Modern Research in Catalysis
Vol.06 No.02(2017), Article ID:75986,21 pages
10.4236/mrc.2017.62008
Simple Preparation of Halogen-Substituted α-Diimine Nickel Complexes Immobilized into Clay Interlayer as Catalysts for Ethylene Oligo-/Polymerization
Miru Yoshida-Hirahara, Shiori Fujiwara, Hideki Kurokawa
Graduate School of Science and Engineering, Saitama University, Saitama, Japan

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: February 7, 2017; Accepted: April 27, 2017; Published: April 30, 2017
ABSTRACT
In the practical use for the production of the α-olefins, it is highly desired to develop a novel heterogeneous catalyst system. The metal complexes immobilized into the clay interlayers show a great potential as heterogeneous catalysts due to their excellent processability. In this study, nine types of heterogeneous procatalyst Ln/Ni2+-micas were synthesized via a one-pot preparation method, which includes both the condensation reaction of the ligand derivatives and the intercalation of the ligands into the Ni2+ ion-exchanged fluorotetrasilicic mica interlayer. The ligand structures of the prepared procatalysts were [Ln: R-N = C(Nap)-C(Nap) = N-R] [(Nap = 1,8-naphthdiyl) (L1, R = 2-MePh; L2, R = 2-FPh; L3, R = 2-BrPh; L4, R = 4-MePh; L5, R = 4-FPh; L6, R = 4-BrPh; L7, R = 2,4-F2Ph; L8, R = 2,4-Br2Ph; L9, R = 2,6-F2Ph). At 50˚C and 0.7 MPa- ethylene pressure, the triisobutylaluminum-activated L1-L6/Ni2+-mica showed a catalytic activity for the ethylene oligo-/polymerization in the range of 334 - 549 g-ethylene∙g-cat−1∙h−1. A high catalyst activity was obtained when the substituent having a larger steric bulk than that of a methyl substituent was introduced at the ortho-position of the aryl rings. The introduction of the fluorine substituent as a strong electron-withdrawing group to the para-position also increased the catalytic activity. The L2, L4, L5, and L6/Ni2+-micas showed moderate selectivities to oligomers consisting of C4-C20 in the range of 19.9 - 41.6 wt% at 50˚C. The calculated Schulz-Flory constants α based on the mole fraction of C12 and C14 were within 0.61 - 0.78.
Keywords:
Ethylene Oligo-/Polymerization, Heterogeneous Catalyst, Halogen Substituted α-Diimine Nickel Complex, α-Olefin Oligomer, Layered Clay Mineral

1. Introduction
The oligomerization of ethylene is a major industrial process to produce α-ole- fins, which are used in the ethylene polymerization as a co-monomer and the preparation of a variety of economically important chemicals. Since Brookhart and coworkers found that the high electrophilicity of the cationic species of the α-diimine Ni(II) and Pd(II) complexes afforded a high activity for the polymerization of α-olefins, late transition-metal technology has attracted great attention in both academic and industrial research [1] - [8] .
Both the high activity of the catalysts and their ability to control the molecular weight of the products are attributed to the following key features of the catalyst design. A major research direction in this field is modification of the α-diimine ligand or bis(imino)pyridine ligand structures [9] [10] [11] . For example, ortho- substituents of the aryl rings on the imino groups play an important role in controlling the molecular weight of the products. In general, the molecular weight of polyethylene depends on the rate ratio of the chain transfer process relative to the chain propagation process [12] [13] [14] . The ortho-substituents on the aryl rings positioned at the axial sites for the triangle consisting of N-Ni-N in the complex can block the chain transfer reaction, such as β-hydrogen transfer to a monomer, because the rate of the chain transfer mainly depended on the steric bulk of the substituents [10] . Therefore, the molecular weight and the selectivity to oligomers or polymer can be controlled by tuning the steric bulk of the ortho- substituent. Another effective strategy is the tuning of the electronic state of the complexes. Qian et al. reported that the steric bulk and electro-withdrawing property of the halogen substituents have significant effects on the catalyst activity and the molar mass of the product [15] [16] . The charge control of the metal center in the complex could be achieved by introducing electro-donating groups or electro-withdrawing groups [17] [18] [19] . Modification of the ligand backbone structure is also known to be an important parameter in the catalyst design [20] [21] [22] .
Although homogeneous catalysts contain uniform and well-defined active sites, there are some drawbacks when these catalysts were used for industrial applications, especially the difficulty in separating the catalysts, products, and solvent [23] [24] . To apply the metal complexes for practical use in the polyolefin production, the complexes are commonly heterogenized by supporting these on solid carriers, which can be used in the slurry and gas-phase processes. Recently, several research groups have reported heterogeneous catalysts which were prepared by attaching the ligand to the carrier silica surface through a covalent bond because silica have high surface area, good porosity, and good mechanical properties [25] [26] [27] [28] [29] . While these supported catalysts were effective for the slurry polymerization, the preparation of the catalysts required a tedious process such as synthesis of a linker structure in the ligand and the reaction between the linker and surface silanol groups. In addition, the combination of the silica-supported methylalumoxane (MAO) with a metal complex is much easier, but often causes a decrease on the activity comparing to the corresponding homogeneous systems. Therefore, a novel heterogeneous catalyst system with both a simple preparation method and high catalyst performance is desired the practical use.
Recently, we developed novel heterogeneous catalyst precursors (procatalyst) immobilizing late transition-metal complexes in clay mineral interlayers, which were prepared by the direct reaction of a cation-exchange clay mineral (host material) and α-diimine or bis(imino)pyridine ligands (guest material) [30] - [36] . These procatalysts were readily assembled by intercalation of the guest into the host interlayers and subsequent coordination of the guest with the interlayer metal cations. The significance of these procatalysts was the wide selectivity to activators involving not only MAO, but also alkyl aluminum compounds and excellent processability, e.g., no bleaching of the active species from the clay mineral during the slurry polymerization. Moreover, the steric and/or electronic properties of these heterogeneous catalysts can be changed by modification of the ligand structures, which are performed in the homogeneous catalysts. We also developed a one-pot preparation method of the heterogeneous procatalysts by the reaction of Fe3+ ion-exchanged montmorillonite, ketone derivatives, and aniline derivatives [35] . While the obtained catalysts showed a high activity for the olefin oligo-/polymerization, the details of these catalyst systems, e.g., the electronic and steric effects on the catalytic properties, have not yet been clarified.
In this study, we report the synthesis of a series of novel heterogeneous catalyst immobilizing α-diimine Ni(II) complexes with an acenaphthyl backbone via a one-pot preparation method. Their catalytic properties for the ethylene oligo-/ polymerization were also investigated.
2. Experimental
2.1. General Information
The chemicals used for the procatalyst preparation and the ethylene oligo-/po- lymerization were purchased from Kanto Chemical Co., Inc., and Tokyo Chemical Industry Co., Ltd. The fluorotetrasilicic mica (Na+-mica) was supplied by COOP Chemical Co., Ltd. The solvent used for the procatalyst preparation and the ethylene oligo-/polymerization was degassed by N2 bubbling, and then dehydrated over 400˚C-dried molecular sieves (MS-13X) before use. The preparation and handling of the procatalysts were performed by a standard Schlenk technique under a N2 atmosphere.
2.2. Preparation of Procatalyst (Ln/Ni2+-mica)
As shown in Figure 1, we synthesized procatalysts (Ln/Ni2+-mica) via the following two steps: 1) cation-exchange reaction of Na+-mica and 2) one-pot preparation of the procatalyst. To overcome the problem in synthesizing unstable ligands or forming ligands in low yields, the one-pot preparation is an efficient methodology to form a metal complex [37] . Our one-pot process includes the condensation reaction of acenaphthenequinone and an aniline derivative, the


Figure 1. Schematic drawing of Ln/Ni2+-mica preparation.
intercalation of the formed ligands into the Ni2+-mica interlayer, and the reaction of the ligand and metal ions in the interlayer to form the corresponding metal complexes.
2.2.1. Preparation of Ni2+-Mica
Five grams of Na+-mica (amount of Na = 10.6 mmol) was added to a solution of nickel nitrate hexahydrate (5.45 g, 18.74 mmol) dissolved in deionized water (125 ml) using a 200-ml Erlenmeyer flask. The resulting suspension was maintained at 30˚C for 24 h. The solid part was recovered by filtration, and these consecutive manipulations were repeated. The crude product was washed five times with ethanol and dried for about two weeks at ambient temperature. The product was calcined at 200˚C for 4 h, and then dried in a vacuum at 200˚C for 4 h to obtain the Ni2+-mica. The composition of Ni2+-mica was determined by an X-ray fluorescence (XRF) analysis (PW2400; PANalytycal B.V.).
2.2.2. Preparation of Ln/Ni2+-Mica
Nine types of Ln/Ni2+-mica with a series of substituted acenaphthenequinone ligands (Ln) were prepared by the one-pot preparation method with 1 equivalent of acenaphthenequinone, 2.5 equivalents of aniline derivatives, and a predetermined amount of Ni2+-mica. The reaction ratio of the ligand derivatives and Ni2+-mica was controlled in order to obtain the Ln/Ni2+-mica with the Ln amount of 0.40 mmol・g-mica−1 because the Ni2+ content of the Ni2+-mica was 0.54 mmol・g-mica−1. Our previous study concluded that acetonitrile was the best preparation solvent due to its strong polarity which is necessary to swell the Ni2+-mica [31] .
L1/Ni2+-mica: An acetonitrile (10 ml) solution of acenaphthenequinone (21.9 mg, 0.12 mmol), o-toluidine (32.2 mg, 0.30 mmol), and Ni2+-mica (300 mg, amount of Ni = 0.16 mmol) was stirred under reflux conditions for 24 h using a Schlenk flask. After the preparation, almost all of the solvent was removed by decantation using a syringe, the solid product was washed with toluene three times, and then finally washed with hexane two or more times by the same decantation manner to remove the free ligand. The residual solvent was removed under vacuum for 4 h. The L1/Ni2+-mica was obtained as a yellow powder.
L2-L9/Ni2+-mica: Based on the above procedure, the procatalysts were prepared with acenaphthenequinone (21.9 mg, 0.12 mmol) and various aniline derivatives (0.30 mmol).
Control: The ligand L5 (bis (4-fluorophenylimino) acenaphthene) and the conventional Brookhart-type nickel dibromide complex L5-NiBr2 were used as the controls. The ligand L5 and the complex L5-NiBr2 were prepared according to the procedures described in the literature with a slight modification [38] [39] .
Characterization of procatalysts: The FT-IR spectra of the procatalysts were recorded by an FT-IR spectrometer in the range of 2000 - 1300 cm−1 (FT/IR 4100; JASCO Corporation). The specimen was prepared by the following method. The procatalyst was mixed with dried SiO2 as a binder (procatalyst/SiO2 = 2 by weight, total amount = 10 mg), and then molded into a 10 mm-φ wafer in a glovebox under a N2 atmosphere. The wafer was placed in a specially designed specimen holder and used for FT-IR measurement. The basal spacing of the procatalyst was determined by the X-ray diffraction (XRD) measurement (Ultima III; Rigaku Corporation) with the following operating conditions: X-ray = CuKα line (λ = 0.154 nm), scan rate = 1.0 degree・min−1, scan angle = 3 - 15 degree, voltage = 40 kV, and current = 40 mA. To avoid exposure to air during the XRD measurement, the well-mixed paste of the procatalyst and a small amount of dry liquid-paraffin were placed on a glass specimen holder and covered with a polyester film under a N2 atmosphere.
2.3. Ethylene Oligo-/Polymerization
The ethylene oligo-/polymerization was conducted using a 120-ml autoclave equipped with a magnetic stirrer. n-Heptane (50 ml), n-tridecane as the internal standard (0.5 ml, 10 vol.% in toluene), the prepared procatalyst (4 mg, theoretical amount of Ni complex = 1.6 μmol), and an alkylaluminum compound as the activator (480 μmol, Al/Nicomplex = 300) were successively added to the autoclave under a N2 atmosphere. The autoclave was placed in a water bath that was maintained at the reaction temperature (50˚C or 70˚C). Ethylene was continuously supplied with the ethylene pressure maintained at 0.7 MPa. After 1.5 h, 1.0 ml of a gas sample was extracted from the gas-phase in the autoclave and analyzed by a gas chromatograph equipped with a thermal conductivity detector (GC-8A; SHIMADZU Corporation) to determine the amount of C4 products in the gas-phase. The autoclave was cooled to 0˚C in an ice bath and the produced oligomers (C6-C20) were quantitatively determined by a gas chromatograph equipped with a flame ionization detector (GC-14A; SHIMADZU Corporation). The reaction was terminated by the addition of ethanol, and the produced solid polyethylene was recovered by filtration, dried, and weighed. During the reaction, the ethylene flow into the reactor was measured by a mass flow meter connected to the ethylene supply line. The activities in all the catalytic runs were determined by the total amount of the ethylene consumption.
3. Results and Discussion
3.1. Characterization of the Procatalysts
The physicochemical properties of the prepared Ni2+-mica were determined by an X-ray fluorescence (XRF) measurement as summarized in Table 1. The Ni2+ content and BET surface area of the prepared Ni2+-mica were 0.54 mmol・g-mica−1 and 3.11 m2・g−1, respectively. The Ln/Ni2+-micas were obtained as yellow-(L1-L3, L7-L9) or orange-colored (L4-L6) powders. No elution of the ligand and/or the metal complex was confirmed by measuring the 1H NMR spectrum of the supernatant solution of the Ln/Ni2+-mica slurry. This insolubility of the metal complex formed in the Ln/Ni2+-mica interlayers is derived from the strong interaction between the clay sheet having a negative charge and the nickel center.
The Ln/Ni2+-mica was characterized by FT-IR and X-ray diffraction (XRD) measurements. Figure 2 shows the FT-IR spectra of the L5/Ni2+-mica, L5-NiBr2, L5, and acenaphthenequinone/Ni2+-mica. The FT-IR spectrum of L5/Ni2+-mica shows an absorption band at 1626 cm−1 corresponding to the C=N stretching vibration. The L5-NiBr2 shows a similar absorption band at 1628 cm−1. In the spectrum of the ligand L5, the adsorption band derived from the C=N vibrations was observed at the higher frequencies around 1635 cm−1. The shift in the characteristic C=N vibrations in the L5/Ni2+-mica was caused by the effective coordination of the Ni2+ ions with the imino nitrogen atoms [18] [40] . Compared with the FT-IR spectrum of the acenaphthenequinone/Ni2+-mica, the formation of the ligand and its coordination to the Ni2+ center was confirmed by the disappearance of the adsorption bands at 1720 cm−1 corresponding to the C=O stretching of acenaphthenequinone in the spectrum of the L5/Ni2+-mica.
Mica-type clay minerals are constructed by stacking of 2:1 layers of which negative charge is compensated by interlayer cations [41] . The intercalation of the ligands into the interlayers causes an increase in the basal spacing (d001), resulting in the shift of the diffraction peak to a lower angle. Figure 3 shows the XRD profiles of the Ln/Ni2+-mica, the dried Ni2+-mica, and that treated with only the acetonitrile solventin the range of 3 - 13 degrees (diffraction angle, 2θ).

Table 1. Physicochemical properties of clay mineralsa.
aThe amount of fluorine was fixed at a constant value (2.48%). bDetermined by XRF. cAmount of exchanged Ni2+ ions = mmol・g-mica−1. dBET surface area determined by N2 adsorption.

Figure 2. FT-IR spectra of L5/Ni2+-mica, L5 NiBr2, L5, and acenaphthenequinone/Ni2+-mica.

Figure 3. XRD profiles of L1-L9/Ni2+-mica, the dried Ni2+- mica, and that treated with only the acetonitrile solvent.
The observation range of 2θ was determined with reference to the basal spacing of the anhydrous mica [30] - [36] [41] . The basal spacing of the dried Ni2+-mica was 1.01 nm corresponding to the thickness of a mica sheet. After treating it with acetonitrile, the distance between the adjacent layers expanded to 1.25 nm, indicating that acetonitrile was intercalated into the mica interlayer. The procatalysts L1-L9/Ni2+-mica showed the basil spacing values of 1.20 - 1.30 nm. The L2, L5, L7, and L9/Ni2+-micas containing one or two fluoro substituents exhibited a slightly smaller basil spacing than the other procatalysts, which were presumably due to the decreasing steric hindrance of the aryl groups. Interestingly, no clear relationship among the basal spacings was observed for the procatalysts having mono- or di-substituted aryl rings. Compared with the Ni2+- mica without any treatments, we observed expansion of the basal spacing for all the procatalysts. These results indicated that we could successfully prepare the Ln/Ni2+-mica by a one-pot preparation method.
3.2. Optimization of Oligo-/Polymerization Conditions
The procatalyst L5/Ni2+-mica was used for the ethylene oligo-/polymerization, and the parameters, such as an effective activator (co-catalyst) and Al/Ni molar ratio, were optimized due to the high catalytic activity. The ethylene oligo-/po- lymerization trials were conducted using four types of alkylaluminum compounds, such as poly(methylalumoxane) (PMAO), modified methylalumoxane (MMAO), triethylaluminum (TEA), and triisobutylaluminum (TIBA), with variations in the molar ratio of the Al and Ni complex (Al/Ni) from 300 to 900 at 50˚C under 0.7 MPa ethylene pressure.
The results are summarized in Table 2. The product shares (wt%) in the tables were calculated from the amounts of the C4-C20 oligomers, the solid product (polyethylene), and the total ethylene consumption. The commonly used activator in this field, PMAO, efficiently activated the procatalyst with high activities. On the other hand, when MMAO was used as the activator, we could not obtain any products. The L5/Ni2+-mica combined with TEA or TIBA exhibited a moderate activity toward the ethylene oligo-/polymerization at 50˚C. In view of the safety and industrial applications, readily available TEA and TIBA with a mild reactivity are better activators than MAO. Therefore, we used TEA and TIBA for further investigation. Variation in the Al/Ni ratio from 300 to 900 was conducted. When the Al/Ni ratio was 600, the highest catalytic activity of 656 g-ethylene・g-cat−1・h−1 was observed. The activity then slightly decreased with the higher Al/Ni ratio of 900. There were no obvious differences in the selectivity to the α-olefin. Due to the handling ability in the laboratory, we fixed the Al/Ni ratio at 300 for further investigation.
In this study, the catalyst activities were determined by the total amount of the
Table 2. Selection of a suitable reaction condition based on L5/Ni2+-mica procatalystsa.
aReaction conditions; temperature = 50˚C, time = 1.5 h, ethylene pressure = 0.7 MPa (gauge), solvent = n-heptane 50 ml. procatalyst = 4 mg (Entry 1-4), 2 mg (Entry 5 and 6). bActivity determined by total amount of ethylene consumption = g-ethylene・g-cat−1・h−1. c“Products” represents the product shares. d“C4-C20” represents the amount of hydrocarbons having carbon numbers 4 - 22. e“Solid” indicates the amount of solid part recovered by filtration of reaction mixture. fSelectivity to linear α-olefin determined by amounts of C4-C20 products).
ethylene consumption. About 70 - 100 wt% of the consumed ethylene was converted into the C4-C20 (oligomers) and the solid (polyethylene) products. The residual ethylene (~30 wt%) was consumed during the formation of the soluble product such as polyethylene wax having a moderate chain length.
3.3. Ethylene Oligomerization
The objective in this study is to clarify the influence of the substituted ligand structure on the catalyst performance with regard to the activity, the selectivity to the α-olefins, and the branching densities of the products. The Ln/Ni2+-mica procatalysts were used for the ethylene oligomerization at the Al/Ni ratio of 300 and 0.7 MPa ethylene pressure along with varying the activator and reaction temperature. The results are summarized in Table 3 (data obtained at 50˚C) and Table 4 (data obtained at 70˚C).
Table 3. Oligo-/polymerization of ethylene using Ln/Ni2+-mica catalysts at 50˚Ca.
aReaction conditions; temperature = 50˚C, time = 1.5 h, ethylene pressure = 0.7 MPa (gauge), solvent = n-heptane 50 ml, Al/Ni = 300, procatalyst = 4 mg. Abbreviations in the table are the same as those in Table 2. bThe share of the branched α-olefins in the oligomer products determined by GC. cReaction conditions; temperature = 50˚C, time = 1 h, ethylene pressure = 0.7 MPa (gauge), solvent = hexane 50 ml, procatalyst = 4 mg. dReaction conditions; temperature = 50˚C, time = 1 h, ethylene pressure = 0.7 MPa (gauge), solvent = n-heptane 50 ml, procatalyst = 4 mg.
Table 4. Oligo-/polymerization of ethylene using Ln/Ni2+-mica catalysts at 70˚Ca.
aReaction conditions; temperature = 70˚C, time = 1.5 h, ethylene pressure = 0.7 MPa (gauge), solvent = n-heptane 50 ml, Al/Ni = 300, procatalyst = 4 mg. Abbreviations in the table are the same as those in Table 3.
3.3.1. Effects of ortho-Substituent on Catalyst Properties
We first studied the effects of the ortho-substituents of the aryl rings on the performance of the catalyst (Table 3, Entries 7 - 12 and Table 4, Entries 23 - 28). At 50˚C, the L1-L3/Ni2+-mica with TIBA had catalysts activities in the range of 376 - 482 g-ethylene・g-cat−1・h−1. The activities of the L1/Ni2+-mica (2-CH3 as the substituent) were slightly higher than those of the L2 (2-F as the substituent) and L3/Ni2+-mica (2-Br as the substituent). Interestingly, though there were no significant differences between the activities of the L1-L3/Ni2+-mica, only the L2/Ni2+-mica exhibited a high selectivity to the α-olefin oligomers. The shares of the C4-C20 oligomers obtained by the L2/Ni2+-mica were 37.5 - 41.6 wt% at 50˚C.
Generally, the catalysts activities and the product distribution are dependent on both the steric bulk and the electronic effect. The sterically bulky ortho-subs- tituents of the aryl rings were considered to block the axial sites of the metal center. The mechanism of ethylene oligo/polymerization by the α-diimine nickel complex possessing ortho-substituents is represented in Scheme 1. During the chain propagation process, both the growing polymer chain and the coordinated ethylene monomer occupy the equatorial sites of the active nickel center. Meanwhile, the chain transfer process proceeds through the 5-coordinate transition state which undergoes the chain transfer with the incoming ethylene monomer from the axial sites of the nickel center [9] . The steric crowding at the axial sites is critical for suppressing the associative chain transfer process with the ethylene monomer at the axial site and results in the formation of a high molecular-weight polymer [12] [13] [14] . Thus, the products prepared by the L1 and L3/Ni2+-mica possessing relatively bulky ortho-substituents were composed
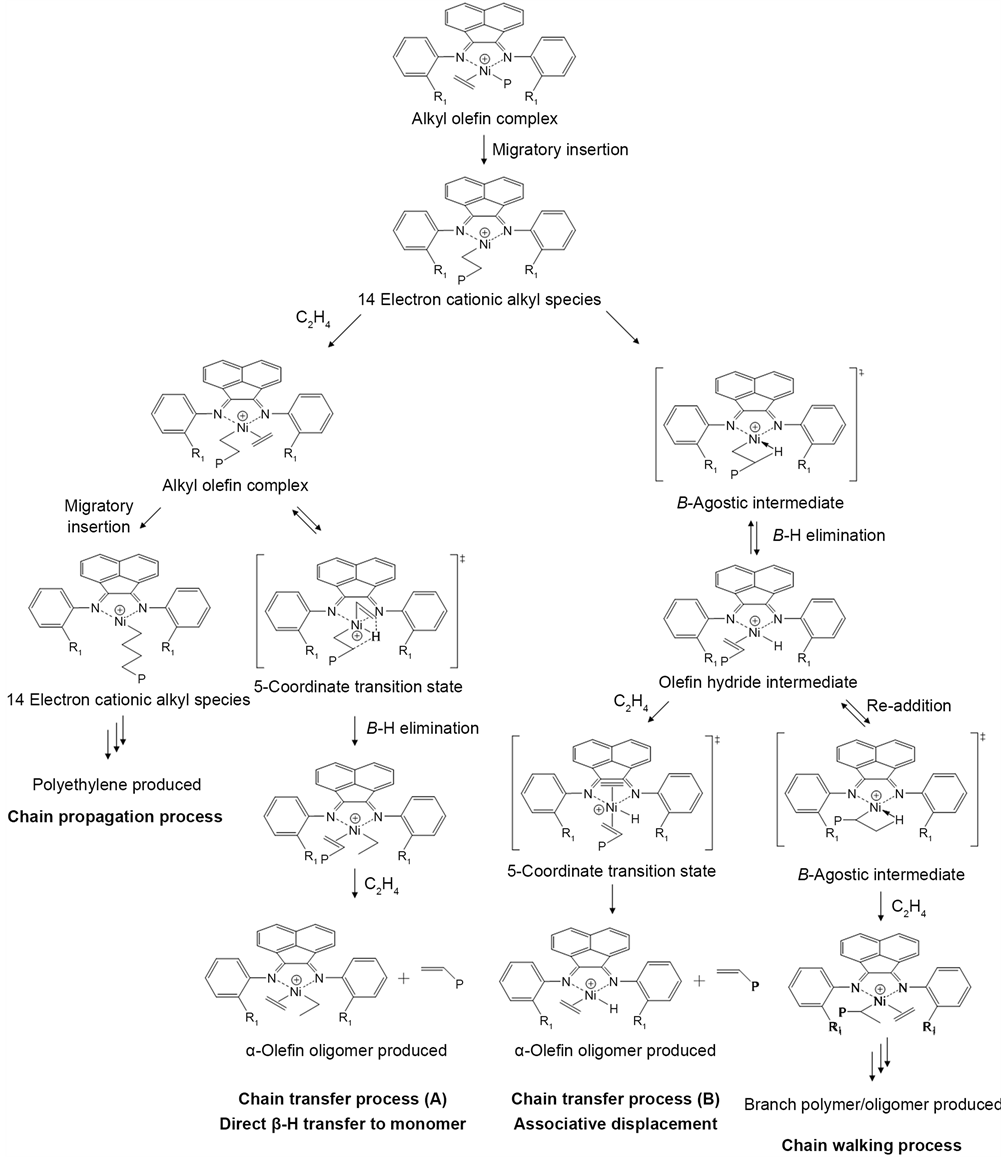
Scheme 1. Mechanism for ethylene oligo-/polymerization with α-diimine nickel complex.
of a large quantity of the solid product with a small amount of oligomers. In addition, the sterically bulky substituent was considered to increase the ground- state energy of the resting-state species [11] [13] . Thus, the larger methyl substituent of the L1/Ni2+-mica was assumed to cause destabilization of the ground state species, resulting in a higher catalyst activity than those of the L2 and L3/Ni2+-mica.
Compared with the hydrogen, the halogen substituents afforded both the steric effects and the electron-withdrawing effects on the active metal center. The introduction of the electron-withdrawing substituents increased the electrophilicity of the metal center. Recently, Zhang et al. calculated the net charge on the metal center in the bis(imino)pyridine iron complexes by the charge equilibration method [17] . The net charge value on the iron in the complexes with di-fluorine substituents was in the range of 0.7549 - 0.8310, which is lower than that of the alkyl-substituted complexes. The L2/Ni2+-mica with the ortho- fluorine substituent was active for both the ethylene polymerization and oligomerization. Meanwhile, the L3/Ni2+-mica with the ortho-bromine substituent showed a significantly lower oligomer selectivity and catalytic activity compared to the L2/Ni2+-mica. The electronegativities of the fluorine and bromine atoms are 4.0 and 2.8, while the covalent radii are 0.72 and 1.14 Å, respectively [16] . The strong electron-withdrawing effect of the fluorine substituent was considered to stabilize the active species and promote the coordination of the ethylene monomer to the nickel center, resulting in the high catalyst activity [42] [43] . Furthermore, the low steric bulk of the fluorine group in the L2/Ni2+-mica was insufficient for the accelerating ethylene insertion during the chain transfer process. This strong electron-withdrawing effect and the small size of the ortho- fluorine substituent produced both the high catalyst activity and the oligomer selectivity. A similar behavior was reported by Alt et al. [44] .
3.3.2. Effects of para-Substituent on Catalyst Properties
We next examined the influence of the para-substituents of the aryl rings on the catalyst properties (Table 3 and Table 4). At 50˚C, the L4-L6/Ni2+-micacombined with TIBA had catalytic activities in the range of 334 - 549 g-ethylene・g-cat−1・h−1. The L5/Ni2+-mica (4-F as the substituent) showed the highest activity and the L4/Ni2+-mica (4-CH3 as the substituent) showed the lowest activity among the catalysts with para-substituents. The L4/Ni2+-mica showed about a 30% lower activity than the L1/Ni2+-mica having ortho-methyl substituents. Meanwhile, the L5/Ni2+-mica showed about a 30% higher activity than the L2/Ni2+-mica, and the L6/Ni2+-mica (4-Br as the substituent) showed an activity similar to the L3/Ni2+- mica (2-Br as the substituent). The catalysts with para-halogen substituents exhibited higher activities than those of the ortho-halogen substituted catalysts.
The results indicated that the catalyst activities of the catalysts with para- substituents mainly depended on the electronic effects relative to those of the steric bulk. Alt et al. reported that the bis(arylimino)pyridine iron complexes with 4-halogen-2-methyl substituents showed much higher activities than the complexes with 2,4-dimethyl substituents [45] . The activities of their catalysts increased in the order from bromine-, chlorine-, iodine-, to fluorine-substituted complexes, which were in good agreement with the order of the electronegativities except for iodine. Thus, it is suggested that the electro-withdrawing para- halogen substituents in the L5 and L6/Ni2+-mica reduced the electron density of the cationic nickel center and enhance the coordination of the ethylene monomer to the nickel center, resulting in the increasing catalytic activity.
In terms of the oligomer selectivity, the L4-L6/Ni2+-mica showed a moderate selectivity to the α-olefins. The shares for the C4-C20 oligomers obtained by the L4-L6/Ni2+-mica were 19.9 - 27.6 wt% at 50˚C. Compared to the ligand having a sterically hindered ortho-substituent in the L1 and L3/Ni2+-mica, the less steric ligands in the L4-L6/Ni2+-mica was considered to affect the low suppression effect on the chain transfer process, resulting in the moderate oligomer selectivity. On the other hand, the L5/Ni2+-mica showed an oligomer selectivity lower than the L2/Ni2+-mica. This is presumably due to the prevention of the chain transfer process caused by the electron-withdrawing effect ofthe para-fluorine substituent. The L4/Ni2+-mica with the electro-donating methyl substituent showed slightly higher oligomer selectivity than the L5 and L6/Ni2+-mica. These results indicated that the para-halogen substituents led to an increase in the catalyst activity and slightly decreased the oligomer selectivity.
When the reaction temperature increased to 70˚C, the catalytic activity decreased and the oligomer selectivity increased in all the catalytic runs. For example, the oligomer share obtained by the L4/Ni2+-mica with TIBA increased from 27.6 to 41.7 wt% as the reaction temperature increased from 50˚C to 70˚C. The higher reaction temperature was considered to enhance the rotation of the aryl group in the complex, and thus, the chain transfer rate increased [21] [43] . Also, the branch rates (represents by the share of the branched α-olefins in the oligomer products determined by GC) tended to slightly increase with the increasing reaction temperature. For example, the branch rates of the L5/Ni2+-mica with TEA increased from 4 to 9 mol% as the reaction temperature increased from 50˚C to 70˚C. The branch rates of the oligomer products depended on the rate of the “chain growth” and “chain walking” which competitively proceeded [3] [11] .
In all the runs, our procatalysts tended to exhibit a lower oligomer selectivity in comparison to the previous reports based on the homogeneous catalysts. For example, Alt et al. reported that the homogeneous α-diimine nickel catalysts with halogen substituents afforded a suitable selectivity to ethylene oligomers with a high activity [44] . The shares for the oligomers obtained by their catalysts with para-halogen substituents were almost 100 wt%, and even the ortho- bromine-substituted catalyst showed a 76 wt% oligomer selectivity. Compared to these homogeneous catalyst systems, the nickel complexes in our procatalyst were immobilized into the mica interlayer. Thus, it was suggested that Ln/Ni2+- mica had less free space at the axial position of the nickel complex than the homogeneous catalyst due to the existence of the mica sheets near the nickel complexes. The narrow space around the nickel center was assumed to prevent the ethylene insertion from the axial site and suppress the chain transfer process, resulting in the lower oligomer selectivity. In addition, the bulky acenaphthene backbone of our procatalysts was considered to afford the lower oligomer selectivity. In a previous study using the Ni2+-mica procatalysts having α-diimine- type ligands, the catalyst with fluorine groups at the ortho-position of the aryl rings showed a higher selectivity to α-olefins with the value of 96.5 wt% at 60˚C in comparison to those of the L2/Ni2+-mica [33] . These results indicated that the Ln/Ni2+-mica having an acenaphthene backbone were preferable to forming polyethylene rather than the oligomers.
3.3.3. Effects of di-Substituent on Catalyst Properties
A striking feature observed in the data is that the procatalysts with di-halogen substituents on the aryl rings did not show any significant activity (Table 3). In a previous study with the Ni2+-mica-based procatalyst having the acenaphthene backbone, the high molecular weight polyethylenes (Mn = 3.90 - 4.76 × 104) were obtained with a sufficient catalytic activity (170 g-ethylene・g-cat−1・h−1 at 50˚C) when two methyl substituents were introduced at both ortho-positions on the aryl rings [35] . Thus, the significant decrease in the catalytic activities was due to the introduction of the two electron-withdrawing groups on the aryl rings. The introduction of an excess amount of the electron-withdrawing groups enhanced the interaction between the nickel center and the coordinated ethylene, and the strong interaction prevented the ethylene insertion into the metal-alkyl bond, resulting in the active species being more easily decomposed [46] .
3.3.4. Properties of Oligomer Products
To investigate the distribution of the oligomer, the molar fraction of each product was measured by a gas chromatograph, as shown in Table 5. The oligomers obtained using the L2, L4, L5, and L6/Ni2+-micas, which produced a sufficient yield of the oligomers, exhibited a Schulz-Flory distribution. The natural logarithm (ln) of the mole fraction of the oligomer in the product exhibited a linear decrease with the increasing carbon number of the oligomers, as shown in Figure 4. The mole fraction of the oligomers was at a maximum for C4 in all the runs, and the mole fraction of C20 was in the range of 0.6% - 2.2%. The content of C4 obtained by the L5/Ni2+-mica increased from 35% to 43% as the reaction

Figure 4. Schulz-Flory distributions of oligo-/polymerization using L2, L4, L5, and L6/Ni2+-micas in the presence of TIBA at 50˚C.
Table 5. Oligomer distribution of ethylene oligo-/polymerization in the presence of TIBA.
aSchulz-Flory constant α, αn = Cn+2 (mol)/Cn (mol).
temperature was elevated from 50˚C to 70˚C, indicating that the β-hydrogen elimination reaction, which is the major chain-transfer reaction and gives an unsaturated chain end, favorably occurredat the higher reaction temperature in comparison to the chain propagation reaction.
The probability of the chain propagation can be described by the Schulz-Flory constant α [12] . A higher α value directly corresponds to an increase in the chain propagation probability, resulting in the formation of higher molecular-weight products. The α12 values calculated from the mole fractions of C12 and C14 were within 0.61 - 0.78 in all the runs. At 70˚C, the L5/Ni2+-mica had a higher α12 value than that obtained at 50˚C due to the dominant chain transfer as already mentioned.
In terms of the ligand structure, the α12 values increased from the L2, L5, L4 to L6/Ni2+-micas, which was in the reverse order for both the electron-withdrawing ability and the steric bulk of the substituents on the aryl rings. For example, the L2/Ni2+-mica containing the strong electron-withdrawing fluorine groups at the ortho-positions showed the lowest α12 values of 0.61. The highest α12 values were observed for the L6/Ni2+-mica which contains the weaker electron-withdrawing bromine groups at the para-positions. Moreover, the lower α12 values tended to afford higher yields of the oligomers. These results indicated that the introduction of fluorine groups, especially at the ortho-positions of the aryl rings, led to the increasing rate of the chain transfer reaction.
Noteworthy, in the oligomers formed by the L2 and L5/Ni2+-micas, two different values of the chain propagation probability α were observed. The L2 and L5/Ni2+-micas afforded significantly lower α18 values than the α18 values, while the α18 values obtained by the L4 and L6/Ni2+-micas showed similar α values. For example, the α18 value obtained by the L5/Ni2+-mica with TEA at 50˚C was 0.37, which was about half the value of α12. These phenomena could be interpreted by the existence of active centers having different structures [43] .
3.3.5. Effects of Activator and Temperature on Catalyst Lifetimes
One of the interesting characteristics of our heterogeneous procatalyst systems was the long lifetime of the active species during the reaction in the presence of TIBA. The catalyst lifetimes of the L1-L6/Ni2+-mica with activators were investigated by measuring the ethylene consumption profiles by a mass flowmeter. Figure 5(a) shows the consumption profiles of ethylene during the oligo-/poly- merization using the L1-L6/Ni2+-mica in the presence of TIBA at 50˚C. The ethylene consumption during the initial 5 - 10 minutes was attributed to the process of ethylene saturation of the n-heptane solvent. Under this reaction condition, all the catalysts showed similar behaviors of the ethylene consumption, i.e., a long induction period was observed in the first 20 minutes, and then the ethylene consumption rate gradually increased with the reaction time. After reaching the maximum point of the ethylene consumption rate, it slowly decreased due to deactivation of the active species. The maximum ethylene-consumption rates obtained by the L1-L6/Ni2+-mica were in the range of 41.2 - 76.7 ml・min−1

Figure 5. Profiles of ethylene consumption during oligo-/polymerization using (a) L1-L6/ Ni2+-mica in the presence of TIBA activator at 50˚C and (b) L2/Ni2+-mica in the presence of TEA or TIBA at 50˚C or 70˚C.
(as standard temperature and pressure), and the periods for the full activation of the procatalysts required 52 - 72 minutes after supplying the ethylene.
The effects of the activator and the temperature on the profile were investigated. Figure 5(b) shows the ethylene flow profiles during the oligomerization using the L2/Ni2+-mica at the various reaction conditions. When we used TEA at 50˚C, the activation period shortened and the maximum ethylene consumption rate decreased in comparison to those using TIBA, resulting in the decreasing catalytic activity as shown in Table 2. The slow activation of the procatalysts by TIBA was presumably due to the lower Lewis acidity of TIBA which indicates a weaker reactivity than TEA. At 70˚C, the lifetimes of the L2/Ni2+-mica in the presence of both TEA and TIBA became shorter than those observed at 50˚C. The maximum consumption rates observed by the L2/Ni2+-mica were similar at both 50˚C and 70˚C. These results indicated that about 30% - 80% lower yields of the product at 70˚C were mainly due to the shorter lifetimes of the active species in comparison to those obtained at 50˚C.
4. Conclusion
We synthesized nine types of novel heterogeneous procatalyst Ln/Ni2+-micas with a series of substituted ligand structures via a one-pot preparation method. The basal spacings of the obtained L1-L9/Ni2+-mica were in the range of 1.20 - 1.30 nm, indicating the immobilization of the Ni complex into the mica interlayers. In the presence of the TEA or TIBA, the L1-L6/Ni2+-mica, which has one ortho- or para-substituent on the aryl ring, was active for the ethylene oligo-/ polymerization reaction, while the L7-L9/Ni2+-mica having two halogen substituents were inactive. The catalyst activity and oligomer selectivity of the procatalysts depended on the position, steric bulk, and electronegativity of the substituents on the aryl rings. The strong electron-withdrawing groups, the ortho- fluorine and para-fluorine substituents, increased the catalyst activity due to increased electrophilicity of the nickel center. The decreasing steric bulk of the ortho-substituent increased the selectivity to the oligomer fraction. Also, the procatalysts having one para-substituent afforded moderate yields of the oligomers.
In conclusion, our heterogeneous procatalyst system exhibited a moderate catalyst activity with a long lifetime as well as that of the conventional homogeneous system. In this system, the substituent and the backbone in the ligand structures are easily controlled by changing the structures of the aniline derivatives and the ketone derivatives, which are the reactants of the one-pot preparation method. Therefore, we can control the activity and the product selectivity by tuning the ligand structure. Our findings suggest that a complex with more free space at the axial position of the nickel center is necessary for increasing the oligomer selectivity. The less bulky structure of the ligand backbone is also considered to enhance the oligomer production.
Acknowledgements
This work was financially supported by theJSPS KAKENHI Grant Number 15K05514. We acknowledge Masa-aki Oshima for use of FT/IR 4100 spectrometer with the original equipment.
Cite this paper
Yoshida-Hirahara, M., Fujiwara, S. and Kurokawa, H. (2017) Simple Preparation of Halogen-Substituted α-Diimine Nickel Complexes Immobilized into Clay Interlayer as Catalysts for Ethylene Oligo-/Polymerization. Modern Research in Catalysis, 6, 100-120. https://doi.org/10.4236/mrc.2017.62008
References
- 1. Johnson, L.K., Killian, C.M. and Brookhart, M. (1995) New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and α-Olefins. Journal of the American Chemical Society, 117, 6414-6415.
https://doi.org/10.1021/ja00128a054 - 2. Britovsek, G.J.P., Gibson, V.C. and Wass, D.F. (1999) The Search for New-Generation Olefin Polymerization Catalysts: Life beyond Metallocenes. Angewandte Chemie, International Edition, 38, 428-447.
https://doi.org/10.1002/(SICI)1521-3773(19990215)38:4<428::AID-ANIE428>3.0.CO;2-3 - 3. Guan, Z., Cotts, P.M., McCord, E.F. and McLain, S.J. (1999) Chain Walking: A New Strategy to Control Polymer Topology. Science, 283, 2059-2062.
https://doi.org/10.1126/science.283.5410.2059 - 4. Leatherman, M.D., Svejda, S.A., Johnson, L.K. and Brookhart, M. (2003) Mechanistic Studies of Nickel(II) Alkyl Agostic Cations and Alkyl Ethylene Complexes: Investigations of Chain Propagation and Isomerization in (α-diimine)Ni(II)-Catalyzed Ethylene Polymerization. Journal of the American Chemical Society, 125, 3068-3081.
https://doi.org/10.1021/ja021071w - 5. Rojas, R.S., Barrera G.G., Wu, G. and Bazan, G.C. (2007) Single-Component α-Iminocarboxamide Nickel Ethylene Polymerization and Copolymerization Initiators. Organometallics, 26, 5339-5345.
https://doi.org/10.1021/om070155g - 6. Yu, J., Zeng, Y., Huang, W., Hao, X. and Sun, W-H. (2011) N-(5,6,7-Trihydroquinolin-8-ylidene)arylaminonickel Dichlorides as Highly Active Single-Site Pro-Catalysts in Ethylene Polymerization. Dalton Transactions, 40, 8436-8443.
https://doi.org/10.1039/c1dt10541h - 7. Weberski, M.P., Chen, C., Delferro, M., Zuccaccia, C., Macchioni, A. and Marks, T.J. (2012) Suppression of β-Hydride Chain Transfer in Nickel(II)-Catalyzed Ethylene Polymerization via Weak Fluorocarbon Ligand-Product Interactions. Organometallics, 31, 3773-3789.
https://doi.org/10.1021/om3002735 - 8. Wang, S., Sun, W.-H. and Redshaw, C. (2014) Recent Progress on Nickel-Based Systems for Ethylene Oligo-/Polymerization Catalysis. Journal of Organometallic Chemistry, 751, 717-741.
- 9. Camacho, D.H. and Guan, Z. (2010) Designing Late-Transition Metal Catalysts for Olefin Insertion Polymerization and Copolymerization. Chemical Communications, 46, 7879-7893.
https://doi.org/10.1039/c0cc01535k - 10. Mu, H., Pan, L., Song, D. and Li, Y. (2015) Neutral Nickel Catalysts for Olefin Homo- and Copolymerization: Relationships between Catalyst Structures and Catalytic Properties. Chemical Reviews, 115, 12091-12137.
https://doi.org/10.1021/cr500370f - 11. Guo, L., Dai, S., Sui, X. and Chen, C. (2016) Palladium and Nickel Catalyzed Chain Walking Olefin Polymerization and Copolymerization. ACS Catalysis, 6, 428-441.
https://doi.org/10.1021/acscatal.5b02426 - 12. Killian, C.M., Johnson, L.K. and Brookhart, M. (1997) Preparation of Linear α-Olefins Using Cationic Nickel(II) α-Diimine Catalysts. Organometallics, 16, 2005-2007.
https://doi.org/10.1021/om961057q - 13. Ittel, S.D., Johnson, L.K. and Brookhart, M. (2000) Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chemical Reviews, 100, 1169-1203.
https://doi.org/10.1021/cr9804644 - 14. Britovsek, G.J.P., Mastroianni, S., Solan, G.A., Baugh, S.P.D., Redshaw, C., Gibson, V.C., White, A.J.P., Williams, D.J. and Elsegood, M.R.J. (2000) Oligomerisation of Ethylene by Bis(imino)pyridyliron and -Cobalt Complexes. Chemistry—A European Journal, 6, 2221-2231.
https://doi.org/10.1002/1521-3765(20000616)6:12<2221::AID-CHEM2221>3.0.CO;2-U - 15. Chen, Y., Qian, C. and Sun, J. (2003) Fluoro-Substituted 2,6-Bis(imino)pyridyl Iron and Cobalt Complexes: High-Activity Ethylene Oligomerization Catalysts. Organometallics, 22, 1231-1236.
https://doi.org/10.1021/om020818o - 16. Chen, Y., Chen, R., Qian, C., Dong, X. and Sun, J. (2003) Halogen-Substituted 2,6-Bis(imino)pyridyl Iron and Cobalt Complexes: Highly Active Catalysts for Polymerization and Oligomerization of Ethylene. Organometallics, 22, 4312-4321.
https://doi.org/10.1021/om0302894 - 17. Zhang, T., Sun, W-H., Li, T. and Yang, X. (2004) Influence of Electronic Effect on Catalytic Activity of Bis(imino)pyridyl Fe(II) and Bis(imino)pyrimidyl Fe(II) Complexes. Journal of Molecular Catalysis A: Chemical, 218, 119-124.
- 18. Zhang, Z., Chen, S., Zhang, X., Li, H., Ke, Y., Lu, Y. and Hu, Y. (2005) A Series of Novel 2,6-bis(imino)pyridyl Iron Catalysts: Synthesis, Characterization and Ethylene Oligomerization. Journal of Molecular Catalysis A: Chemical, 230, 1-8.
- 19. Popeney, C.S. and Guan, Z. (2010) Effect of Ligand Electronics on the Stability and Chain Transfer Rates of Substituted Pd(II) α-Diimine Catalysts. Macromolecules, 43, 4091-4097.
https://doi.org/10.1021/ma100220n - 20. Guo, L., Gao, H., Guan, Q., Hu, H., Deng, J., Liu, J., Liu, F. and Wu, Q. (2012) Substituent Effects of the Backbone in α-Diimine Palladium Catalysts on Homo- and Copolymerization of Ethylene with Methyl Acrylate. Organometallics, 31, 6054- 6062.
https://doi.org/10.1021/om300380b - 21. Liu, J., Chen, D., Wu, H., Xiao, Z., Gao, H., Zhu, F. and Wu, Q. (2014) Polymerization of α-Olefins Using a Camphyl α-Diimine Nickel Catalyst at Elevated Temperature. Macromolecules, 47, 3325-3331.
https://doi.org/10.1021/ma5004634 - 22. Zou, W. and Chen, C. (2016) Influence of Backbone Substituents on the Ethylene (Co)polymerization Properties of α-diimine Pd(II) and Ni(II) Catalysts. Organometallics, 35, 1794-1801.
https://doi.org/10.1021/acs.organomet.6b00202 - 23. Rossetto, E., Caovilla, M., Thiele, D., de Souza, R.F. and Bernardo-Gusmao, K. (2013) Ethylene Oligomerization Using Nickel-β-diimine Hybrid Xerogels Produced by the Sol-Gel Process. Applied Catalysis A: General, 454, 152-159.
- 24. Comito, R.J., Fritzsching, K.J., Sundell, B.J., Schmidt-Rohr, K. and Dinca, M. (2016) Single-Site Heterogeneous Catalysts for Olefin Polymerization Enabled by Cation Exchange in a Metal-Organic Framework. Journal of the American Chemical Society, 138, 10232-10237.
https://doi.org/10.1021/jacs.6b05200 - 25. Preishuber-Pflugl, P. and Brookhart, M. (2002) Highly Active Supported Nickel Diimine Catalysts for Polymerization of Ethylene. Macromolecules, 35, 6074-6076.
https://doi.org/10.1021/ma020230t - 26. Wegner, M.M., Ott, A.K. and Rieger, B. (2010) Gas Phase Polymerization of Ethylene with Supported α-Diimine Nickel(II) Catalysts. Macromolecules, 43, 3624- 3633.
https://doi.org/10.1021/ma9025256 - 27. Okada, M., Nakayama, Y. and Shiono, T. (2014) Heterogenization of an Anilinonaphthoquinone-Chelated Nickel Complex for Ethylene Polymerization. Macromolecular Chemistry and Physics, 215, 1792-1796.
- 28. Semikolenova, N.V., Zhang, W., Zakharov, V.A., Bryliakov, K.P. and Sun, W.-H. (2015) Ethylene Polymerization with Homogeneous and Heterogeneous Catalysts Based on Bis(4-fluorophenyl)methyl-Substituted Bis(imino)pyridyliron Complexes. Journal of Applied Polymer Science, 132, Article No. 42674.
https://doi.org/10.1002/app.42674 - 29. Andrei, R.D., Popa, M.I., Cammarano, C. and Hulea, V. (2016) Nickel and Molybdenum Containing Mesoporous Catalysts for Ethylene Oligomerization and Metathesis. New Journal of Chemistry, 40, 4146-4152.
https://doi.org/10.1039/C5NJ02586A - 30. Kurokawa, H., Matsuda, M., Fujii, K., Ishihama, Y., Sakuragi, T., Ohshima, M.-A. and Miura, H. (2007) Bis(imino)pyridine Iron and Cobalt Complexes Immobilized into Interlayer Space of Fluorotetrasilicic Mica: Highly Active Heterogeneous Catalysts for Polymerization of Ethylene. Chemistry Letters, 36, 1004-1005.
https://doi.org/10.1246/cl.2007.1004 - 31. Fujii, K., Ishihama, Y., Sakuragi, T., Ohshima, M-A., Kurokawa, H. and Miura, H. (2008) Heterogeneous Catalysts Immobilizing α-Diimine Nickel Complexes into Fluorotetrasilicic Mica Interlayers to Prepare Branched Polyethylene from Only Ethylene. Catalysis Communications, 10, 183-186.
- 32. Kurokawa, H., Miura, K., Yamamoto, K., Sakuragi, T., Sugiyama, T., Ohshima, M.-A. and Miura, H. (2013) Oligomerization of Ethylene to Produce Linear α-Olefins Using Heterogeneous Catalyst Prepared by Immobilization of α-Diiminenickel(II) Complex into Fluorotetrasilicic Mica Interlayer. Catalysts, 3, 125-136.
https://doi.org/10.3390/catal3010125 - 33. Kurokawa, H., Ogawa, R., Yamamoto, K., Sakuragi, T., Ohshima, M.-A. and Miura, H. (2014)Nickel(II) Complex Bearing Fluorine-Substituted α-Diimine Ligand Immobilized in Fluorotetrasilicic Mica Interlayer as Heterogeneous Catalysts for Ethylene Oligomerization. Journal of the Japan Petroleum Institute, 57, 146-154.
https://doi.org/10.1627/jpi.57.146 - 34. Kurokawa, H., Ishikawa, S., Yamamoto, K., Sakuragi, T., Ohshima, M.-A. and Miura, H. (2014) Ethylene Oligomerization Using Bis(imino)pyridinecobalt(II) Complexes Immobilized in Fluorotetrasilicic Mica Interlayer as Heterogeneous Catalysts. Chemistry Letters, 43, 1365-1367.
https://doi.org/10.1246/cl.140319 - 35. Kurokawa, H., Hayasaka, M., Yamamoto, K., Sakuragi, T., Ohshima, M.-A. and Miura, H. (2014) Self-Assembled Heterogeneous Late Transition-Metal Catalysts for Ethylene Polymerization; New Approach to Simple Preparation of Iron and Nickel Complexes Immobilized in Clay Mineral Interlayer. Catalysis Communications, 47, 13-17.
- 36. Yamanaka, H., Yamamoto, K., Sakuragi, T., Ohshima, M.-A., Nagashima, S., Kurokawa, H. and Miura, H. (2016)Ethylene Oligomerization Using Quinoline-Imine Nickel(II) Complex Immobilized in Fluorotetrasilicic Mica Interlayer by One-Pot Preparation Method. Journal of Molecular Catalysis A: Chemical, 425, 275-282.
- 37. Yu, J., Hu, X., Zeng, Y., Zhang, L., Ni, C., Hao, X. and Sun, W.-H. (2011) Synthesis, Characterization and Ethylene Oligomerization Behaviour of N-(2-substituted- 5,6,7-trihydroquinolin-8-ylidene)arylaminonickel Dichlorides. New Journal of Chemistry, 35, 178-183.
https://doi.org/10.1039/C0NJ00516A - 38. Svejda, S.A. and Brookhart, M. (1999) Ethylene Oligomerization and Propylene Dimerization Using Cationic (α-Diimine)nickel(II) Catalysts. Organometallics, 18, 65-74.
https://doi.org/10.1021/om980736t - 39. Fan, L., Yue, E., Du, S., Guo, C.-Y., Hao, X. and Sun, W-H. (2015) Enhancing Thermo-Stability to Ethylene Polymerization: Synthesis, Characterization and the Catalytic Behavior of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacena-phthylnickel Halides. RSC Advances, 5, 93274-93282.
https://doi.org/10.1039/C5RA18257C - 40. Yue, E., Zhang, L., Xing, Q., Cao, X.-P., Hao, X., Redshaw, C. and Sun, W.-H. (2014) 2-(1-(2-Benzhydrylnaphthylimino)ethyl)pyridylnickel Halides: Synthesis, Characterization, and Ethylene Polymerization Behavior. Dalton Transactions, 43, 423-431.
https://doi.org/10.1039/C3DT52234B - 41. Kim, J.H., Kim, H.-J. and Park, M. (2014) Stabilization of Alkali Earth Metal Cations in Na-4-Mica. Applied Clay Science, 101, 272-276.
- 42. Gates, D.P., Svejda, S.A., Onate, E., Killian, C.M., Johnson, L.K., White, P.S. and Brookhart, M. (2000) Synthesis of Branched Polyethylene Using (α-Diimine)nickel (II) Catalysts: Influence of Temperature, Ethylene Pressure, and Ligand Structure on Polymer Properties. Macromolecules, 33, 2320-2334.
https://doi.org/10.1021/ma991234+ - 43. Huang, Z., Song, K., Liu, F., Long, J., Hu, H., Gao, H. and Wu, Q. (2008) Synthesis and Characterization of a Series of 2-Aminopyridine Nickel(II) Complexes and Their Catalytic Properties toward Ethylene Polymerization. Journal of Polymer Science, Part A: Polymer Chemistry, 46, 1618-1628.
https://doi.org/10.1002/pola.22499 - 44. Helldorfer, M., Backhaus, J., Milius, W. and Alt, H.G. (2003) (α-Diimine)nickel(II) Complexes Containing Chloro Substituted Ligands as Catalyst Precursors for the Oligomerization and Polymerization of Ethylene. Journal of Molecular Catalysis A: Chemical, 193, 59-70.
- 45. Goerl, C. and Alt, H.G. (2007) Influence of the Para-Substitution in Bis(arylimino) Pyridine Iron Complexes on the Catalytic Oligomerization and Polymerization of Ethylene. Journal of Organometallic Chemistry, 692, 4580-4592.
- 46. Xie, G., Li, T. and Zhang, A. (2010) Highly Active and Selective Ethylene Oligomerization Catalysts: Asymmetric 2,6-bis(imino)pyridyl Iron (II) Complexes with Alkyl and Halogen Substitutients. Inorganic Chemistry Communications, 13, 1199- 1202.