Identification of plateled-derived growth factor-bb as cardiogenesisinducing factor in mouse embryonic stem cells under serum-free conditions. Cellular Physiology and Biochemistry, 13, 423-429. doi:10.1159/000075130
- Price, M.M., Kapitonov, D., Allegood, J., Milstien, S., Oskeritzian, C.A. and Spiegel, S. (2009) Sphingosine-1- phosphate induces development of functionally mature chymase-expressing human mast cells from hematopoietic progenitors. FASEB Journal, 23, 3506-3515. doi:10.1096/fj.08-128900
- Blom, T., Slotte, J.P., Pitson, S.M. and Törnquist, K. (2005) Enhancement of intracellular sphingosine-1-phosphate production by inositol 1,4,5-trisphosphate-evoked calcium mobilisation in HEK-293 cells: Endogenous sphingosine-1-phosphate as a modulator of the calcium response. Cellular Signalling, 17, 827-836. doi:10.1016/j.cellsig.2004.11.022
- Van Brocklyn, J.R., Lee, M.J., Menzeleev, R., Olivera, A., Edsall, L.C., Cuvillier, O., et al. (1998) Dual actions of sphingosine-1-phosphate: Extracellular through the Gi- coupled receptor Edg-1 and intracellular to regulate proliferation and survival. The Journal of Cell Biology, 142, 229-240. doi:10.1083/jcb.142.1.229
- Spiegel, S., Milstien, S. and Grant, S. (2012) Oncogene 31, 537-551.
- Kohama, T., Olivera, A., Edsall, L., Nagiec, M.M., Dickson, R. and Spiegel, S. (1998) Molecular Cloning and Functional Characterization of Murine Sphingosine Kinase. The Journal of Biological Chemistry, 273, 23722- 23728. doi:10.1074/jbc.273.37.23722
- Liu, H., Sugiura, M., Nava, V.E., Edsall, L., Kono, K., Poulton, S., et al. (2000) Molecular cloning and functional characterization of a novel mammalian sphin-gosine kinase type 2 isoform. The Journal of Biological Chemistry, 275, 19513-19520. doi:10.1074/jbc.M002759200
- Liu, H., Chakravarty, D., Maceyka, M., Milstien, S., and Spiegel, S. (2002) Sphingosine kinases: A novel family of lipid kinases. Progress in Nucleic Acid Research and Molecular Biology, 71, 493-511. doi:10.1016/S0079-6603(02)71049-0
- Liu, H., Toman, R.E., Goparaju, S.K., Maceyka, M., Nava, V.E., Sankala, H., et al. (2003) Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. The Journal of Biological Chemistry, 278, 40330- 40336. doi:10.1074/jbc.M304455200
- Igarashi, N., Okada, T., Hayashi, S., Fujita, T., Jahangeer, S. and Nakamura, S. (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. The Journal of Biological Chemistry, 278, 46832-46839. doi:10.1074/jbc.M306577200
- Mizugishi, K., Yamashita, T., Olivera, A., Miller, G.F., Spiegel, S. and Proia, R.L. (2005) Essential role for sphingosine kinases in neural and vascular development. Molecular and Cellular Biology, 25, 11113-11121. doi:10.1128/MCB.25.24.11113-11121.2005
- Hashimoto, T., Igarashi, J. and Kosaka, H. (2009) Sphingosine kinase is induced in mouse 3T3-L1 cells and promotes adipogenesis. Journal of Lipid Research, 50, 602- 610. doi:10.1194/jlr.M800206-JLR200
- Jun, D.J., Lee, J.H., Choi, B.H., Koh, T.K., Ha, D.C., Jeong, M.W., et al. (2006) Sphingosine-1-phosphate modulates both lipolysis and leptin production in differentiated rat white adipocytes. Endocrinology, 147, 5835- 5844. doi:10.1210/en.2006-0579
- Rosen, H., Gonzalez-Cabrera, P.J., Sanna, M.G. and Brown, S. (2009) Sphingosine 1-phosphate receptor signaling. Annual Review of Biochemistry, 78, 743-768. doi:10.1146/annurev.biochem.78.072407.103733
- Olivera, A., Kohama, T., Tu, Z., Milstien, S. and Spiegel, S. (1998) Purification and characterization of rat kidney sphingosine kinase. The Journal of Biological Chemistry, 273, 12576-12583. doi:10.1074/jbc.273.20.12576
- Mastrandrea, L.D., Sessanna, S.M. and Laychock, S.G. (2005) Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells response to cytokines. Diabetes, 54, 1429-1436. doi:10.2337/diabetes.54.5.1429
- Livak, K.J., Flood, S.J., Marmaro, J., Giusti, W. and Deetz, K. (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. Genome Research, 4, 357-362. doi:10.1101/gr.4.6.357
- Laychock, S.G., Tian, Y. and Sessanna, S.M. (2003) Endothelial differentiation gene receptors in pancreatic islets and INS-1 cells. Diabetes, 52, 1986-1993. doi:10.2337/diabetes.52.8.1986
- Tang, Q.Q. and Lane, M.D. (1999) Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Development, 13, 2231-2241. doi:10.1101/gad.13.17.2231
- Machwate, M., Rodan, S.B., Rodan, G.A. and Harada, S.I. (1998) Sphingosine kinase mediates cyclic AMP suppression of apoptosis in rat periosteal cells. Molecular Pharmacology, 54, 70-77.
- Zhang, J.W., Klemm, D.J., Vinson, C. and Lane, M.D. (2004) Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. The Journal of Biological Chemistry, 279, 4471- 4478.
- Johnson, K.R., Becker, K.P., Facchinetti, M.M., Hannun, Y.A. and Obeid, L.M. (2002) PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. The Journal of Biological Chemistry, 277, 35257-35262. doi:10.1074/jbc.M203033200
- Pitson, S.M., Xia, P., Leclerq, T.M., Moretti, P.A., Zebol, J.R., Lynn, H.E., et al. (2005) Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. Journal of Experimental Medicine, 201, 49-54. doi:10.1084/jem.20040559
- Prusty, D., Park, B.H., Davis, K.E. and Farmer, S.R. (2002) Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor γ(PPARγ) and C/EBPα gene expression during the differentiation of 3T3-L1 preadipocytes. The Journal of Biological Chemistry, 277, 46226-46232. doi:10.1074/jbc.M207776200
- Buehrer, B.M., Bardes, E.S. and Bell, R.M. (1996) Protein kinase C-dependent regulation of human erythroleukemia (HEL) cell sphingosine kinase activity. Biochimica et Biophysica Acta, 1303, 233-242. doi:10.1016/0005-2760(96)00092-6
- Meyer zu Heringdorf, D., Lass, H., Alemany, R., Laser, K.T., Neumann, E., Zhang, C., et al. (1998) Sphingosine kinase-mediated Ca2+ signalling by G-protein-coupled receptors. EMBO Journal, 17, 2830-2837. doi:10.1093/emboj/17.10.2830
- Mastrandrea, L.D., Sessanna, S.M., Del Toro, A. and Laychock, S.G. (2010) ATP-independent glucose stimulation of sphingosine kinase in rat pancreatic islets. Journal of Lipid Research, 51, 2171-2180. doi:10.1194/jlr.M000802
- Nakade, Y., Banno, Y., T-Koizumi, K., Hagiwara, K., Sobue, S., Koda, M., et al. (2003) Regulation of sphingosine kinase 1 gene expression by protein kinase C in a human leukemia cell line, MEG-O1. Biochimica et Biophysica Acta, 1635, 104-116. doi:10.1016/j.bbalip.2003.11.001
- Imagawa, M., Chiu, R. and Karin, M. (1987) Transcription factor AP-2 mediates induction by two different signal-transduction pathways: Protein kinase C and cAMP. Cell, 51, 251-260. doi:10.1016/0092-8674(87)90152-8
- Murakami, M., Ito, H., Hagiwara, K., Kobayashi, M., Hoshikawa, A., Takagi, A., et al. (2011) Sphingosine kinase 1/S1P pathway involvement in the GDNF-induced GAP43 transcription. Journal of Cellular Biochemistry, 112, 3449-3458. doi:10.1002/jcb.23275
- French, K.J., Schrecengost, R.S., Lee, B.D., Zhuan, Y., Smith, S.N., Eberly, J.L., et al. (2003) Cancer Research, 63, 5962-5969.