Advances in Alzheimer's Disease
Vol.2 No.1(2013), Article ID:28624,18 pages DOI:10.4236/aad.2013.21003
Biomarkers for the early diagnosis of Alzheimer’s disease: The challenge of XXI century
![]()
Department of Pharmacy, University of Bari, Bari, Italy; *Corresponding Author: marialessandra.contino@uniba.it
Received 25 October 2012; revised 1 December 2012; accepted 12 December 2012
Keywords: Alzheimer’s Disease; Early Diagnosis; Genetic Biomarkers; Non Genetic Biomarkers; PET Probes
ABSTRACT
AD is the most common form of dementia among the aging population. The neuropathological alterations of AD are represented by the neurofibrillary tangles and extracellular amyloid plaques formation. These two hallmarks are routinely used as biomarkers for AD diagnosis and can allow the identification of the pathology in a late phase. The urgent need to develop probes for PET analysis that can be used in an early diagnosis of this disorder opened a new scenario in which new biomarkers involved in the first step of AD can be easily detected. Recently, an increasing number of studies indicated as new biomarkers P-gp, TLR4, MIR and free serum copper that are involved in the onset of AD. It has been extensively reported that a P-gp dysfunction in brain can be considered one of the causes of the AD accumulation in brain parenchyma and that the up-regulation of inflammatory gene expression and inflammatory signaling due to MIR and TLR4 modulated the development and the progression of AD.
1. INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by irreversible cognitive decline, memory impairment, and behavioral changes. Studies performed postmortem on AD patients’ brains showed, as neuropathological features, the presence of senile plaques (SPs) and neurofibrillary tangles (NFTs), characterized by β-amyloid (Aβ) peptides and highly phosphorylated tau proteins [1]. Indeed, the “Aβ hypothesis” suggests that the development of AD is due to the accumulation and deposition of Aβ peptide aggregates in the brain. The amyloid precursor peptide (APP) is degraded by three different enzymes (α-, β-, and γ-secretases) as depicted in Figure 1 and an imbalance between the production and the clearance of Aβ40-42 fragments by β- and γ-secretases leads to the accumulation of Aβ peptide monomers, oligomers, and finally of Aβ plaques that “gum up” the parenchymal space between neurons in the brain. Currently, since the definitive confirmation of AD is on the base of postmortem histopathologic examination of Aβ deposits in the brain, Aβ plaques are useful biomarkers for the AD diagnosis, and their detection in vivo by positron-emission tomography (PET) or single-photon emission computed tomography (SPECT) should ameliorate diagnosis and improve the discovery of new therapeutic agents. To date, FDA approved five medications for AD therapy (donepezil, rivastigmine, galantamine, tacrine, and memantine) but these agents are only useful to treat the symptoms and not to stop the disease progression. Therefore, to remove and prevent excess build up of Aβ peptides in the brain (β- and γ-secretases inhibitors) can be suggested as a possible goal in AD therapy even if the results of clinical trials in this area have been disappointing. Moreover, also in the diagnostic field, since the used probes are able to target the Aβ plaques, they do not allow an early diagnosis.
2. EPIDEMIOLOGY
The current estimated prevalence in the United States is 5.3 million individuals, 5.1 million of whom are over 65 years [2]. The global prevalence of the disease is estimated around 24 million in 2001, and predicted to double every 20 years, affecting more than 80 million people by 2040 [3]. The number of patients will rise from 2001 to 2040 by 80% - 190% in Europe, North America and the developed Western Pacific region, while in Latin America, India, China, North Africa and the Middle Eastern Crescent more than 300%. Incidence rates between approximately 0.4% in people aged 65 - 69 years
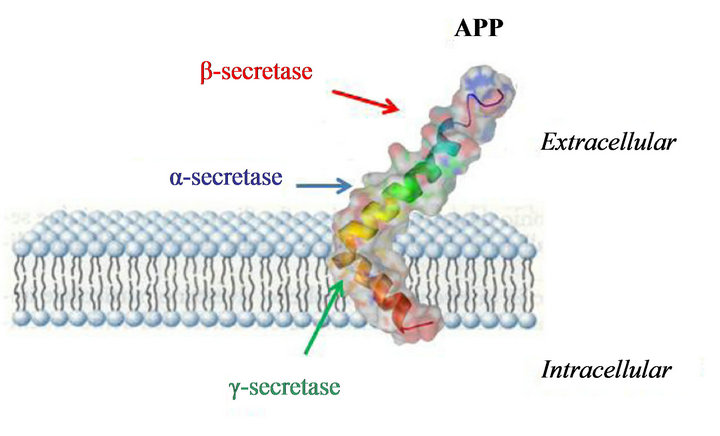
Figure 1. Amyloid precursor peptide (APP) degraded by three different enzymes (α-, β-, and γ-secretases).
up to 10% in people aged over 90 years have been reported, and a prevalence between approximately 2% in people aged 65 - 69 years up to more than 25% in the group aged over 90 years [3]. The prevalence of AD is greater in women than men because of the greater life expectancy of women. The economic impact of dementia was $172 billion, including $123 billion for Medicare and Medicaid. By 2050, the estimated cost of AD in the United States will exceed $1 trillion per year. The total cost from 2011 through 2050 is estimated at $20 trillion [2]. This health problem is not limited to the United States but estimates of global costs of AD in 2009 were $604 billion per year. All these alarming data indicate that AD could be considered as a modern epidemy. Therefore an urgent need for preventive, diagnostic and therapeutic measures exists in order to control the impact of this devastating disorder.
3. PATHOPHYSIOLOGY AND CLINICAL MANIFESTATION
AD pathophysiology related processes and mechanisms begin several decades before the clinical onset (late stage) of the disease. For several years, the Aβ plaques and NFTs have been identified and largely studied as the two neuropathological hallmarks of the disorder [1]. NFTs are intracellular aggregates, while Aβ plaques aggregated in the extracellular space. Other pathologic features are represented by the periplaque inflammatory reaction and neurodegeneration (synapse loss, neuron loss, and cerebral atrophy) [2]. The neurofibrillary pathologic process begins in the transentorhinal area and progresses to the hippocampus, to paralimbic and adjacent medial-basal temporal cortex, to neocortical association areas, and then to primary sensorimotor and visual areas [2]. Tau protein is a component of the cytoskeletal microtubule system and in AD onset it becomes hyperphosphorylated, disassociates from microtubules, assumes a paired helical filament configuration, leading to insoluble NFTs in the neurons. The Aβ
plaques, second hallmark of the disorder, are made up by a dense central Aβ core with inflammatory cells and dystrophic neurites in its periphery. Normal proteolytic metabolism of amyloid precursor protein by secretases enzymes leads to several peptide fragments and, among them, Aβ40 (more abundant) and Aβ42 (more prone to aggregate) fragments are the most important [2]. The limitation of these biomarkers is that AD can be confirmed with certainty only postmortem, and a clinical diagnosis is usually possible a long time after the onset of the pathology. An early detection can be considered pivotal for the prevention and the therapy of AD. To date several efforts have been aimed to validate and qualify biomarkers at the prodromal stages of the disease with the attempt to allow an early prediction, diagnosis and treatment, and monitor the disease progression and treatment response [3-5]. The prodromal stage of AD is the phase when pathophysiological alterations are developing with beginning mild (symptomatic) clinically significant cognitive impairment. The clinically silent asymptomatic stage before the prodromal AD, is studied by pathophysiological mechanistic indicators such as the Aβ in CSF and/or its brain increase by PET analysis and by particular risk factors such as genetic predisposition, defined either by genetic determinants (e.g. APP), and/or by genetic susceptibility factors (APOE-ε4) [6]. The subsequent neurodegenerative stage with reduced reversibility is characterized by disintegration of white matter fiber tracts (microstructural changes), and clinically, by the first mild cognitive symptoms (prodromal phase). The late neurodegenerative (dementia) stage shows progressive macrostructural volume loss (i.e. regional brain atrophy on magnetic resonance imaging, MRI), and progressive clinical dementia syndrome [7]. The clinical manifestations of AD useful to monitor the progression of the disease could be classified into three progressive steps [8]: a) recent memory loss, mild coordination problems, depression and apathy; b) pervasive and persistent memory loss (forgetfulness about personal history and inability to recognize friends and family), sleep disturbances, changes in mood and behavior, rambling speech, unusual reasoning, and confusion about current events, time, and place. Mobility and coordination is affected by slowness, rigidity, and tremors; c) severe or total loss of ability to remember and/or to communicate, immobility and extreme problems with mood, behavior, hallucinations, and delirium. The pathophysiological alterations associated with clinical AD manifestations are: inflammation, oxidative stress, protein misfolding, changes in brain microenvironment and metabolism, and neurodegeneration. These alterations have been characterized at the level of gene expression (transcriptomic), proteins (proteomic), and lipid signaling (lipidomic). Since the clinical diagnosis of AD, on the base of the standard diagnostic criteria, does not take into account the long preclinical and prodromal phases, biomarkers involved in steps occurring before the appearance of the clinical dementia syndrome, could support and improve the early detection, prediction and classification (differential diagnosis) of the disorder.
4. GENETICS
AD can be classified as early onset or late onset. Late onset (after 65 years) accounts for over 95% of cases while the early onset (before 65 years) is much rarer. Few cases (around 1%) displayed autosomal dominant mutations in one of the three genes: 1) APP gene on chromosome 21; 2) the presenilin-1 (PRE1) gene on chromosome 14; 3) the presenilin-2 (PRE2) gene on chromosome 1 [2]. These autosomal dominant mutations are all involved in the production or metabolism of amyloid protein [2]. Deterministic genetic mutations for late onset AD have not been found, but APOE-ε4 gene is the major genetic risk factor. The APOE gene encodes a protein involved in cholesterol metabolism. Three normally occurring alleles of APOE exist: ε2, ε3, and ε4. APOE-ε3 is the most abundant allele in the population (frequency of 80%), while APOE-ε4 increases the risk of developing AD lowering the mean age at onset of the disease [2-6]. APOE-ε4 is involved in AD pathogenesis by the modulation of the metabolism, aggregation, and clearance of Aβ peptide and probably by directly regulating brain lipid metabolism and synaptic functions. Therefore, APOE-ε4 could be considered as biomarker being a risk factor for brain Aβ deposition [9,10]. The APOE-ε2 decreases the risk to develop AD but this protective effect is not as strong as the risk conferred to carriers of the APOE-ε4.
5. BIOMARKERS INVOLVED IN THE ONSET AND PROGRESSION OF AD
The pathological cascade in AD begins with amyloid and tau pathology, followed by neural injury and dysfunction and finally with structural alterations [11]. Recent findings demonstrated that brain and cognitive changes occur up to 10 years prior to the AD diagnosis and so the combination of genetic and structural brain biomarkers, together with imaging quantitative traits in the medialtemporal lobe (MTL), could be considered a helpful outcome for the diagnosis. To date, there is an urgent need not only to develop an aimed therapy for the AD but also to identify reliable AD-specific biomarkers. The “ideal” AD biomarker should: a) detect a fundamental feature of neuropathology; b) have a sensitivity > 80% in AD detection; c) have a specificity > 80% in the discrimination from other dementias; d) be reliable; e) be non-invasive; f) be easy to perform; g) inexpensive. In AD two types of biomarkers are defined: 1) Genetic biomarkers and 2) Non-Genetic biomarkers.
6. GENETIC BIOMARKERS
Five leading genes have been identified as causative elements of AD: 1) APP; 2) PSEN1; 3) PSEN2; 4) APOE; 5) TOMM40. Mutations in the first three genes lead to the early onset of the disorder, while APOE is the only gene marked as risk factor for late-onset disease [12].
6.1. APP
APP gene, localized on chromosome 21, encodes a transmembrane protein, named APP, ubiquitously expressed, that contains an internal 39 - 43 amino acid sequence corresponding to the Aβ peptide. Indeed, Aβ peptide is generated from APP by two endoproteolytic cleavages catalyzed by β and λ secretases (Figure 1) [13]. Aβ peptides are the principal component of amyloid deposits or plaques in brain parenchyma and in the walls of brain vessels. To date, 24 APP single nucleotide mutations as AD causes are reported and all these mutations are clustered within a 54 amino acid segment, localized near or within the sequence encoding Aβ peptides. The Swedish mutation is a double substitution that induces a change in two amino acids immediately before the beginning of the Aβ peptide sequence: The normal sequence, lysine-methionine is replaced by asparagineleucine. The amount of Aβ peptide produced is two-three fold higher than that produced from non-mutated APP, presumably by affecting the efficiency of β-secretase cleavage. Other APP mutations are at or after the C-terminal amino acids of Aβ peptides, and alter the activity of λ-secretase cleavage. When the proteolysis of normal APP occurs by λ-secretase, the predominant formed species is 40 amino acids long (Aβ40) with smaller amounts of Aβ that is 42 amino acids long (Aβ42). APP mutations at the C-terminal end of Aβ produce more Aβ42 that is more amyloidogenic and more prone to aggregate than shorter forms of Aβ. Another mutation is p.Glu693Gly also known as the Arctic mutation. This missense mutation increases the aggregation rate of the mutant peptide [13].
6.2. PSEN1 and PSEN2
PSEN1 and PSEN2 genes encode two closely related proteins that are part of the λ-secretase complex as depicted in Figure 2. Therefore, mutations on both genes induce a modification in the λ-secretase that is still able to catalyze the cleavage of APP, but the proteolytic site is altered. Mutant λ-secretase produces Aβ42, more amyloidogenic and more prone to aggregate than Aβ40 normally produced by the normal enzyme.

Figure 2. γ-secretase complex.
6.3. APOE
APOE gene is located on chromosome 19 and encodes Apolipoprotein E (APOE), a component of lipoprotein particles binding the cell surface receptors. APOE translates into three common allelic variations ε2, ε3, and ε4 [14]. Polymorphisms within the APOE gene have been strongly associated with AD. The ε2 allele encodes a cysteine (Cys) at amino acid positions 112 and 158; allele ε3 encodes a Cys at 112 and an arginine (Arg) at 158, while the ε4 allele encodes an Arg both at 112 and at 158. The ε4 allele is the ancestral and high-risk form, the ε3 allele is the most common in humans and the neutral allele, and the ε2 allele is associated with decreased risk of AD [14]. APOE plays an essential role in the lipid metabolism within the Central Nervous System (CNS) and its allelic variations modulate neural repair, lipid homeostasis, oxidative stress, and Aβ deposition. APOE, as the main cholesterol transport lipoprotein, exerts a pivotal role in maintaining brain integrity since lipids are essential for myelination of axons. Indeed, the protein seems to modulate the cholesterol delivery to neurons. The ε4 allele decreases the delivery of cholesterol, consequently disturbing lipid homeostasis in the CNS leading to Aβ depositions. Moreover, APOE polymorphism may influence not only the amyloid cascade but also the neuronal repair mechanisms and the maintenance of synaptic connections [14].
6.4. TOMM40
Translocase of Outer Mitochondrial Membrane 40 (TOMM40) is a promising gene in AD onset and is responsible for mitochondrial survival [14]. Mitochondrial dysfunction has been associated with several pathological processes in AD, such as brain hypometabolism, synaptic pathology, accumulation of APP, and Aβ influx to the cell [14-16]. Damaged mitochondria exert a specific influence on the AD pathophysiology through interplay with Aβ and its precursor, AβPP. The encoded protein, localized at the outer mitochondrial membrane, is one of the primary pores by which proteins can readily enter the mitochondria. In AD, mitochondria exert neurotoxic influence by allowing the influx of Aβ to the cell through the TOMM40 import pore. The influx TOMM40- mediated of Aβ increases Reactive Oxygen Species (ROS)
resulting in apoptosis. Moreover, it has been demonstrated that in AD mitochondria show high intracellular Aβ accumulation and this precedes extracellular Aβ deposition [14]. This finding supports the role of mitochondria in the pathogenesis of AD [14]. A genetic involvement of TOMM40 on AD-type cognitive deficits is due to TOMM40 length variation that was found to influence episodic memory, and the integrity of the medial ventral precuneus and posterior cingulated, [17] sites of early amyloid burden in AD [18]. Three TOMM40 risk alleles have been reported (rs157580; rs2075650; rs11556505) that are overexpressed in the AD population. These three alleles have recently been associated with CSF biomarkers including Aβ42, T-tau, and P-tau [18]. In the “adapted mitochondrial disconnection model” TOMM40 can act through two mechanisms: 1) APOEindependent mechanism and 2) APOE-dependent mechanism. In the first mechanism, TOMM40 modulates Aβ influx to the mitochondria through the TOMM40 outer membrane pore. In the second mechanism, an interaction between APOE and TOMM40 exists, and both carriers in turn influence the Aβ influx. The proposed model suggests mitochondria as an early and accurate biomarker for preclinical AD since mitochondria dysfunction begins the pathophysiological cascade in AD. Moreover, mitochondrial Aβ aggregation precedes and induces extracellular Aβ aggregation supporting the mitochondrial cascade hypothesis rather than the amyloid cascade hypothesis, as the primary event in the biomarker timeline of AD.
7. NON GENETIC BIOMARKERS
Among these biomarkers, two types are defined: 1) direct biomarkers and 2) indirect biomarkers. Among direct biomarkers, the mostly reported and overall studied are: 1) Aβ42 peptide; 2) total (T)-tau protein; and 3) the phosphorylated (P)-tau protein. These are reliable biomarkers of AD progression that, by contrast, do not allow an early diagnosis because they are detectable when the brain functionality is irreversibly compromised. The detection of the accumulation of Aβ or tau in the interstitial fluid of brain parenchyma (Figure 3) can be combined with the measure of the cerebrospinal fluid biomarkers also defined “core CSF markers” [19,20]. A decrease of Aβ42 levels in the CSF well correlates with the positive amyloid PET imaging findings in AD patients and, therefore, with the clinical diagnosis of AD and Aβ pathophysiology [19,20]. CSF tau is an indicator of tau pathophysiology and associated neuronal injury while phosphorylated tau is a more specific indicator of AD. High CSF tau is detectable in stroke and head trauma, and therefore is not AD specific. In AD, the high level of tau protein in the CSF is a direct result of a tau
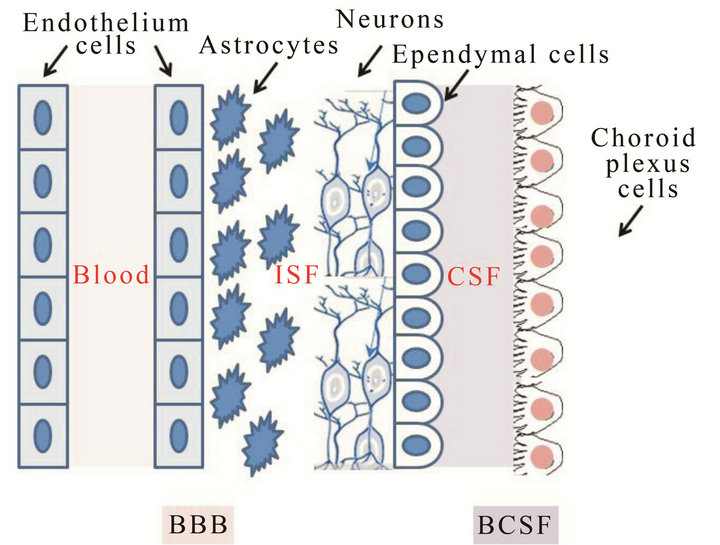
Figure 3. Blood Brain Barrier (BBB) and Blood Cerebro-, Spinal Fluid (BCSF) structures.
pathophysiology that disrupts neurons, particularly axons, inducing the release of cytoskeletal elements, such as tau, into the extracellular space. High level of CSF tau correlates with neurofibrillary pathophysiology at autopsy. These three CSF biomarkers are reliable markers for AD diagnosis, even at early stages, particularly vs healthy controls but their limitation is due to their inability to monitor the progression of the disorder since they are not able to distinguish AD from other dementias. An indirect biomarker is represented by the measure of glucose metabolism that could allow a support to AD diagnosis. The measure of glucose metabolism by 2-deoxy-2-[18F] fluoroglucose (FDG) in PET analysis is an indicator of brain metabolism, which reflects synaptic activity. In AD, the decrease of FDG uptake shows an impaired synaptic function in a lateral temporo-parietal band a posterior cingulated-precuneus distribution.
Recently, new biomarkers have been suggested as useful for the detection of the pathology onset and can be useful for the early diagnosis. These new biomarkers can be direct biomarkers, such as P-glycoprotein, “free” serum copper and microRNAs, and indirect, such as Tolllike Receptors 4 (TLR4).
8. PET DIAGNOSTIC USED FOR AD DIAGNOSIS
8.1. PET Probes for Imaging Beta-Amyloid Plaques
During the last decade, a number of Aβ plaque-specific imaging agents for PET and SPECT have been re ported. Among them [11C]4-N-methylamino-4’-hydroxystilbene (SB-13) [21], N-Methyl-[11C]2-(4’-methylaminophenyl)-6-hydroxybenzothiazole ([11C]PIB) [22], [11C]2-2(-[2-dimethylaminothiazol-5-yl]ethenyl)-6-(2-(fluoro)ethoxy)benzoxazole (BF-227) [23], and (6-[123I] iodo-2-(4’-N,N-dimethylamino)-phenylimidazo-[1,2-a]
pyridine) [24] (IMPY), have been widely tested clinically demonstrating differential uptake and retention in AD brain patients as compared to controls and have proved their potential for imaging Aβ aggregates in vivo (Figure 4). Recently, four PET imaging agents have been studied in humans: [11C]PIB, 4-{(E)-2-[6-(2-{2-[2-(18F)fluoroethoxy]ethoxy}ethoxy)-3-pyridinyl]vinyl}-N-methylaniline ([18F]florbetapir, AV-45, Amyvid) recently approved by FDA, trans-4-(N-methyl-amino)-4’-{2-[2-(2- [18F]fluoroethoxy)-ethoxy]-ethoxy}-stilbene ([18F]florbetaben, BAY94-9172) and 2-{3-[18F]fluoro-4-(methylamino)phenyl}-1,3-benzothiazol-6-ol ([18F]flutemetamol) (Figure 4) [25]. [18F]Florbetabir (Phase III) has been approved by FDA for routine clinical uses. It is a safe radiotracer for studying Aβ distribution in human brains and it is a sensitive marker of amyloid load in cortical gray matter in elderly individuals; it can differentiate among subjects with the standard diagnostic AD criteria by patients with mild cognitive impairment (MCI) and normal cognitive function. Therefore, [18F]florbetabir has an higher specificity for AD then PIB for both diagnosis and monitoring of progression also in the early stages of the pathology [26]. Results from Phase III clinical studies—with [18F]florbetapir, have demonstrated a close correlation between in vivo amyloid PET imaging by [18F]florbetapir and postmortem histopathological findings (www.clinicaltrials.gov/NCT00857415). Recently, [18F]florbetapir has been used to visualize amyloid deposition in a transgenic murine model of amyloidosis (APP/PS1-21) [27]. Studies have been performed ex vivo by autoradiography and in vivo by microPET. MicroPET imaging demonstrated an increased [18F] florbetapir uptake in APP/PS1-21 as compared to control mice. This study opens the possibility of [18F]florbetapir coupled with microPET, to visualize and quantitatively measure amyloid deposits in the brain of living APP/PS1 mice.
Therefore, [18F]florbetapir could be considered the front runner for daily routine use in early diagnosis of amyloid plaques in living human brain.
[11C]PIB is presently the one most extensively evaluated, and it is a neutral analog of Thioflavin T, an established histological stain for detecting Aβ. [11C]PIB binds with high affinity and specificity to neuritic Aβ plaques and in particular, significant retention of this probe was observed in the frontal, temporal, parietal and occipital cortices and in the striatum of AD patients. The strong correlation between in vivo [11C]PIB retention and quantitative analysis of Aβ plaques in tissue brain after autopsy validated the use of [11C]PIB for in vivo Aβ plaques detection. Furthermore, increased [11C]PIB binding may also be predictive of conversion of MCI to AD. Recently, a feasible protocol for PET imaging of Aβ in mouse brain with [11C]PIB and specific activities

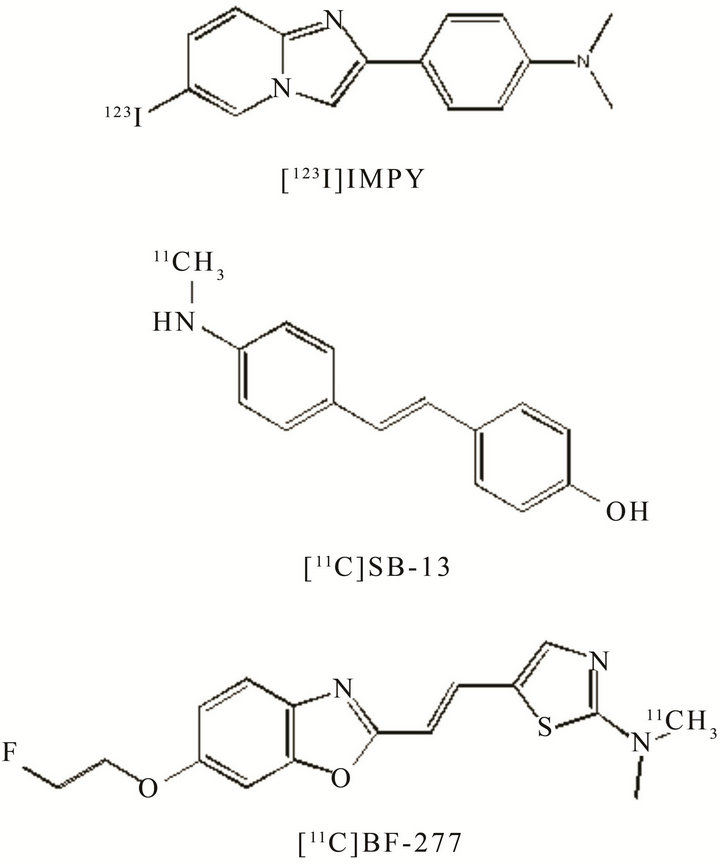
Figure 4. PET probes for imaging Aβ.
commonly used in human studies has been proposed [28]. APP/PS1 mouse transgenic model was employed to measure Aβ at different disease stages allowing longitudinal imaging studies with follow-up periods of approximately one and a half years. In a recent 5-year follow-up PET study with [11C]PIB, it was evident at the individual level that increased, stable and decreased [11C]PIB retention was observed and the disease progression was reflected in significant decline in cerebral regional cerebral glucose metabolism (rCMRglc) and cognition [29]. In a recent 20-month follow-up study, it was reported a 5.7% increase in fibrillar Aβ in AD patients [30]. These studies support the hypothesis that the Aβ levels in the AD brain reach a maximal level at the early clinical stage of the disease, although both increase and decline in later stages of the disease cannot be excluded. Finally, a trial evaluating the biodistribution of [11C]PIB using PET imaging in normal elderly volunteers and relevant patient groups is ongoing (www.clinicaltrials.gov/NCT00811122. Recent work has suggested that [18F]flutemetamol, 18F-labeled PIB derivative, can give similar imaging results in patients with probable AD and MCI relative to healthy controls and demonstrated a high correlation with [11C]PIB when tested in the same patients [31]. Moreover in a recent report the concordance of [18F]flutemetamol PET imaging with histopathology has been demonstrated supporting its sensitivity to detect amyloid and potential use in the detection of AD. To date, two clinical trials are ongoing. The first is aimed to test the equivalency of 11C]PIB and [18F]flutemetamol because, although similarity in their distribution in AD or MCI subjects was demonstrated, no comparison data in normal subjects have been published
(www.clinicaltrials.gov/NCT01607476). Phase II is aimed to study the biodistribution and binding of [18F] flutemetamol in comparison with FDG-PET to correlate metabolic changes and for anatomic co-registration of [18F]PIB images for group analysis
(www.clinicaltrials.govNCT01202994). [18F] Florbetaben [32] is a stilbene derivative with some structural similarities to PIB. The results obtained from a first proof of mechanism trial in Europe in AD patients and healthy controls indicated that florbetaben was an efficacious and safe Aβ tracer with favorable brain kinetics that yielded valuable visual and quantitative brain PET data capable of supporting the clinical AD diagnosis. An operator-independent voxel-based analysis approach was also developed which allows quantification of the individual brain volume affected by Aβ. In another recent report, participants with a wide range of dementing and non-dementing neurodegenerative diseases were evaluated with [18F]florbetaben. Results demonstrated that [18F]florbetaben can reliably detect Aβ deposition in the brain and was thereby useful in the early and differential diagnosis of AD from other dementing or non-dementing neurodegenerative conditions. It provided images similar to those provided by [11C]PIB, and, as for [11C]PIB, [18F]florbetaben PET appears to be an excellent test for distinguishing FTLD (FrontoTemporal Lobar Degeneration) from AD. Further, a trial in subjects with MCI has been completed (www.clinicaltrials.gov/NCT01138111). In this trial, the potential of the tracer to predict the conversion to AD is being examined. Phase III is currently in the recruitment step (www.clinicaltrials.gov/NCT01020838) and it aims to investigate the association between the in vivo florbetaben brain uptake and the postmortem brain Aβ deposition, as determined by histopathological investigation. Moreover another study (Phase I), has been completed, and it was aimed to investigate [18F]florbetaben ability to distinguish patients with MCI progressing to AD from those with MCI not progressing to AD (www.clinicaltrials.gov/NCT01138111). [18F]BF277 is a benzoxazole derivative developed on the basis of the structure of [11C]PIB. It is able to bind to Aβ42 fibrils with low nanomolar affinity and Aβ plaques in AD brain sections. This tracer was considered a promising PET imaging agent for the in vivo detection of Aβ but recently, it has been demonstrated that BF277 labelled both Aβ plaques and Lewy bodies typical of Parkinson’s disease, so it was not Aβ-selective [33]. [123I]IMPY is a radioiodinated derivative of thioflavin-S, with high selectivity for Aβ plaques in the brain in transgenic animal models of AD. This probe has been studied also as [124I]IMPY and high binding ability and specificity to hippocampus and frontal cortex of two AD mouse models (Tg2576 and TT transgenic mice) has been found by in vitro autoradiography [34]. Moreover, although IMPY binds to the Aβ protein, some studies suggested that it could also bind to proteins associated with prion disease.
8.1.1. Benzothiazole Derivatives
Recently, on the basis of PIB, 18F-fluoroethylated benzothiazole derivatives have been developed [35] and one of these compounds, [18F]-6-FEtO-BTA-1, was studied in normal SCID (Severe Combined Immunodeficiency) mice. It showed a suitable biokinetics profile and good binding affinity for amyloid plaques (Ki = 7.2 nM) so that it represents a promising new diagnostic probe. Starting from PIB and [123I]IMPY, a new series of 2-aryl-imidazo[2,1-b]benzothiazole derivatives has been recently developed and tested for their ability to bind to amyloid Aβ1-40. This series was developed combining benzothiazole part of PIB fused with the 2-arylimidazo moiety of IMPY. These compounds showed acceptable binding affinity for synthetic Aβ40 fibril, ranging from 133 nM to 6.09 nM, and could be labeled with [11C]
CH3 for in vivo evaluation. These results support the hypothesis that mix-condensed analogues of PIB-IMPY could achieve similar potency to those of the individual compounds [36].
8.1.2. Flavones Derivatives
Recently fluoro-pegylated flavones have been developed, showing high affinity for plaques (from 5 to 321 nM) by in vitro binding experiments carried out in Tg2576 transgenic mice brain section and high uptake (from 2.9% to 4.2% ID/g) and fast washout (1.3% - 2.0% ID/g) in the brain of normal mice after injection [37].
8.1.3. Aurones Derivatives
[123I]IMPY was the first SPECT probe tested in humans. However, preliminary clinical data showed poor signal-to-noise ratio, most likely due to high lipophilicity and low stability, making it difficult to distinguish AD patients. Recently, a new fluorinated and iodinated aurone derivative, (Z)-2-(4-(2-fluoroethoxy)benzylidene) -5-iodobenzofuran-3(2H)-one, has been synthesized and evaluated as a PET/SPECT dual imaging probe for Aβ. This compound showed high affinity for Aβ aggregates by in vitro binding experiments and both [125I]- and [18F]-derivative showed good uptake and washout from the brain by biodistribution experiment using normal mice [38]. These results suggested that the fluorinated and iodinated aurone derivative may function as a PET/ SPECT probe for detecting Aβ plaques in AD brain.
8.1.4. Indole-Chalcone Derivatives
Another class of derivatives recently developed bears an indole-chalcone scaffold, among these compounds the radioiodinated probe [125I]-(E)-3-(1H-indol-5-yl)-1-(4- iodophenyl)prop-2-en-1-one, by autoradiography in sections of brain tissue from an animal model of AD showed that it specifically labeled Aβ plaques [39]. However, experiments with normal mice indicated that compound exhibited a low uptake into the brain in vivo (0.41% ID/g at 2 min). Indole-chalcone scaffold with additional chemical modifications may lead to more useful imaging agents for detecting Aβ plaques in AD brain. Currently, the relationship between fibrillar Aβ (plaques) and soluble Aβ oligomers remains still unknown. Presently there is no information on how the smaller soluble Aβ oligomers, could be visualized in vivo with the available Aβ tracers. A challenge could be to develop PET tracers able to visualize these smaller forms of Aβ in the brain, although the probably lower content of oligomers in AD brains compared with fibrillar Aβ might be a limiting factor. The soluble Aβ oligomers are important since they probably could induce and interfere with the neurotransmission in the brain.
8.2. PET Probes to Measure Glucose Metabolism
PET studies employing [18F]FDG, (Figure 5) a probes useful to study CMRglc in humans, demonstrated that in AD metabolic deficits are presented in different neocortical association areas. In particular, by longitudinal studies a significant reduction of CMRglc in the frontal, parietal, temporal and posterior cingulate regions in AD patients respect to healthy subjects have been demonstrated. In early stages of the disease, accumulation of Aβ is an ongoing event while minor metabolic changes occur. As the clinical course of AD progresses, the amyloid curve flattens and evident generalized glucose hypometabolism arises. [18F]FDG and [11C]PIB tracers track molecular changes in different stages of AD. [11C]PIB is useful to detect prodromal AD-earlier stage, while [18F]FDG is useful to detect the clinical progresssion of pathology. For diagnostic purposes, [18F]FDG and [11C]PIB PET provide complementary information and can be used in combination for early and accurate detection of AD [40]. Moreover, [18F]FDG PET is a sensitive and specific biomarker for early differential diagnosis of the forms of dementia. A significant further advancement in the use of [18F]FDG PET for suspected AD is the capacity to distinguish other neurodegeneration, such as DLB (Dementia with Lewy bodies) and FTLD, in initial clinical evaluation [41]. Moreover, [18F]FDG PET scans have been approved in the United States for Medicare reimbursement to aid in the distinction of AD from FTLD.
8.3. PET Probes to Measure Neurotransmitter Activity
Several neurotransmission, especially the cholinergic system but also the dopaminergic and serotonergic, is
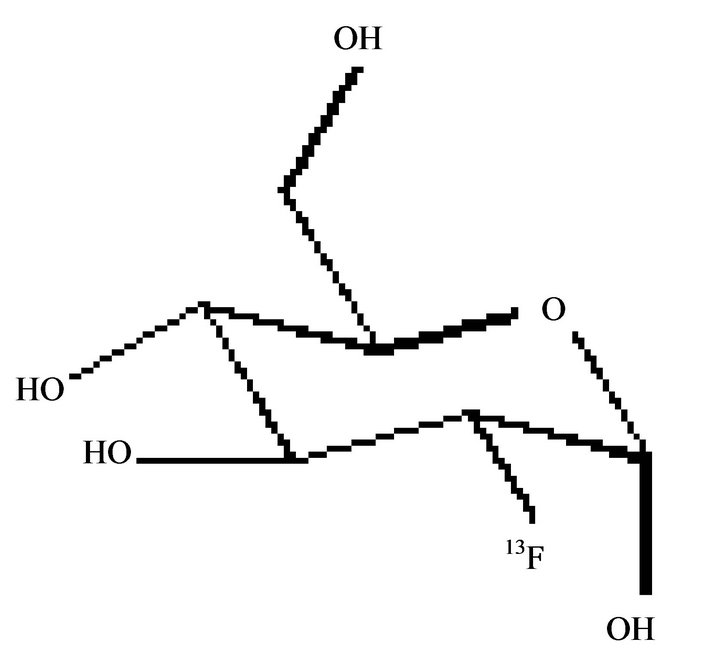
Figure 5. [18F]FDG.
impaired in AD. PET tracers as N-[11C]-methylpiperidine-4-yl-propionate ([11C]PMP)41 and N-[11C]-methylpiperidyl-4-yl-acetate ([11C]MP4A) [42] have been employed to measure acetylcholinesterase activity and a reduction was found in AD patients, respect to healthy controls. Reduction in acetylcholinesterase activity has also been reported in MCI patients who later convert to AD, suggesting that acetylcholinesterase changes might precede the development of clinical AD. Furthermore, PET studies using (S)-(2)-[11C]-methylnicotine have shown a decrease in the number of cortical nAChRs (Nicotinic acetylcholine receptors) in AD patients and this reduction significantly correlates with impaired cognitive function, such as impaired attention. 2-[18F]- Fluoro-A-85380 is another PET tracer developed to visualize a specific subtype of nAChRs, α4β2, a reduction of this receptors was found in both AD and MCI patients. Recently, there is a great interest to develop selective PET tracers for the imaging of the α7 nicotinic receptors in the brain since these receptors interact with Aβ and might therefore be a new target for AD therapy [43].
8.4. PET Probes for Imaging Tau Protein
2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl}-ethylidene)malononitrile ([18F]FDDNP) (Figure 6) has been the first PET tracer employed to visualize NFTs as well as amyloid plaques in living humans [44]; it is the only probe able to visualize AD in the hippocampal region of living humans. (FDDNP) PET results demonstrated that FDDNP binding is higher in medial temporal cortical then in neocortical area in AD. Moreover, previous postmortem histological studies demonstrated that in AD, Aβ plaque deposition is high in neocortical regions but relatively low in some medial temporal cortical substructures as hippocampus. By contrast, tangle accumulation is high in the medial temporal cortex at all levels of cognitive impairment with subsequent neocortical increases with disease progression. Starting from these findings, FDDNP could be considered a sensitive in vivo marker for NFTs. FDDNP could be employed with PIB in the same subjects to visualize different aspects of AD progression. AD patients showed low PIB but high FDDNP uptake in the medial temporal cortex and sig-
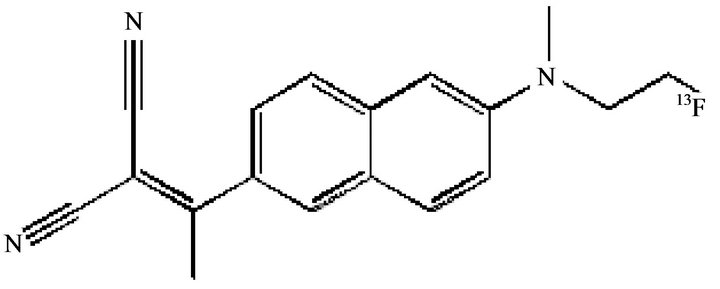
Figure 6. [18F]FDDNP.
nificant increased uptake of both agents in neocortical areas; in agreement with this result, the medial temporal cortex represents the most significant differential brain region in the voxel mapping. Moreover, medial temporal cortex plays a central role in modulating episodic memory so that FDDNP binding, in contrast to PIB, is also related to episodic memory impairment that is one of the most prevalent cognitive deficits in patients with AD and in non-demented older adults who eventually develop AD. FDDNP provides unique information on AD pathology relevant to disease progression. Recently, Zhang et al. studied a new compound, [18F]-T808 that by in vitro preclinical and in vivo assays, displayed a high level of binding affinity and good selectivity for tau aggregates [44] suggesting that [18F]-T808 possesses suitable properties and characteristics to be a specific and selective PET probe for the imaging of paired helical filament tau in human brains [45].
8.5. PET Probes to Measure Reactive Microglia
Activated microglia, involved in immune response of the brain to neurodegeneration, represents a useful marker in AD diagnosis. In particular, amyloid has been demonstrated to mobilize and activate microglia that was found in autopsy brain tissue at sites of aggregated Aβ deposition of AD patients. 1-[2-Chlorophenyl]-NmethylN-[1-methylpropyl]-3-isoquinolinecarboxamide ([11C]- (R)- PK11195) (Figure 7), a selective TSPO (translocator protein) radioligand, has been employed to measure the transition of microglia from a resting state to an activated state in the brain, due to an increase in the number, rather than the affinity, of TSPO. An increase in [11C]- (R)-PK11195 binding was found in the temporoparietal,
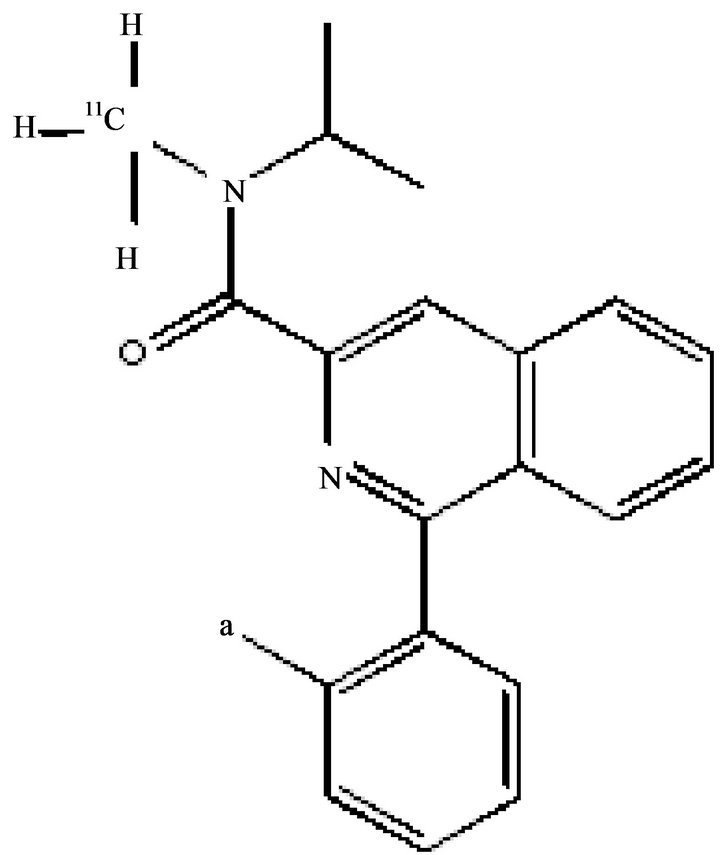
Figure 7. [11C]-(R)-PK11195.
cingulated and entorhinal cortices of AD patients, means a strong microglia activation [46]. [11C]-PK11195 apication displays some limitation including a high level of non-specific binding and a poor signal to noise ratio, for this reason novel PET tracers with improved capacities to quantify TSPO expression have been developed and among them [11C]-[11C]N-(2,5-dimethoxybenzyl)-N- 5-luoro-phenoxyphenyl)-aceamide (DAA-1106) [47], which has shown increased binding in several brain reons including the frontal, parietal, temporal cortices and striatum of AD patients compared to controls. Activated astrocytes participate in the inflammatory processes ocrring around the Aβ plaques. An increased number of astrocytes have been measured in autopsy brain tissue from AD patients. N-[11C]-methyl-N-deuterodeprenyl ([11C]-DED) bind to the enzyme monoaminoxidase B expressed in reactive astrocytes and has been tested as a PET ligand for measurement of activated astrocytes. An increased [11C]DED binding was found in the cortical and subcortical brain regions of MCI patients compared with AD patients and controls suggesting that astrocysis might be a very early event in the time course of pathological processes in AD [48].
9. BIOMARKERS FOR THE EARLY DIAGNOSIS OF AD: A NEW SCENARIO
9.1. P-glycoprotein (P-gp)
P-glycoprotein (P-gp) is an ATPase belonging to the ATP Binding Cassette (ABC) transporters family, transmembrane proteins able to efflux xenobiotics out of cells using the energy by ATP hydrolysis [49]. This protein is highly expressed at the luminal surface of brain capillary endothelium and is the major component of BBB; because of this strategic localization, P-gp exerts a protecttive function in the BBB. Several studies reported a potential correlation between P-gp activity and/or expression and the onset of some CNS disorders such as AD. In AD brains the Aβ levels inversely correlated with the P-gp level in brain vasculature. This finding suggested that P-gp exerts a critical role in the clearance of Aβ from the brain. Aβ proteins are usually secreted from neurons into the interstitial fluid and can be eliminated in three different ways: 1) proteolytic degradation; 2) passive efflux; 3) active efflux through BBB. P-gp is responsible for the active efflux of Aβ from the brain, and its decreased expression and/or activity leads to increased Aβ levels in the ISF (Figure 3) [48]. These findings support the hypothesis that a P-gp dysfunction in the BBB could play a pivotal role in AD onset. Moreover, it was demonstrated that Aβ is a P-gp substrate [49]. Therefore, P-gp ligands could be used as PET tracers to detect P-gp activity and expression allowing an early diagnosis of AD. Currently, AD diagnosis by PET is performed using probes able to measure amyloid deposition such as [11C]PIB and [18F]-FDDNP that characterize the pathology when it is already in an advanced phase.
9.2. MicroRNA
MicroRNAs (miRNAs) are a class of small (22 nucleotides) non-coding RNAs, generated from a long primary transcript (primiRNA), cleaved in the nucleus by an enzymatic complex to a shorter hair-pin structure (50-120 nucleotides) constituting the miRNA precursor (pre-miRNA) [50]. The pre-miRNA is then exported to the cytoplasm and processed by the RNAse III enzyme Dicer to a 22-nucleotide duplex. Mature miRNAs bind to partially complementary sites within the 3’ untranslated region (UTR) of target mRNAs, targeting them for degradation or translation arrest. The identification of miRNAs significantly increased the knowledge on the regulatory mechanisms of gene expression in the CNS. Approximately 70% of the identified miRNAs are expressed in the CNS with a specific localization. Several papers identified an AD-specific miRNA altered expression. Lukiw et al. studying the miRNA abundance in fetal and adult hippocampal neurons, reported differenttial expression patterns between fetal and adult brains for six different miRNAs (miR-9, miR-124a, miR-125b, miR-128, miR-132, and miR-219) [51]. In particular, miR-9 was upregulated in AD brains compared to the adult brain, while miR128 was overexpressed compared both to adult and fetal samples. AD brains also displayed a significant upregulation of miRNA-146a, coupled to downregulation of the target gene complement factor H (CFH), an important repressor of the inflammatory response in the brain. Moreover, it has been reported that: 1) some of the AD-modulated miRNAs (miR-9 and MiR-132) were involved in the neurogenesis and neuronal differentiation; 2) other miRNAs were involved in metabolic pathway (e.g. miR-29a and -b, overexpressed in models of insulin resistance). Aβ fibrils are generated from APP by the beta-siteAPP-Cleaving Enzyme (BACE1). In AD brains, a dysregulation of some miRNAs targets APP (let-7, miR-101, miR-15a, and miR-106b) or BACE1 (miR-15a, miR-29b-1, miR-9, miR-19b, miR-107, miR-298 and miR-328), regulating BACE1 expression in vitro and so interfering with Aβ disposition in cell culture. Overall, these findings strongly suggest miRNAs as novel biomarkers for AD pathogenesis [51]. Recently, increasing evidences support the hypothesis that the progressive up-regulation of inflammatory gene expression and inflammatory signaling due to miR-146 induces the development and the progression of AD [52].
9.3. Free Copper ion
The ‘metal causative role’ in AD was supported by the following evidences: 1) APP is able to bind and reduce copper from Cu(II) to Cu(I), responsible for the copper-induced toxicity and oxidative stress; 2) Aβ and metals are packed together in plaques; 3) chelating agents sequestering metals within Aβ dissolves the plaque. In a series of clinical studies, AD patients showed an altered metabolism of a specific fraction of serum copper, copper non-bound to ceruloplasmin (also called “free” copper). 85% - 95% of the serum copper is tightly bound to ceruloplasmin while the resting “free” copper is bound to albumin, alpha 2 macroglobulin, and low-molecularweight compounds such as peptides and amino acids. The difference between the two copper pools is that the “free” copper overcomes BBB [52,53]. Moreover, in AD patients “free” copper is increased in CSF [54] and in brain parenchyma. It has been hypothesised that the increased “free” copper found in AD was due to a defect in copper incorporation into ceruloplasmin. This hypothesis was supported by: 1) the increased levels of serum apoceruloplasmin derived by the failure of copper binding into ceruloplasmin during its biosynthesis in the liver; 2) liver dysfunction due to “free” copper disturbance of hepatocytes; 3) evidence of the move of “free” copper from the serum to the CSF, overcoming BBB, and of “free” copper interaction with CSF markers of AD [52-57].
9.4. TLR4
Aβ peptides trigger microglial activation by interacting with several TLRs, including TLR4. TLRs are molecules involved in the innate immune response. TLRs are type I transmembrane receptors characterized by an extracellular domain with leucine-rich repeats and a carboxy terminal intracellular domain, designated the Toll/IL-1R (TIR) domain, containing about 200 amino acids and three conserved regions essential for the signaling cascade to downstream adapter molecules [58]. A recent study reported that resveratrol, a natural polyphenol found in abundance in red wine, displaying antiamyloidogenic and neuroprotective properties in vitro and in vivo currently in clinical trials for AD, prevented the activation of microglia by interfering with TLR4 oligomerization upon receptor stimulation. Moreover, in a mouse model of cerebral amyloid deposition, resveratrol inhibited microglial activation associated with cortical amyloid plaque formation. Song and co-workers studied the TLR4 signaling in two AD animal models, an AD mouse model homozygous for a loss-of-function mutation of TLR4 and a TLR4 wild-type AD mouse model demonstrating a TLR4-dependent activation of microglia at the early stage of AD. This activation at the onset of the Aβ disposition, is able to lower Aβ deposits preserveing cognitive function. The link existing between TLR4 expression and AD could suggest TLR4 as new biomarkers to diagnose AD onset and progression [58,59].
10. PERSPECTIVES
10.1. P-gp Ligands: Inhibitors and Substrates
P-gp ligands have been proposed for their ability to hit the protein responsible of the first step of the pathology progression as new potent probes for the early diagnosis of neurodegenerative disorders such as AD but also PD and epilepsy by PET technique [48]. Indeed, a P-gp dysfunction is the principal cause of the accumulation of Aβ in brain parenchyma. Therefore, the detection of this dysfunction, due to a decreased activity or expression of the pump, can be a useful strategy to early diagnosis AD. Four distinct interaction sites have been identified in P-gp structure: three sites (I-III) for the transport of substrates (vinblastine, paclitaxel, rhodamine-123, and Hoechst33342) and modulators (XR9576, XR9051); site IV, a regulatory site where elacridar and nicardipine act as modulators. These binding sites were able to allosterically communicate in a negative heterotropic manner, and the binding to one of sites switched the other sites to a low-affinity conformation [59]. P-gp ligands can be classified as: substrates, inhibitors and modulators. Substrates are actively transported by P-gp in an ATP-dependent manner and therefore their concentration is higher outside the cell with respect to the cytosol (Figure 8(a)) [49]. Modulators are able to alter substrate binding in a non competitive manner by interacting with P-gp within a distinct site from that of the substrate (Figure 8(b)). Moreover, an allosteric communication between substrate and modulator binding sites exists [60]. Inhibitors block P-gp translocation by interacting with the ATP binding site and therefore preventing drug transport by the pump (Figure 8(c)). In neurodegenerative diseases, substrates, modulators, and inhibitors are suitable tools to investigate the expression and functionality of P-gp, respectively, in the biological barriers. Substrates, as 11Cor 18F-radiolabelled, are useful probes to evaluate P-gp activity whereas inhibitors to measure P-gp expression; modulators, as cold ligands, are useful to define the specificity of the probe for the target in PET analysis [61]. As previously described, the importance to develop probes for detecting the functionality of P-gp is due to the possibility to early diagnose the onset of AD since an impairment of the efflux protein is the cause of the onset of the pathology (β-amyloid accumulation). To date, several PET tracers have been developed to detect the function and expression of P-gp at the BBB. Among them, [11C]verapamil [61,62] and [11C]N-desmethyllo-
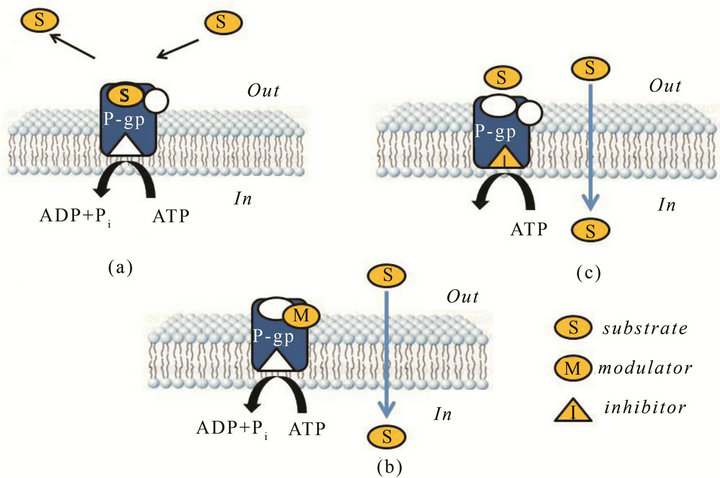
Figure 8. Interaction of P-gp with: substrate (a); modulator (b); inhibitor (c).
peramid [63] have been reported as P-gp substrates whereas [11C]laniquidar [64] [11C]elacridar [65], [11C] tariquidar [66] and [11C]MC18 [67] as P-gp inhibitors (Figure 9). Among substrates, (R)- and (S)-verapamil exerted similar P-gp activity, but (R) [11C]verapamil showed a low basal brain uptake and its distribution volume (DV) and kinetic constant k1 were increased after co-administration with cyclosporin A (CsA) or tariquidar; [11C]loperamide metabolized into [N-methyl- 11C]N-desmethylloperamide ([11C]dLop) via demethylation and this metabolite also behaved as P-gp substrate. However, measures of changes in P-gp function are crucial because of the very large capacity of the pump, which induces a very low brain uptake of radiolabeled substrates. Indeed, since radiolabeled substrates such as [11C]loperamide and [11C]verapamil displayed a low brain uptake in vivo, new P-gp ligands with improved kinetics are still required. Among P-gp inhibitors, [11C] elacridar and [11C]tariquidar displayed lower brain uptake than [11C]verapamil and their brain uptake was increased by pretreatment with the corresponding cold compound. Moreover, both [11C]radiotracers displayed high metabolic stability, and a comparable inhibition activity towards P-gp and breast cancer resistance protein (BCRP, ABCG2). Therefore, they are not specific for P-gp. 6,7-dimethoxy-2-{3-[4-methoxy-3,4-dihydro-2Hapthalen-(1E)-ylidene]-propyl}-1,2,3,4 tetrahydroisoquinoline (also named MC18) has been characterized as potent P-gp inhibitor (EC50 = 1.6 μM) in vitro [64] and, as [11C]MC18, has been proposed as the first PET tracer for the measurement of P-gp expression [65]. [11C] MC18 showed a good uptake in control brain that was significantly reduced after pretreatment of rats with cold compound and higher cerebral distribution volume (DV) in untreated rats than [11C]verapamil. After pretreatment of animals with cold MC18, the uptake of [11C]MC18 was reduced in many brain areas and in peripheral target organs (colon, duodenum, ileum and spleen). These find-
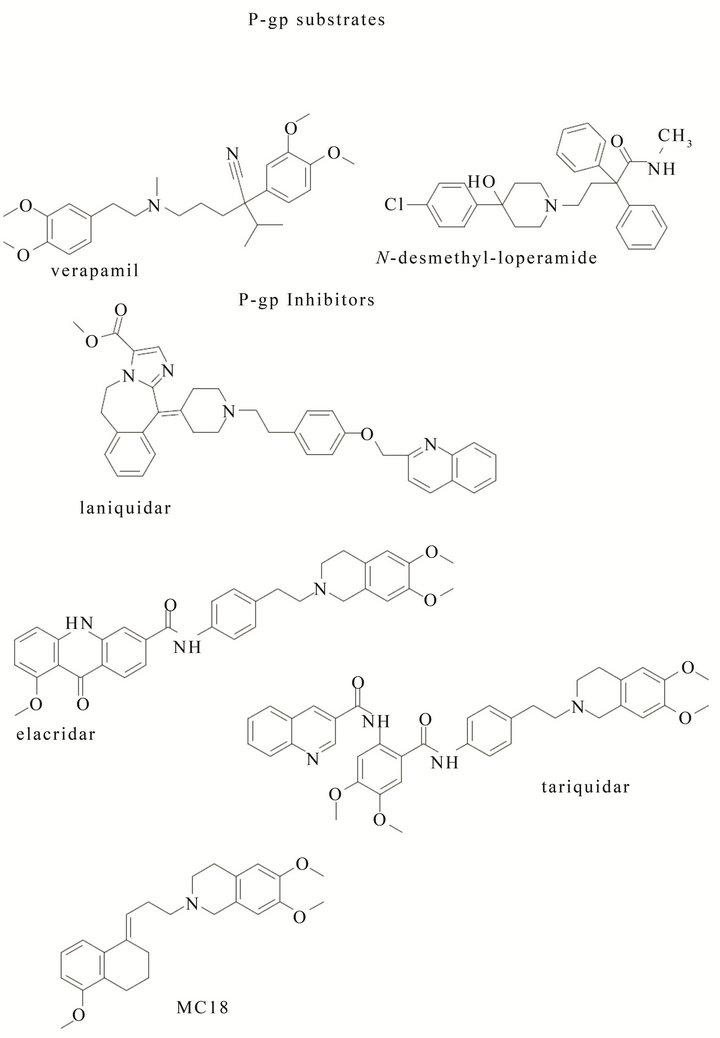
Figure 9. P-gp substrates and inhibitors.
ings suggest [11C]MC18 as unique PET tracer to measure P-gp expression, showing specific binding in the brain and in peripheral target organs [68]. The importance to develop 18F-probes for PET analysis is due to the their longer half-life with respect to [11C]labeled compounds. Studies performed in vivo on [18F]fluoroethyl elacridar and tariquidar as PET probes displayed an increased uptake of [18F]elacridar and [18F]tariquidar in mice brain comparable to that of their [11C]labeled analogues [69,70]. Moreover, since [18F]tariquidar displayed a metabolic stability higher than [18F]elacridar, it could be considered a promising probe for the evaluation of P-gp taking into account their low selectivity towards BCRP that needs the optimization of the used concentration [71,72]. New studies on modified [18F]MC18 are ongoing in clinical phase in AD patients and the results are topic of patents [73,74].
10.2. TLR4 and MIR
The development of radiotracers useful to detect biomarkers directly or indirectly involved in the first stages of the pathology could give important outcomes in AD diagnosis and therapy. In this scenario, TLR4 and MIRs can be targeted with probes with high specificity. TLR4 is linked to the microglia activation, a consequence of Aβ deposits in the brain parenchyma. In AD, activated microglia can be beneficial since it is able to protect neurons from toxic substances, such as aggregated Aβ by taking up and degrading them and by secreting neurotrophic factors. On the other hand, activated microglia can be synaptoand neuro-toxic by initiating and advancing the disease. Thus, since the activated microglia via TLR4 in the early stages of AD pathogenesis exerts a neuroprotective effect, TLR4 signaling pathways offer a pivotal and potential therapeutic and diagnostic target for a early diagnosis and therapy of the disease. The same aim could be hit with the design of probes for the detection of MIRs in AD pathology since these are new potential biomarkers of the disorder. Therefore, the development of ligands, as PET probes, able to detect an overexpression of TLR4 or MIRs can be useful in the first stage of the disease and can be helpful for an early diagnosis. The innovation is due to the lack of TLR4 or MIR ligands bearing a simple structure since to date LPS and oligonucletides are the major molecules interacting with TLR4 and MIRs, respectively.
10.3. Free Copper
The increase of “free” copper in the serum of AD patients, in the early stage of the pathology, candidates copper as new target for the development of probes for the early diagnosis of AD. Indeed, studies for the development and biological evaluation of fluorescent probes able to bind the ion are ongoing and in this scenario two are the possibilities: 1) “switch on” probes; 2) “switch off” probes. The “switch on” probes are pro-fluorescent molecules that increase their fluorescent signal after interacting with the target; the “switch off” probes are fluorescent molecules that decrease their fluorescent signal after interacting with the target. By the development of fluorescent probes specific for the free copper, its quantification in some biological fluids such as CSF or serum can be supportive to early diagnose AD.
The development of probes for the detection of these new biomarkers (P-gp, TLR4, MIR and free copper) could be a useful strategy that can be added to the use of tracers for amyloid-β plaques in brain parenchyma.
11. CONCLUSION
The treatment of AD represents the challenge of XXI century, since to date it is possible to treat only the symptoms of the disorder. This limitation is due to the difficulty to hit the onset of the disease by an early diagnosis. As data stand, the identification of high sensitive and specific biomarkers involved in the first step of AD could help AD diagnosis and address more efficaciously the pharmacological treatment to date employed. Aim of this paper was the in vitro characterization of the biomarkers involved in AD onset and progression and the development of PET probes useful to detect in vivo these biomarkers. Beside the two hallmarks routinely used in AD diagnosis, neurofibrillary tangles and beta-amyloid plaques, specific targets detectable in the late phase of the pathology, new biomarkers (P-gp, TLR4, MIR and free serum copper) involved in the first step of AD have been identified. A P-gp dysfunction in brain can be considered one of the cause of the Aβ accumulation in brain parenchyma and the up-regulation of inflammatory gene expression and inflammatory signaling due to MIR and TLR4 modulated the development and the progression of AD. PET probes for these biomarkers will be the tools to face the challenge of XXI century.
REFERENCES
- Kung, H.F. (2012) The β-amyloid hypothesis in Alzheimer’s disease: Seeing is believing. ACS Medicinal Chemistry Letters, 3, 265-267. doi:10.1021/ml300058m
- Jack, C. R. Jr. (2012) Alzheimer disease: New concepts on its neurobiology and the clinical role imaging will play. Radiology, 263, 344-361. doi:10.1148/radiol.12110433
- Hampel, H., Prvulovic, D., Teipel, S., Jessen, F., Luckhaus, C., Frolich, L., Riepe, M.W., Dodel, R., Leyhe, T., Bertram, L., Hoffmann, W. and Faltraco, F. (2011) The future of Alzheimer’s disease: The next 10 years. Progress in Neurobiology, 95, 718-728. doi:10.1016/j.pneurobio.2011.11.008
- Blennow, K., Hampel, H., Weiner, M. and Zetterberg, H. (2010) Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nature Reviews: Neurology, 6, 131-144. doi:10.1038/nrneurol.2010.4
- Mattsson, N., Zetterberg, H., Hansson, O., Andreasen, N., Parnetti, L., Jonsson, M., Herukka, S.K., Van der Flier, W. M., Blankenstein, M.A., Ewers, M., Rich, K., Kaiser, E., Verbeek, M., Tsolaki, M., Mulugeta, E., Rosén, E., Aarsland, D., Visser, P.J., Schröder, J., Marcusson, J., De Leon, M., Hampel, H., Scheltens, P., Pirttilä, T., Wallin, A., Jönhagen, M.E., Minthon, L., Winblad, B. and Blennow, K. (2009) CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. The Journal of the American Medical Association (JAMA), 302, 385-393. doi:10.1001/jama.2009.1064
- Bekris, L.M., Galloway, N.M., Montine, T.J., Schellenberg, G.D. and Yu, C.E. (2010) APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. American Journal of Medical Genetics B: Neuropsychiatric Genetic, 153B, 409-417.
- Prvulovic, D., Bokde, A.L., Faltraco, F. and Hampel, H. (2011) Functional magnetic resonance imaging as a dynamic candidate biomarker for Alzheimer’s disease. Progress in Neurobiology, 95, 557-569. doi:10.1016/j.pneurobio.2011.05.008
- Winslow, B.T., Onysko, M.K., Stob, C.M. and Hazlewood, K.A. (2011) Treatment of Alzheimer disease. American Family Physician, 83, 1403-1412.
- Morris, J.C., Roe, C.M., Xiong, C., Fagan, A.M., Goate, A.M., Holtzman, D.M. and Mintun, M.A. (2010) APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology, 67, 122- 131. doi:10.1002/ana.21843
- Vemuri, P., Wiste, H.J., Weigand, S.D., Knopman, D.S., Shaw, L.M., Trojanowski, J.Q., Aisen, P.S., Weiner, M., Petersen, R.C. and Jack, C.R. Jr. (2010) Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Annals of Neurology, 67, 308-316.
- Jack, C.R. Jr., Knopman, D.S., Jagust, W.J., Shaw, L.M., Aisen, P.S., Weiner, M.W., Petersen, R.C. and Trojanowski, J. Q. (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology, 9, 119-128. doi:10.1016/S1474-4422(09)70299-6
- Masoodi, T.A., Al Shammari, S.A., Al-Muammar, M.N. and Alhamdan, A.A. (2012) Screening and evaluation of deleterious SNPs in APOE gene of Alzheimer’s disease. Neurology Research International, 2012, Article ID: 480609.
- Schellenberg, G.D. and Montine, T.J. (2012) The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathologica, 124, 305-323. doi:10.1007/s00401-012-0996-2
- Ferencz, B., Karlsson, S. and Kalpouzos, G. (2012) Promising genetic biomarkers of preclinical Alzheimer’s disease: The influence of APOE and TOMM40 on brain integrity. Int. J. Alzheimers Dis., 15 pages.
- Baloyannis, S.J., et al. (2011) Mitochondria are related to synaptic pathology in Alzheimer’s disease. International Journal of Alzheimer’s Disease, 2011, Article ID: 421452.
- Hong, M.G., Alexeyenko, A., Lambert, J.C., Amouyel, P. and Prince, J.A. (2010) Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. Journal of Human Genetics, 55, 707-709. doi:10.1038/jhg.2010.92
- Johnson, S.C., La Rue, A., Hermann, B.P., Xu, G., Koscik, R.L., Jonaitis, E.M., Bendlin, B.B., Hogan, K.J., Roses, A. D., Saunders, A.M., Lutz, M.W., Asthana, S., Green, R.C. and Sager, M.A. (2011) The effect of TOMM40 poly-T length on gray matter volume and cognition in middleaged persons with APOE epsilon3/epsilon3 genotype. Alzheimer’s & Dementia, 7, 456-465. doi:10.1016/j.jalz.2010.11.012
- Roses, A.D. (2010) An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Archives of Neurology, 67, 536-541. doi:10.1001/archneurol.2010.88
- Kim, S., Swaminathan, S., Shen, L., Risacher, S. L., Nho, K., Foroud, T., Shaw, L.M., Trojanowski, J.Q., Potkin, S. G., Huentelman, M.J., Craig, D.W., DeChairo, B.M., Aisen, P.S., Petersen, R.C., Weiner, M.W. and Saykin, A.J. (2011) Genome-wide association study of CSF biomarkers A β1-42, t-τ, and p-τ181p in the ADNI cohort. Neurology, 76, 69-79. doi:10.1212/WNL.0b013e318204a397
- Rosenmann, H. (2012) CSF biomarkers for amyloid and tau pathology in Alzheimer’s disease. Journal of Molecular Neuroscience, 47, 1-14. doi:10.1007/s12031-011-9665-5
- Ono, M., Wilson, A., Nobrega, J., Westaway, D., Verhoeff, P., Zhuang, Z.P., Kung, M.P. and Kung, H.F. (2003) 11C-Labeled stilbene derivatives as Aβ-aggregate-specific PET imaging agents for Alzheimer’s disease. Nuclear Medicine and Biology, 30, 565-571. doi:10.1016/S0969-8051(03)00049-0
- Mathis, C.A., Wang, Y., Holt, D.P., Huang, G.F., Debnath, M.L. and Klunk, W.E. (2003) Synthesis and evaluation of 11C-Labeled 6-Substituted 2-Arylbenzothiazoles as amyloid imaging agents. Journal of Medical Chemistry, 46, 2740-2755. doi:10.1021/jm030026b
- Kudo, Y., Okamura, N., Furumoto, S., Tashiro, M., Furukawa, K., Maruyama, M., Itoh, M., Iwata, R., Yanai, K. and Arai, H. (2007) 2-(2-[2-Dimethylaminothiazol-5-yl] ethenyl)-6-(2-[fluoro]ethoxy)benzoxazole: A novel PET agent for in vivo detection of dense amyloid plaques in Alzheimer’s disease patients. Journal of Nuclear Medicine, 48, 553-561. doi:10.2967/jnumed.106.037556
- Kung, M.P., Hou, C., Zhuang, Z.-P., Zhang, B., Skovronsky, D.M., Trojanowski, J.Q., Lee, V.M.-Y. and Kung, F. (2002) HIMPY: An improved thioflavin-T derivative for in vivo labeling of beta-amyloid plaques. Brain Research, 956, 202-210. doi:10.1016/S0006-8993(02)03436-4
- Chopra, A. (2012) 2-{3-[18F]Fluoro-4-(methylamino) phenyl}-1,3-benzothiazol-6-ol. Molecular Imaging and Contrast Agent Database (MICAD). http://www.ncbi.nlm.nih.gov/books/NBK115136/
- Saint-Aubert, L., Planton, M., Hannequin, D., Albucher, J., Delisle, M., Payoux, P., Hitzel, A., Viallard, G., Péran, P., Campion, D., Laquerriere, A., Barbeau, E., Puel, M., Raposo, N., Chollet, F. and Parient, J. (2012) Amyloid imaging with AV45 ((18)F-florbetapir) in a cognitively normal AβPP duplication carrier. Journal of Alzheimer’s Disease, 28, 877-883.
- Poisnel, G., Dhilly, M., Moustié, O., Delamare, J., Abbas, A., Guilloteau, D. and Barredoi, L. (2011) PET imaging with [18F]AV-45 in an APP/PS1-21 murine model of amyloid plaque deposition. Neurobiology of Aging, 3, 2561-2571.
- Manook, A., Yousefi, B.H., Willuweit, A., Platzer, S., Reder, S., Voss, A., Huisman, M., Settles, M., Neff, F., Velden, J., Schoor, M., Von der Kammer, H., Wester, H., Schwaiger, M., Henriksen, G. and Drzezga, A. (2012) Small-animal PET imaging of amyloid-beta plaques with [11C]PiB and its multi-modal validation in an APP/PS1 mouse model of Alzheimer’s disease. PLoS One, 7, e31310. doi:10.1371/journal.pone.0031310
- Kadir, A., Almkvist, O., Forsberg, A., Wall, A., Engler, H., Långström, B. and Nordberg, A. (2012) Dynamic changes in PET amyloid and FDG imaging at different stages of Alzheimer’s disease. Neurobiology of Aging, 33, 198.e1- 198.e14. doi:10.1016/j.neurobiolaging.2010.06.015
- Villemagne, V.L., Pike, K.E., Chételat, G., Ellis, K.A., Mulligan, R.S., Bourgeat, P., Ackermann, U., Jones, G., Szoeke, C., Salvado, O., Martins, R., O’Keefe, G., Mathis, C.A., Klunk, W.E., Ames, D., Masters, C.L. and Rowe, C.C. (2011) Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Annals of Neurology, 69, 181-192. doi:10.1002/ana.22248
- Vandenberghe, R., Laere, K., Ivanoiu, A., Salmon, E., Bastin, C., Triau, E., Hasselbalch, S., Law, I., Andersen, A., Korner, A., Minthon, L., Garraux, G., Nelissen, N., Bormans, G., Buckley, C., Owenius, R., Thurfjell, L., Farrar, G. and Brooks, D. (2010) 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: A phase 2 trial. Annals of Neurology, 68, 319-329. doi:10.1002/ana.22068
- Villemagne, V.L., Ong, K., Mulligan, R.S., Holl, G., Pejoska, S., Jones, G., O’Keefe, G., Ackerman, U., TochonDanguy, H., Chan, J.G., Reininger, C.B., Fels, L., Putz, B., Rohde, B., Masters, C.L. and Rowe C.C. (2011) Amyloid imaging with 18F-florbetaben in Alzheimer disease and other dementias. Journal of Nuclear Medicine, 52, 1210- 1217. doi:10.2967/jnumed.111.089730
- Fodero-Tavoletti, M.T., Mulligan, R.S., Okamura, N., Furumoto, S., Rowe, C.C., Kudo, Y., Masters, C.L., Cappai, R., Yanai, K., Villemagne, V.L. (2009) In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. European Journal of Pharmacology, 617, 54-58. doi:10.1016/j.ejphar.2009.06.042
- Chang, K.W., Chen, C.C., Lee, S.Y., Shen, L.H. and Wang, H.E. (2009) The synthesis and characterization of [124I] IMPY, a thioflavin-S derivative, in transgenic mouse models of Alzheimer’s disease. Applied Radiation and Isotopes, 67, 1397-1400. doi:10.1016/j.apradiso.2009.02.039
- Neumaier, B., Deisenhofer, S., Sommer, C., Solbach, C., Reske, S.N. and Mottaghy, F. (2010) Synthesis and evaluation of 18F-fluoroethylated benzothiazole derivatives for in vivo imaging of amyloid plaques in Alzheimer’s disease. Applied Radiation and Isotopes, 68, 1066-1072. doi:10.1016/j.apradiso.2009.12.044
- Alagille, D., DaCosta, H., Baldwin, R.M., Tamagnan, G. D. (2011) 2-Arylimidazo[2,1-b]benzothiazoles: A new family of amyloid binding agents with potential for PET and SPECT imaging of Alzheimer’s brain. Bioorganic & Medicinal Chemistry Letters, 21, 2966-2968. doi:10.1016/j.bmcl.2011.03.052
- Ono, M., Watanabe, R., Kawashima, H., Kawai, T., Watanabe, H., Haratake, M., Saji, H. and Nakayama, M. (2009) 18F-labeled flavones for in vivo imaging of beta-amyloid plaques in Alzheimer’s brains. Bioorganic & Medicinal Chemistry, 17, 2069-2072. doi:10.1016/j.bmc.2009.01.025
- Watanabe, H., Ono, M., Kimura, H., Kagawa, S., Nishii, R., Fuchigami, T., Haratake, M., Nakayama, M., Saji and H. (2011) A dual fluorinated and iodinated radiotracer for PET and SPECT imaging of β-amyloid plaques in the brain. Bioorganic & Medicinal Chemistry Letters, 21, 6519-6522. doi:10.1016/j.bmcl.2011.08.063
- Cui, M., Ono, M., Kimura, H., Liu, B.L. and Saji, H. (2011) Synthesis and biological evaluation of indolechalcone derivatives as β-amyloid imaging probe. Bioorganic & Medicinal Chemistry Letters, 21, 980-982. doi:10.1016/j.bmcl.2010.12.045
- Nordberg, A. (2011) Molecular imaging in Alzheimer’s disease: New perspectives on biomarkers for early diagnosis and drug development. Alzheimer’s Research & Therapy, 3, 34-43. doi:10.1186/alzrt96
- Bohnen, N. I., Djang, D.S.W., Herholz, K., Anzai, Y. and Minoshima, S. (2012) Effectiveness and safety of 18F-FDG PET in the evaluation of dementia: A review of the recent literature. Journal of Nuclear Medicine, 53, 59-71. doi:10.2967/jnumed.111.096578
- Kadir, A. and Nordberg, A. (2010) Target-specific PET probes for neurodegenerative disorders related to dementia. Journal of Nuclear Medicine, 51, 1418-1430. doi:10.2967/jnumed.110.077164
- Parri, R.H. and Dineley, T.K. (2010) Nicotinic acetylcholine receptors interact with beta amyloid: Molecular, cellular and physiological consequences. Current Alzheimer Research, 7, 27-39. doi:10.2174/156720510790274464
- Shin, J., Kepe, V., Barrio, J.R. and Small, G.W. (2011) The merits of FDDNP-PET imaging in Alzheimer’s disease. Journal of Alzheimer’s Disease, 26, 135-145.
- Zhang, W., Arteaga, J., Cashion, D.K., Chen, G., Gangadharmath, U., Gomez, L.F., Kasi, D., Lam, C., Liang, Q., Liu, C., Mocharla, V.P., Mu Sinha, A., Szardenings, A. K., Wang, E., Walsh, J.C., Xia, C., Yu, C., Zhao, T. and Kolb, H.C.A. (2012) Highly selective and specific PET tracer for imaging of tau pathologies. Journal of Alzheimer’s Disease, 31, 601-612.
- Cagnin, A., Brooks, D.J., Kennedy, A.M., Gunn, R.N., Myers, R., Turkheimer, F.E., Jones, T. and Banati, R.B. (2001) In-vivo measurement of activated microglia in dementia. Lancet, 358, 461-467. doi:10.1016/S0140-6736(01)05625-2
- Yasuno, F., Ota, M., Kosaka, J., Ito, H., Higuchi, M., Doronbekov, T.K., Nozaki, S., Fujimura, Y., Koeda, M., Asada, T. and Suhara, T. (2008) Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with [11C] DAA1106. Biological Psychiatry, 64, 835-841. doi:10.1016/j.biopsych.2008.04.021
- Carter, S.F., Schöll, M., Almkvist, O., Wall, A., Engler, H., Långström, B. and Nordberg, A. (2012) Evidence for astrocytosis in prodromal Alzheimer’s disease provided by 11C-deuterium-L-deprenyl: A multi-tracer PET paradigm combining 11C-PIB and 18F-FDG. Journal of Nuclear Medicine, 53, 37-46. doi:10.2967/jnumed.110.087031
- Colabufo, N.A., Berardi, F., Cantore, M., Contino, M., Inglese, C., Niso, M. and Perrone, R. (2010) Perspectives of P-glycoprotein modulating agents in oncology and neurodegenerative diseases: Pharmaceutical, biological and diagnostic potentials. Journal of Medicinal Chemistry, 53, 1883-1897. doi:10.1021/jm900743c
- Lam, F.C., Liu, R., Lu, P., Shapiro, A.B., Renoir, J.M., Sharom, F.J. and Reiner, P.B. (2001) Beta-amyloid efflux mediated by p-glycoprotein. Journal of Neurochemistry, 76, 1121-1128. doi:10.1046/j.1471-4159.2001.00113.x
- De Smaele, E., Ferretti, E. and Gulino, A. (2010) MicroRNAs as biomarkers for CNS cancer and other disorders. Brain Research, 1338, 100-111. doi:10.1016/j.brainres.2010.03.103
- Wang, L.L., Huang, Y., Wang, G. and Chen, S.D. (2012) The potential role of microRNA-146 in Alzheimer’s disease: Biomarker or therapeutic target? Medical Hypotheses, 78, 398-401. doi:10.1016/j.mehy.2011.11.019
- Capo, C.R., Arciello, M., Squitti, R., Cassetta, E., Rossini, P.M., Calabrese, L. and Rossi, L. (2008) Features of ceruloplasmin in the cerebrospinal fluid of Alzheimer’s disease patients. BioMetals, 21, 367-372. doi:10.1007/s10534-007-9125-4
- Squitti, R., Ventriglia, M., Barbati, G., Cassetta, E., Ferreri, F., Dal Forno, G., Ramires, S., Zappasodi, F. and Rossini, P.M. (2007) “Free” copper in serum of Alzheimer’s disease patients correlates with markers of liver function. Journal of Neural Transmission, 114, 1589-1594. doi:10.1007/s00702-007-0777-6
- Squitti, R., Barbati, G., Rossi, L., Ventriglia, M., Dal Forno, G., Cesaretti, S., Cesaretti, S., Moffa, F., Caridi, I., Cassetta, E., Pasqualetti, P., Calabrese, L., Lupoi, D. and Rossini, P.M. (2006) Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF beta-amyloid, and h-tau. Neurology, 67, 76-82. doi:10.1212/01.wnl.0000223343.82809.cf
- Squitti, R. (2012) Copper dysfunction in Alzheimer’s disease: From meta-analysis of biochemical studies to new insight into genetics. Journal of Trace Elements in Medicine and Biology, 26, 93-96. doi:10.1016/j.jtemb.2012.04.012
- Ventriglia, M., Bucossi, S., Panetta, V. and Squitti, R. (2012) Copper in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. Journal of Alzheimer’s Disease, 30, 981-984.
- Capiralla, H., Vingtdeux, V., Zhao, H., Sankowski, R., Al-Abed, Y., Davies, P. and Marambaud, P. (2012) Resveratrol mitigates lipopolysaccharideand Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/ STAT signaling cascade. Journal of Neurochemistry, 120, 461-472. doi:10.1111/j.1471-4159.2011.07594.x
- Song, M., Jin, J., Lim, J.E., Kou, J., Pattanayak, A., Rehman, J.A., Kim, H.D., Tahara, K., Lalonde, R. and Fukuchi, K. (2011) TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Journal of Neuroinflammation, 8, 92. doi:10.1186/1742-2094-8-92
- [61] Martin, C., Berridge, G., Mistry, P., Higgins, C., Charlton, P. and Callaghan, R. (2000) Drug binding sites on Pglycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry, 39, 11901-11906. doi:10.1021/b i000559b
- [62] Colabufo, N.A. and Van Waarde, A. (2010) Imaging of ABC transporter function and expression. Current Topics in Medicinal Chemistry, 10, 1701-1702.
- [63] Luurtsema, G., Molthoff, C.FM., Windhorst, A.D., Smit, J. W., Keizer, H., Boellaard, R., Lammertsma, A.A. and Franssen, E.J. (2003) (R)- and (S)-[11C]verapamil as PETtracers for measuring P-glycoprotein function: In vitro and in vivo evaluation. Nuclear Medicine and Biology, 30, 747-751. doi:10.1016/S0969-8051(03)00078-7
- [64] Lazarova, N., Zoghbi, S.S., Hong, J., Seneca, N., Tuan, E., Gladding, R.L., Liow, J.S., Taku, A., Innis, R.B. and Pike, V.W. (2008) Synthesis and evaluation of [N-methyl-11C] N-desmethyl-loperamide as a new and improved PET radiotracer for imaging P-gp function. Journal of Medicinal Chemistry, 51, 6034-6043. doi:10.1021/jm800510m
- [65] Luurtsema, G., Schuit, R.C., Klok, R.P., Verbeek, J., Leysen, J.E., Lammertsma, A.A. and Windhorst A.D. (2009) Evaluation of [11C]laniquidar as a tracer of P-glycoprotein: Radiosynthesis and biodistribution in rats. Nuclear Medicine and Biology, 36, 643-649. doi:10.1016/j.nucmedbio.2009.03.004
- [66] Dörner, B., Kuntner, C., Bankstahl, J.P., Stanek, J., Wanek, T., Stundner, G., Mairinger, S., Löscher, W., Müller, M., Langer, O. and Erker, T. (2009) Synthesis and smallanimal positron emission tomography evaluation of [11C]- elacridar as a radiotracer to assess the distribution of Pglycoprotein at the blood-brain barrier. Journal of Medicinal Chemistry, 52, 6073-6082. doi:10.1021/jm900940f
- [67] Bauer, F., Kuntner, C., Bankstahl, J.P., Stanek, J., Mairinger, S., Dörner, B., Löscher, W., Müller, M., Erker, T. and Langer, O. (2010) Synthesis and in vivo evaluation of [11C]tariquidar, a positron emission tomography radiotracer based on a third-generation P-glycoprotein inhibitor. Bioorganic & Medicinal Chemistry, 18, 5489-5497. doi:10.1016/j.bmc.2010.06.057
- [68] Van Waarde, A., Ramakrishnan, N.K., Rybczynska, A., Elsinga, P.H., Berardi, F., De Jong, J.R., Kwizera, C., Perrone, R., Cantore, M., Sijbesma, J.W., Dierckx, R.A. and Colabufo, N.A. (2009) Synthesis and preclinical evaluation of novel PET probes for P-glycoprotein function and expression. Journal of Medicinal Chemistry, 52, 4524-4532. doi:10.1021/jm900485a
- [69] Colabufo, N.A. and Van Waarde, A. (2010) Preclinical evaluation of [11C]MC18, a radiotracer for PET imaging of P-gp expression. Current Topics in Medicinal Chemistry, 10, 1.
- [70] Leung, K. (2010) 6,7-Dimethoxy-2-{3-[4-[11C]methoxy- 3,4-dihydro-2H-naphthalen-(1E)-ylidene]-propyl}-1,2,3,4- tetrahydro-isoquinoline. Molecular Imaging and Contrast Agent Database (MICAD). http://www.ncbi.nlm.nih.gov/books/NBK45979/
- [71] Kawamura, K., Yamasaki, T., Konno, F., Yui, J., Hatori, A., Yanamoto, K., Wakizaka, H., Ogawa, M., Yoshida, Y., Nengaki, N., Fukumura, T. and Zhang, M.R. (2011) Synthesis and in vivo evaluation of 18F-fluoroethyl GF120918 and XR9576 as positron emission tomography probes for assessing the function of drug efflux transporters. Bioorganic & Medicinal Chemistry, 19, 861-870. doi:10.1016/j.bmc.2010.12.004
- [72] Kawamura, K., Yamasaki, T., Konno, F., Yui, J., Hatori, A., Yanamoto, K., Wakizaka, H., Takei, M., Kimura, Y., Fukumura, T. and Zhang, M.R. (2011) Evaluation of limiting brain penetration related to P-glycoprotein and breast cancer resistance protein using [(11)C]GF120918 by PET in mice. Molecular Imaging and Biology (MIB), 13, 152-160.
- [73] Kannan, P., Telu, S., Shukla, S., Ambudkar, S.V., Pike, V. W., Halldin, C., Gottesman, M.M Innis, R.B. and Hall, M.D. (2011) The “Specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for Breast Cancer Resistance Protein (BCRP/ABCG2). ACS Chemical Neuroscience, 2, 82-89. doi:10.1021/cn100078a
- [74] Colabufo, N.A., Perrone, R., Leopoldo, M., Cantore, M., Contino, M. and Niso, M. (2011) Novel tetrahydroisoquinoline compounds for use in the diagnosis and treatment of neurodegenerative diseases. WO2012159666. http://worldwide.espacenet.com/publicationDetails/originalDocument?CC=WO&NR=2012159666A1&KC=A1&FT= D&ND=3&date=20121129&DB=EPODOC&locale=en_EP
- [75] Paradiso, A., Azzariti, A., Berardi, F., Colabufo, N.A. and Perrone, R. (2006) Verapamil analogues with inhibition activity on ABC (ATP Binding cassette) cell extrution pumps. WO2008017588. http://worldwide.espacenet.com/publicationDetails/originalDocument?CC=WO&NR=2008017588A1&KC=A1&FT= D&ND=3&date=20080214&DB=EPODOC&locale=en_EP
Abbreviations and Acronyms
AD, Alzheimer’s diseaseSPs, senile plaques;
NFTs, neurofibrillary tangles;
Aβ, β-amyloid;
APP, amyloid precursor peptide;
PET, positron-emission tomography;
SPECT, single-photon emission computed tomography;
MRI, magnetic resonance imaging;
PRE1, presenilin-1;
PRE2, presenilin-2;
MTL, medialtemporal lobe;
APOE, Apolipoprotein E;
CNS, Central Nervous System;
TOMM40, Translocase of Outer Mitochondrial Membrane 40;
ROS, Reactive Oxygen Species;
FDG, 2-Deoxy-2-[18F]fluoroglucose;
TLR4, Toll-like Receptors 4;
SB-13, [11C]4-N-methylamino-4'-hydroxystilbene;
[11C]PIB, N-Methyl-[11C]2-(4'-methylaminophenyl)-6-hydroxybenzothiazole;
BF-227, [11C]2-2(-[2-dimethylaminothiazol-5-yl]ethenyl)-6- (2-(fluoro)ethoxy)benzoxazole;
IMPY, 6-[123I]iodo-2-(4'-N,N-dimethylamino)-phenylimidazo[1,2-a]pyridine;
florbetapir, 4-{(E)-2-[6-(2-{2-[2-(18F)Fluoroethoxy]ethoxy} ethoxy)-3-pyridinyl]vinyl}-N-methylaniline;
florbetaben, trans-4-(N-methyl-amino)-4’-{2-[2-(2-[18F] fluoro-ethoxy)-ethoxy]-ethoxy}-stilbene;
flutemetamol, 2-{3-[18F]fluoro-4-(methylamino)phenyl}-1,3- benzothiazol-6-ol;
MCI, mild cognitive impairment;
rCMRglc, cerebral regional cerebral glucose metabolism;
FTLD, FrontoTemporal Lobar Degeneration;
SCID, Severe Combined Immunodeficiency;
DLB, Dementia with Lewy bodies;
[11C]PMP, N-[11C]-methylpiperidine-4-yl-propionate;
[11C]MP4A, N-[11C]-methylpiperidyl-4-yl-acetate;
nAChRs, Nicotinic acetylcholine receptors;
FDDNP, 2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl}-ethylidene)malononitrile;
[11C]-(R)-PK11195, 1-[2-Chlorophenyl]-N-methyl-N-[1-methylpropyl]-3-isoquinolinecarboxamide;
TSPO, translocator protein;
[11C]DAA1106, [11C]N-(2,5-dimethoxybenzyl)-N-(5-fluoro- 2-phenoxyphenyl)-acetamide);
[11C]-DED, N-[11C]-methyl-N-deuterodeprenyl;
P-gp, P-glycoprotein;
ABC, ATP Binding Cassette;
miRNAs, MicroRNAs;
UTR, untranslated region;
CFH, complement factor H;
BACE1, beta-site APP-Cleaving Enzyme, TIR, Toll/IL-1R;
CsA, cyclosporin A;
[11C]dLop , N-methyl-[11C]N-desmethylloperamide;
BCRP, breast cancer resistance protein;
DV, distribution volume;
MC18,6,7-dimethoxy-2-{3-[4-methoxy-3,4-dihydro-2H-napthalen-(1E)-ylidene]-propyl}-1,2,3,4 tetrahydroisoquinoline.