Neuroscience and Medicine
Vol.08 No.03(2017), Article ID:79414,8 pages
10.4236/nm.2017.83005
Neurofibromatosis Type 1 in Four Children Cases
C. Mammad1, N. Mekaoui1, F. Z. Ouadghiri1, K. Mammad2, L. Karboubi1, B. S. Benjeloun Dakhama1
1Pediatric Medical Emergency Department, Rabat Children’s Hospital, University Hospital of IbnSina, Faculty of Medicine and Pharmacy, Mohammed V University, Rabat, Morocco
2Unit of Neuroscience and Applied Nutrition, Laboratory of Nutrition, Health and Environment, Department of Biology, Faculty of Science, Kenitra, Morocco
Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: July 27, 2017; Accepted: September 25, 2017; Published: September 28, 2017
ABSTRACT
Neurofibromatosis Type 1 (NF-1 or Von Recklinghausen disease) is an autosomal dominant genetic disease, characterized by an extreme variability of its clinical expression which is also found in the same family. Our work focuses on the exploitation of four cases of patients with NF-1 who were enrolled in the paediatric neurology consultation at Rabat Children’s Hospital. They are two infants and two children. Otherwise the diagnosis was made in front of the existence of café au lait and lentiginous spots in two boys, also the existence of café au lait spots and abnormalities in brain imaging in two girls. Thus an evolution was marked by a favorable outcome for three patients and neurological sequelae in one patient.
Keywords:
Neurofibromatosis Type 1, Children, Café au lait Spots, Lentiginous

1. Introduction
The NF-1 is a condition characterized by changes in skin coloring (pigmentation) and the growth of tumors along nerves in the skin, brain, and other parts of the body. Beginning in early childhood, almost all people with NF-1 have multiple café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. These spots increase in size and number as the individual grows older. Freckles in the underarms and groin typically develop later in childhood. The signs and symptoms of this condition vary widely among affected people. In addition, it is a congenital anomaly of the embryonic development of the neuro-ectodermal tissue, at the origin of particularly cutaneous and neurological tumor malformations [1] . It accounts for 95% of all neurofibromatosis; its global distribution is homogeneous without ethnic predilection and its penetrance is almost complete at the age of 8 years [2] .
The NF1 gene is one of the genes whose spontaneous mutation rate is one of the most important in humans: About half of the people affected by this disease are the result of a de novo mutation [3] .
2. Aim
Clinical and evolutionary and therapeutic evaluation with a focus on the epidemiological specificities of NF-1 in children.
3. Methodology
Our study is a retrospective study of 4 cases with neurofibromatosis type 1, collected during the consultation of pediatric neurology of the Rabat hospital, over a period of 5 years, spread from March 2011 to February 2016.
The inclusion criteria: These are patients whose diagnosis of type 1 neurofibromatosis is certain according to the diagnostic criteria of the NIH (The national institute of health) 1988 (at least 2 of the 7 cardinal diagnostic criteria). Exclusion criteria: 1 case was excluded for insufficient diagnostic criteria.
4. Results
We present hereafter the description of our 4 observations followed by summary tables of the main characteristics of the patients concerned (Table 1, Table 2).

Table 1. Summary of the clinical characteristics of the four observations.
CALM: Café-au-lait spots; CNS: central nervous system.
Table 2. Frequency of clinical signs in our study.
Observation No. 1: S. female, aged 1 year and 8 months, without a particular pathological history, consult for delay motor acquisitions evolving since the age of 10 months, The clinical examination has objectified axial and peripheral hypotonia with the presence of multiple café au lait spots (>6) diffuse all over the body, of different sizes, the largest was 5cm. The remainder of the somatic examination and the paraclinic assessment are unusual. Otherwise the diagnosis of NF-1 was retained on the combination of cutaneous and neurological signs, Initial management consisted of orthopedic rehabilitation sessions. The evolution was marked by a marked improvement, but the skin lesions remained unchanged and the fundus of the eye remained normal.
Observation No. 2: Y, a male, 10 months old, with no significant neonatal history, consults for a marcocrania and motor acquisition delay, and the clinical examination finds CP (Cranial perimeter) = 48 cm (+3SD), a slight hypotonia of the lower limbs, without motor deficit, it presences of multiple cafe au lait (>6) spots scattered over the whole body of different sizes, several of which are higher than 0.5 cm. Thus lenticular spots and in sheet, the thoracic examination makes it possible to palpate a hard formation sitting at the level of the anterior wall, and the rest of the somatic examination was without peculiarities. The thoracic radiography showed a clinically palpable left para-sternal swelling with no radiological translation. In addition, the TDMC and heart Eco are unusual, while the diagnosis of NF-1 has been retained. Initial management consisted of clinical monitoring of the macrocranium and motor rehabilitation. The evolution has been favorable with good psycho-motor development, but macrocrania is still persistent with a CP at 51 cm at 15 months (+2SD).
Observation No. 3: O, a male, 10 years old, consults for psychomotor retardation and epilepsy, The examination noted the presence of severe relational difficulty, hyperactivity, fine motor disorders, and difficulty in the acquisition of words, with the presence allows to note multiple cafe au lait (>6) tasks on the whole body of different sizes of which the largest measured 3/1.5cm, as well as the presence of the lentiginous. Brain MRI has demonstrated an UBOs, the fundus was normal, EEG: without abnormalities, abdominal ultrasound and heart Eco are normal. The diagnosis of a NF-1 was retained on the association of cutaneous, neurological signs, and abnormalities in cerebral imaging. Initial management consisted of Depakine-based antiepileptic therapy at a dose of 30 mg/Kg/d with psychomotor rehabilitation and speech therapy. Thus during its follow-up, the evolution was marked by a pharmaco-resistance of its epilepsy, and the aggravation of the cognitive impairments associated with moderate mental retardation.
Observation No. 4: I, is a female, 11 years old, present for 1 month of headache and vomiting in a context of apyrexia. Clinical examination showed a size at (−3SD), a slurred neck, unequal limbs, and the presence of multiple diffuse café au lait spots (>6) throughout the body of different sizes, the largest was 5 cm (Figure 1). The orthopedic examination showed a hyper lumbar lordosis and confirmed the absence of bony deformations of the upper and lower limbs. Also, the cerebral MRI (Figure 2) showed a large active tri ventricular hydrocephalus, associated with cystic dilatation, and a strong suspicion of Arnold chiari type 2. There is also a nodular contrast enhancement under left frontal cortical and signal abnormalities of the internal substance under internal temporal cortical. The fundus revealed bilateral papillary edema. The diagnosis of NF-1 has a high probability with gliomas of the optical pathways was retained. Its initial management consisted in sending it to the neurosurgery department for possible hydrocephalic bypass, and orthopedic rehabilitation of the lumbar hyperlordosis. Since its follow-up is done in these structures (Table 1).
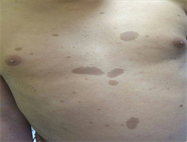
Figure 1. CALMs observed in Case 4.

Figure 2. Cerebral MRI images of Case 4 showing tri-ventricular hydrocephalus associated with cystic dilatation, nodular contrast enhancement under left frontal cortical coil, and signal abnormalities of the internal temporal cortical internal substance.
5. Discussion
The Neurofibromatosis are genetic diseases. They are part of the phacomatoses caused by dysplastic lesions of the neural crest [4] .Most commonly, there are certain skin signs (pigmented spots and tumors) [5] [6] [7] , however, The NF-1 or Von Recklinghausen disease is a genetic condition with autosomal dominant transmission, 50% of which are sporadic cases of de novo mutation. The gene responsible was localized (in 1987) on chromosome 17 by the description of two balanced translocations involving the 17q11.2 band, and to binding studies with markers of this chromosome [8] [9] .
The NF-1 can occur at any age, so we can’t talk about age of onset: it is a congenital condition and children are born with their disease. This is why the initial signs of the disease may vary from one individual to another [10] [11] . Sex-ratio is one in most series [12] , and no predominant geographic distribution has been described in the literature.
In our series, the age of revelation varied between 10 months to 11 years, the sex ratio is 1, without racial or ethnic predominance, and none of our patients have benefited from a mutation research NF1: explained most often for lack of means. Otherwise the clinical picture most often associates multiple café au lait spots, axillary and inguinal lentiginous, cutaneous neurofibromas and Lisch nodules.
The morbidity and mortality associated with NF-1 result from the occurrence of multi-systemic complications. The most frequent complications are neurological complications including learning and attention disorders [13] , also some intracerebral gliomas, in particular the glioma of the optic pathways which mainly affects the optic nerve and/or chiasm, but may also extend along the retrochiasmatic optic pathways and the attainment of both optic nerves is recognized as a specific sign of NF1 [14] , and a difficulty in motor coordination, a recent memory deficit, hydrocephalus, mental retardation, macrocrania, and headache [15] .
The cutaneous complications are represented by cutaneous and plexiform neurofibromas, gelatinous Myxoglioma of the endoneurium, dermatolysis or cutis laxa, and xanthogranulomas. These complications are at the origin of the aesthetic and functional sequelae [16] . There are also orthopedic complications including scoliosis and cyphoscoliosis, bone erosions, osteoporosis, and tumor secondary deformities [17] . Other complications are less frequent such as vascular abnormalities as well as other visceral attacks [18] [19] .
We propose a decision tree for the behavior to be treated before the appearance of CALM in the child (Figure 3):
a) If the Child with TCL <3 without any ATCD (antecedent): reassure the parents and ask for a simple monitoring.
b) If CALM is greater than 6 an exploration should be considered in search of other diagnostic criteria of NF1 (lentiginous, lisch nodules, Asymptomatic Optic Glioma ...).

Figure 3. A decision tree for the behavior to be treated before the appearance of CALM in the child.
c) At the end of this exploration, 2 possibilities are offered:
1) the diagnosis of NF-1 is retained before the association of CALM (> 6) plus an additional diagnostic criterion;
2) the absence of other clinical criteria must make seek the mutation NF-1: if the research is negative move to other differential diagnoses.
6. Conclusion
Unfortunately, the therapeutic strategy remains limited to the treatment of different manifestations. Though with gene therapy, it will be possible in the near future to intervene at the very origin of the disturbances and to compensate for all the pathological consequences. The development of multidisciplinary centers for the care of patients with NF-1 will improve the level of knowledge of professionals and the population, to standardize surveillance and treatment procedures, and to establish phenotype-genotype correlations that facilitate patient prevention, surveillance and support.
Cite this paper
Mammad, C., Mekaoui, N., Ouadghiri, F.Z., Mammad, K., Karboubi, L. and Dakhama, B.S.B. (2017) Neurofibromatosis Type 1 in Four Children Cases. Neuroscience & Medicine, 8, 33-40. https://doi.org/10.4236/nm.2017.83005
References
- 1. Koob, M. and Dietemann, J.-L. (2014) Imagerie de la neurofibromatose type 1. EMC Radiologie et imagerie médicale-musculo squeletique-neurologique maxillofaciale, 9, 1-13.
- 2. Valeyrie, L. and Allanore, P. (2009) Wolkenstein Neurofibromatose 1 et formes variantes. Elsevier Masson SAS, Paris, 17-170-A-65.
- 3. Jett, K. and Friedman, J.M. (2010) Clinical and Genetic Aspects of Neurofibromatosis 1. Genetics in Medicine, 12, 1-11. https://doi.org/10.1097/GIM.0b013e3181bf15e3
- 4. Sales de Gauzy, J., Lemaire, O., Accadbled, F. and Zabraniecki, L. (2008) Manifestations orthopédiques des phacomatoses. [Orthopedic Manifestations of Phacomatoses.] Elsevier Masson SAS, Paris, 14-023-T-10.
- 5. Huson, S.M. and Hughes, R.A.C. (1994) The Neurofibromatoses: A Pathogenetic and Clinical Overview. Chapman and Hall, London.
- 6. Rubenstein, A.E. and Korf, B.R. (1990) Neurofibromatoses: A Handbook for Patients, Families, and Health-Care Professionals. Thieme Medical Publishers, New York.
- 7. Riccardi, V.M. (1982) Neurofibromatosis: Phenotype, Natural History and Pathogenesis. Johns. Hopkins University Press, Baltimore, MD.
- 8. Henry, I. (1995) Type 1 Neurofibromatosis or Von Recklinghausen Disease: Many Questions Remain. médecine/sciences, 11, 93-98. https://doi.org/10.4267/10608/2164
- 9. Schmidt, M.A., Michels, W. and Dewald, G.W. (1987) Cases of Neumfibromatosis with Rearrangements of 17 Involving Band 17q11.2. American Journal of Medical Genetics, 28, 771-777. https://doi.org/10.1002/ajmg.1320280333
- 10. Korf, B.R. (1998) The NF1 Genetic Analysis Consortium. Neurofibromatosis Type 1: From Genotype to Phenotype. Bios Scientific Publishers, Oxford, 57-63.
- 11. Bethesda, M.D. (1988) National Institute of Health Consensus Development Conference Statement: Neurofibromatosis. Neurofibromatosis, 1, 172-178.
- 12. Wolkenstein, P. and Zeller, J. (2002) Et Ismaili N. [Neurofibromatosis.] Elsevier, Paris, 98-755-A-10.
- 13. Nguyen, S., Mahé, J.Y., Coutant, X., Gloanec, Y. and Peuvrel, E. (1997) Troubles de l’apprentissage: Dépistage et prise en charge des enfants NF1. [Learning Disorders: Screening and Management of Children NF1.] Médecine Thérapeutique, 3, 636-639.
- 14. Caron, S., Soto-Ares, G., Vinchon, M., Dhellemes, P. and Pruvo, J.P. (2004) Neuroimagerie des phacomatoses. [Neuroimaging of Phacomatoses.] Radiology Sheets, 44, 241-263.
- 15. Beylot, C.I., Doutre, M.S. and Bioulac, P. (1981) Phakomatoses Neurofibromatosis. Encyclmedchir Paris Dermatology, 12720, A10, 2.
- 16. Morier, P., Merot, Y., Paccaud, D., Beck, D. and Frenk, E. (1990) Juvenile Chronic Granulocytic Leukemia, Juvenile Xanthogranulomas, and Neurofibromatosis. Case Report and Review of the Literature. Journal of the American Academy of Dermatology, 22, 962-965.
- 17. Gilbert, A. and Brockman, R. (1995) Congenital Pseudarthrosis of the Tibia. Long-Term Follow up of 29 Cases Treated by Microvascular Bone Transfer. Clinical Orthopaedics and Related Research, 37-44.
- 18. Lachhab, H. (2010) These en medecine: Neurofibromatose de type1 chez l’enfant: Mise au point clinique et genetique, Rabat Annee, These. [Thesis in Medicine: Neurofibromatosis Type 1 in Children: Clinical and Genetic Development, Rabat,Thesis.] N: 105.
- 19. Ameur, A., Touiti, D., Jira, H., Alami, M.E., Ouahbi, Y. and Abbar, M. (2003) Urinary and Nephrological Appearances in Von Recklinghausen’s Disease. Two Case Reports and Literature Review. Annalesd’urologie, 37, 150-154.