Case Reports in Clinical Medicine
Vol.1 No.2(2012), Article ID:25510,4 pages DOI:10.4236/crcm.2012.12004
Soft tissue neuroendocrine carcinoma of thigh: A case report with literature review
![]()
1Department of Radiotherapy, King George’s Medical University, Lucknow, India; *Corresponding Author: seemagupta02@sify.com
2Department of Pathology, King George’s Medical University, Lucknow, India
Received 6 October 2012; revised 11 November 2012; accepted 22 November 2012
Keywords: Soft Tissue Neuroendocrine Carcinoma; Thigh; Treatment; Chemoradiation; Prognosis
ABSTRACT
A rare case of advanced neuroendocrine carcinoma of thigh in a young adult female, diagnosed with the aid of immunohistochemistry and electron microscopic examinations together with a review of the literature are reported. The primary tumor involved the lateral aspect of thigh without bone involvement. Patient showed significant response with adjuvant chemoradiotherapy following incomplete surgery.
1. INTRODUCTION
Neuroendocrine tumours (NET) are tumours arising from neuroendocrine cells or neurosecretory cells of neural crest origin. These are characterized by the presence of neurosecretory granules often producing biogenic amines and hormones [1].
Neuroendocrine tumors (NETs) are considered rare tumors, annual incidence of these tumors is approximately 6.5 - 5 per 500,000. Majority of them in which about 2/3rd are low grade carcinoid tumors and the rest 1/3rd account for other NETs [2].
Neuroendocrine tumors most commonly occur in the intestine, but may also occur in the lung and other parts of the body [3,4].
Primary soft tissue NET is excessively rare, only few case studies are available in literature [5,6].
Treatment recommendations range from complete excision to combined modality treatment including surgery and chemo radiation [6,7].
We report our experience with such an unusual case.
2. CASE PRESENTATION
A 30 years young female presented to us in April 2011, with large diffuse swelling over lower end of left thigh which had been progressively increasing for 5 months. On examination she had a painless firm swelling involving the lateral aspect of left lower thigh of size about 15 cm × 10 cm. evident on 64 slice contrast enhanced CT scan, which showed a ill defined heterogeneously enhancing soft tissue lesion with nodularity involving the muscles of lateral compartment of lower end of left thigh with stranding and thickening of overlying subcutaneous fat plane and a small sclerotic lesion on condyle of left tibia without bone or knee joint involvement (Figure 1). She had no systemic symptoms such as flushing, diarrhoea or wheeze.
Patient was evaluated for the primary lesions in other sites by CT scans of thorax, abdomen and pelvis, which did not reveal any lesion.
Biopsy from the thigh lesion was suggestive of neuroendocrine carcinoma. Immunohistochemistry was strongly positive for Cytokeratin and Synaptophysin (Figure 2).
Since the primary lesion was unresectable, patient was given 2 cycles of Cisplatin (50 mg/m2 in divided dose day 1, day 2), 5FU (400 mg/m2 in divided dose day 1- day 5), Doxorubicin (40 mg/m2 day 2) chemotherapy at 3 weeks interval to which she showed good response. The tumor size was reduced to about 5 cm × 5 cm evident on contrast enhanced MRI (Figure 3).
The patient then received external beam radiotherapy to a total dose of 66 Gy in 33 fractions in conventional fractionation schedule to the local tumor site which was completed in October 2011.
Further patient received 4 more cycles of same chemotherapy in same schedule started 4 weeks from completion of external radiotherapy. Following completion of treatment patient was kept on regular follow-up. Contrast enhanced MRI which was done in July 2012 revealed significant reduction of tumor size of about 2 cm × 1.5 cm (Figure 4).
When reviewed last, after 6 months from completion of treatment she was well with no cancer-related symptoms at local or other sites.
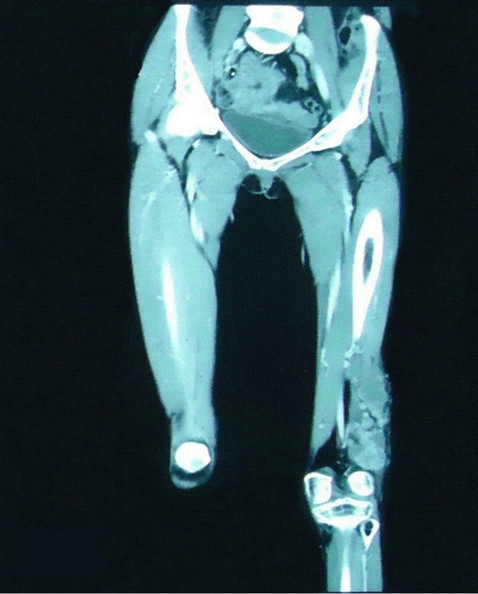
Figure 1. 64 slice contrast enhanced CT scan showing ill defined heterogeneously enhancing soft tissue lesion with nodularity involving the muscles of lateral compartment of lower end of left thigh with stranding and thickening of overlying subcutaneous fat plane without bone or knee joint involvement.
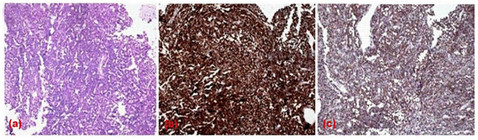
Figure 2. Microphotograph of neuroendocrine carcinoma showing (a) malignant tumor disposed in diffuse sheets, tumor cells are mono-morphic round in shape with focal resetting with some acinar arrangement (H & Ex 100×), staining reaction in immunohistochemistry showing (b) strongly positive for cytokeratin (c) positive for Synaptophysin expression (DAB × 100×).
3. DISCUSSION
Neuroendocrine tumours (NET) most commonly originate in the appendix, small intestine, rectum and bronchus [3,4]. The majority two third are of low-grade malignant potential called carcinoids with an indolent course and favorable prognosis, as recurrences are rare after prolonged follow-up [2,8].
Primary soft tissue NET is extremely rare and there are only few reports available of these tumours originating in soft tissues. Many of these tumors are poorly differenti-
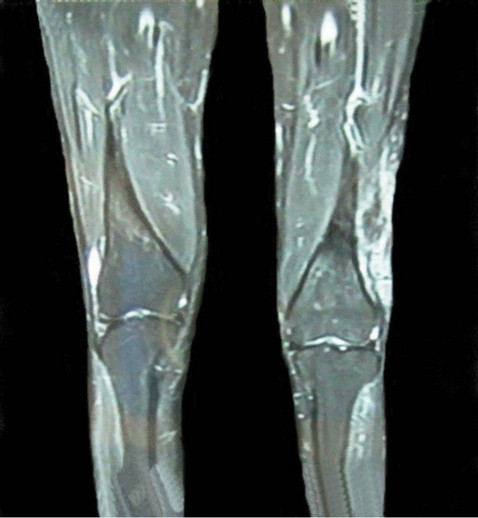
Figure 3. Contrast enhanced MRI of thigh after completion of 2 cycles of chemotherapy showing reduction in tumor size.
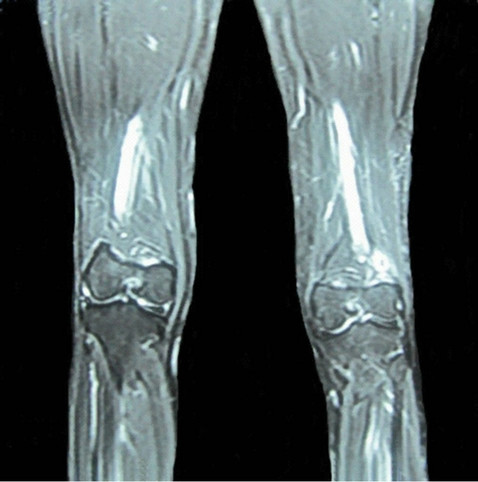
Figure 4. Contrast enhanced MRI of thigh after completion of treatment in follow-up showing significant reduction of tumor size.
ated and aggressive [5,6,9].
Neuroendocrine tumors are classified histologically based on tumor differentiation and tumor grade. They may be well differentiated, low grade (G1) neuroendocrine tumors or Carcinoid tumor; well differentiated, intermediate grade (G2); neuroendocrine tumors or A typical carcinoid; and poorly differentiated, high grade (G3) neuroendocrine tumors or high grade neuroendocrine carcinoma, these are characterized by high mitotic rate and aggressive clinical course and usually require combined multimodality treatment [8].
The presentation of neuroendocrine carcinoma depends on the site of origin. The release of 5-HT and other vasoactive substances into the systemic circulation may give rise to the “carcinoid syndrome”, consisting of flushing, wheezing, diarrhoea [10].
Treatment has improved over the past several decades, and outcomes are improving [7,11].
Treatment aims at curing the disease or relieving symptoms depending on tumor grade, location, invasiveness, release of vasoactive substances, and metastasis.
Surgery is the mainstay of treatment and curative for some neuroendocrine tumors however prognosis depends on anatomic site of tumor, grade, tumor size, presence of multicentric disease, perineural and vascular invasion, margin status, differentiation.
Low grade malignant carcinoid tumours appear to have a good prognosis and have shown median survival of more than eight years with radical surgery; however the presence of metastases may reduce 5 year survival from 70% to less than 50% [4,11]. Adjuvant radiotherapy for these tumors may be helpful when the tumour margin is narrows [6].
The treatment of poorly differentiated high grade unresectable or partially resected neuroendocrine carcinoma has not been clearly defined due to rarity of disease and limited data available; however there are evidences which show encouraging results with surgery and chemoradiation in these patients [6,12-14] as seen in our patient.
The impact of grading and differentiation in neuroendocrine carcinoma on the prognosis and treatment outcome is difficult to assess due limited number of cases and studies available.
Although neuroendocrine carcinoma tends to have a good prognosis, each case is different and varying results are seen. Some investigators have concluded from small clinical studies that poorly differentiated high grade neuroendocrine carcinoma has poor prognosis and have reported 5 year overall survival rate of 13% and survival duration of 13 months with all possible multimodality treatment available [12,15] whereas others believe an aggressive approach of treatment can bring about longterm survival [6,14].
4. CONCLUSION
Neuroendocrine tumors seems to have a good prognosis and adjuvant chemoradiation in unresectable, partially resected or narrow resected margin may form an effective therapeutic approach, however more studies are required to solve the perplexity of neuroendocrine carcinoma.
REFERENCES
- Ramage, J.K., Davies, A.H., Ardill, J., Bax, N., Caplin, M., Grossman, A., et al. (2005) Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut, 54, iv1-iv16. doi:10.1136/gut.2004.053314
- Oberg, K. and Castellano, D. (2011) Current knowledge on diagnosis and staging of neuroendocrine tumors. Cancer and Metastasis Reviews, 30, 3-7. doi:10.1007/s10555-011-9292-1
- Kulke, M.H. and Mayer, R.J. (1999) Carcinoid tumors. The New England Journal of Medicine, 340, 858-868. doi:10.1056/NEJM199903183401107
- Modlin, I.M. and Sandor, A. (1997) An analysis of 8305 cases of carcinoid tumors. Cancer, 79, 813-829. doi:10.1002/(SICI)1097-0142(19970215)79:4<813::AID-CNCR19>3.0.CO;2-2
- Caplin, M.E., Buscombe, J.R., Hilson, A.J., Jones, A.L., Watkinson, A.F. and Burroughs, A.K. (1998) Carcinoid tumour. Lancet, 352, 799-805. doi:10.1016/S0140-6736(98)02286-7
- Hyer, S.L., McAleese, J. and Harmer, C.L. (2007) Neuroendocrine carcinoma arising in soft tissue: Three case reports and literature review. World Journal of Surgical Oncology, 5, 77. doi:10.1186/1477-7819-5-77
- Kim, S.G. and Jang, H.S. (2001) Small cell carcinoma of the oral cavity: Report of a case. Journal of Oral and Maxillofacial Surgery, 59, 680-684. doi:10.1053/joms.2001.23402
- Klimstra, D.S., Modlin, I.R., Coppola, D., Lloyd, R.V. and Suster, S. (2010) The pathologic classification of neuroendocrine tumors: A review of nomenclature, grading, and staging systems. Pancreas, 39, 707-712. doi:10.1097/MPA.0b013e3181ec124e
- Polish, A., Vergo, M.T. and Agulnik, M. (2011) Management of neuroendocrine tumors of unknown origin. Journal of the National Comprehensive Cancer Network, 9, 1397-1402.
- Godwin, J.D. 2nd. (1975) Carcinoid tumors. An analysis of 2837 cases. Cancer, 36, 560-569. doi:10.1002/1097-0142(197508)36:2<560::AID-CNCR2820360235>3.0.CO;2-4
- Oberg, K. (1998) Carcinoid tumors: Current concepts in diagnosis and treatment. Oncologist, 3, 339-345.
- Babin, E., Rouleau, V., Vedrine, P.O., Toussaint, B., de Raucourt, D., Malard, O., et al. (2006) Small cell neuroendocrine carcinoma of the nasal cavity and paranasal sinuses. The Journal of Laryngology & Otology, 120, 289-297. doi:10.1017/S0022215106000594
- Smith, S.R., Som, P., Fahmy, A., Lawson, W., Sacks, S. and Brandwein, M. (2000) A clinicopathological study of sinonasal neuroendocrine carcinoma and sinonasal undifferentiated carcinoma. Laryngoscope, 110, 1617-1622. doi:10.1097/00005537-200010000-00007
- Fitzek, M.M., Thornton, A.F., Varvares, M., Ancukiewicz, M., Mcintyre, J., Adams, J., et al. (2002) Neuroendocrine tumors of the sinonasal tract. Results of a prospective study incorporating chemotherapy, surgery, and combined proton-photon radiotherapy. Cancer, 94, 2623-2634. doi:10.1002/cncr.10537
- Tarozzi, M., Demarosi, F., Lodi, G., Sardella, A. and Carrassi, A. (2007) Primary small cell carcinoma of the nasal cavity with an unusual oral manifestation. Journal of Oral Pathology & Medicine, 36, 252-254. doi:10.1111/j.1600-0714.2007.00486.x