CellBio
Vol.2 No.4(2013), Article ID:40433,6 pages DOI:10.4236/cellbio.2013.24021
Alteration of Major Insulin Signaling Molecules by Chronic Ethanol in Hypertensive Vascular Smooth Muscle Cells of Rats
Department of Biological Sciences, Tennessee State University, Nashville, USA
Email: bwashington@tnstate.edu
Copyright © 2013 Sparkle D. Williams, Benny Washington. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received September 14, 2013; revised October 14, 2013; accepted October 21, 2013
Keywords: HVSMCs; Ethanol; Insulin; ERK 1/2; AKT
ABSTRACT
Insulin resistance is an important risk factor in the development of cardiovascular diseases such as hypertension and atherosclerosis. However, despite its importance, the specific role of insulin resistance in the etiology of these diseases is poorly understood. At the same time, ethanol (ETOH) is a potent vasoconstrictor that primarily induces down regulation of mitogen activated protein kinases (MAPKs) which could exacerbate insulin resistance and possibly lead to cardiovascular diseases. This article describes how chronic ETOH exposure interferes with insulin signaling in hypertensive vascular smooth muscle cells (HVSMCs) which leads to the alteration of MAPKs, the major signaling molecules. Elevated (50 - 800 mM) chronic exposure (24 hr) of HVSMCS to ETOH prior to insulin stimulation decreased insulin-induced ERK 1/2 (MAPKs) and AKT expression. Similar experiments were conducted using normotensive cells from rat. These cells showed reductions in insulin-induced ERK 1/2 phosphorylation as well, but only at higher concentrations of ETOH (400 - 800 mM). These alterations in insulin signaling could provide an alternative molecular mechanism that may increase the risk of insulin resistance, thus increasing the possibility of cardiovascular diseases.
1. Introduction
Insulin resistance can lead to the development of cardiovascular diseases such as hypertension and atherosclerosis [1-3]. However, there is little information known about the role of insulin resistance in cardiovascular diseases. As a result of chronic alcohol consumption, insulin resistance can occur. Insulin release and activation is important in the body to alleviate the outcome of diabetes and in some cases, hypertension [4,5]. Chronic ethanol (ETOH) consumption can also affect insulin’s ability to bind to its receptor, therefore, leading to insulin resistance [4,5]. However, there are not many studies to prove this effect, especially in a hypertensive phenotype. Studies have shown that moderate consumption of alcohol is not a precursor to coronary artery disease, but chronic consumption of alcohol can be detrimental [5].
Insulin signaling is important in the cell for the release of insulin to control glucose intake and lipid metabolism [5]. Insulin has an important role in the biological process because it binds to its receptor (insulin receptor) to initiate glucose metabolism [5]. The insulin receptor is a transmembrane domain spanning tyrosine kinase receptor composed of alpha and beta subunits that mediate the actions of insulin [6-8]. Insulin is closely associated with hypertension, non-insulin dependent diabetes, atherosclerosis, and dyslipidemia [6,7]. As a result, insulin activation of PI3K-AKT (Phosphoinositide 3-Kinase), which mediates neuronal survival, motility, energy metabolism, and plasticity is impaired [6,8,9].
The extracellular signal-regulated kinases (ERKs), also known as extracellular mitogen activated protein kinases (MAPKs), have been found to be altered by ethanol treatment of vascular smooth muscle cells from the aorta of a rat [5,9-11]. This effect of ETOH has been shown to be manifested via several pathways by the use of signaling inhibitors, such as PKC, leading to a cascading effect in treated cells [11,12]. Several studies have shown that insulin activates a complex set of intracellular responses, including the activation of mitogenactivated protein kinases ERK 1/2 [13] and AKT [5].
The normal role of AKT in the cell is to propagate insulin receptor signaling to downstream effectors [14]. AKT is downstream of PI3K, both of which are a part of one of the major pathways in insulin signaling. The other major pathway is the mitogen activated protein kinase pathway. It is also known that AKT has a role in activating insulin response [10]. Alteration of AKT expression is exhibited by ETOH impaired insulin signaling in the body and a decrease in glucose transport of rat cardiac muscle cells [10]. Although insulin response can be altered with chronic ETOH, there are other factors that can alter this response such as genetics and the person’s environment [5].
Chronic ETOH consumption in experimental animal models has been shown to alter insulin signaling events via the mitogen activated protein kinases producing insulin resistance in the liver. These adverse effects of ethanol have been shown to be the result of the inhibition of insulin or insulin-like growth factor which alters mRNA and DNA synthesis and the activation of proapoptotic signals through PI3K and AKT [15-17]. Results from previous studies suggest that ETOH impairment of insulin action is likely to be downstream from PI3K, however, the mechanisms underlying the effects of ETOH on insulin resistance and the effect of insulin resistance on the development of cardiovascular diseases remain to be determined [5,18]. This paper describes how chronic ETOH exposure can alter insulin signaling in HVSMCs using mitogen protein kinases as indicators. Chronic ETOH exposure’s effect on insulin signaling has not been analyzed before in hypertensive cells.
2. Materials and Methods
2.1. Reagents and Antibodies
ERK 1/2 (p44/p42) and AKT antibodies were purchased from Cell Signaling (Beverly, MA); anti-rabbit IgG antibodies (horseradish peroxidase linked) from Amersham Bioscience (Piscataway, NJ); and ECL detection system was obtained from Pierce Biotechnology (Rockford, IL). Other supplies include Dulbecco’s Modified Eagle’s Medium (Amersham), fetal bovine calf serum, penicillin and streptomycin were purchased from Sigma/Aldrich (St. Louis, MO).
2.2. Cell Culture
Vascular smooth muscle cells (VSMCs) were received from Vanderbilt University. Cells were cultured in DMEM containing 10% fetal bovine serum, 2% penicillin and streptomycin. Subcultured passages were between 3 and 12. Cells were maintained at a pH of 7.1 in 75 cm2 flasks under a humidified atmosphere of 5% CO2, 95% O2 at 37˚C and plated in 6-well falcon plates.
2.3. Insulin Treatment
For dose response experiments, cells were stimulated with 1, 2, 4, 8, and 16 µM insulin for 30 min. Insulin was aspirated from each well. For time course experiments, cells were stimulated with 8 µM of insulin for 1, 5 10, 20, and 40 min. Cells were lysed with 300 - 500 µl Laemmli Sample Buffer (2% SDS, 25% glycerol, 0.01% bromophenol blue, and 62.5 mM Tris-HCl pH 6.8). Cells were then scraped from the monolayer surface and collected in microcentrifuge tubes.
2.4. Ethanol Treatment
Hypertensive and normal rat cells were induced with 50, 100, 200, 400, and 800 mM ETOH. Control cells were induced with a DMEM (-) solution containing no serum that aids in cell proliferation. After a 24 hr incubation period, ETOH was aspirated from all wells and cells (except controls) were stimulated with 8 µM of insulin. Insulin was aspirated from each well and the cells were lysed with 300 - 500 µl Laemmli Sample Buffer (2% SDS, 25% glycerol, 0.01% bromophenol blue, and 62.5 mM Tris-HCl pH 6.8). Cells were then scraped from the monolayer surface and collected in microcentrifuge tubes.
2.5. Western Blot Analysis
Whole cell lysates were collected and diluted with sample buffer to equal concentrations of 40 µg/20 µl. Lowry protein assay was conducted to determine standard protein concentration. Protein samples were then separated along with rainbow markers to measure the molecular weight of proteins on a 10% SDS-polyacrylamide gel from Bio-Rad Laboratories (Hercules, CA) at 200 volts for approximately 50 min. Proteins were then transferred to a nitrocellulose membrane from Amersham Biosciences (Piscataway, NJ) using a semi-dry transfer apparatus at 10 volts for 90 min. Blots were blocked with 2% non-fat dry milk in TBS (Tris-Buffered Saline) for at least one hour. After which blots were incubated with primary antibodies ERK 1/2 or AKT overnight followed by anti-rabbit secondary antibody for 1 hour. Blots were immersed in chemiluminscent solution and developed in a dark room.
2.6. Statistical Analysis
All experiments were performed in triplicate and expressed as means ± SE of the density using arbitrary units from three individual experiments. Statistical significance was determined with paired or unpaired onetailed Student’s t-test, with P < 0.05 considered significant.
3. Results
3.1. Insulin Induction Increases ERK 1/2 and AKT Expression in Hypertensive VSMCs
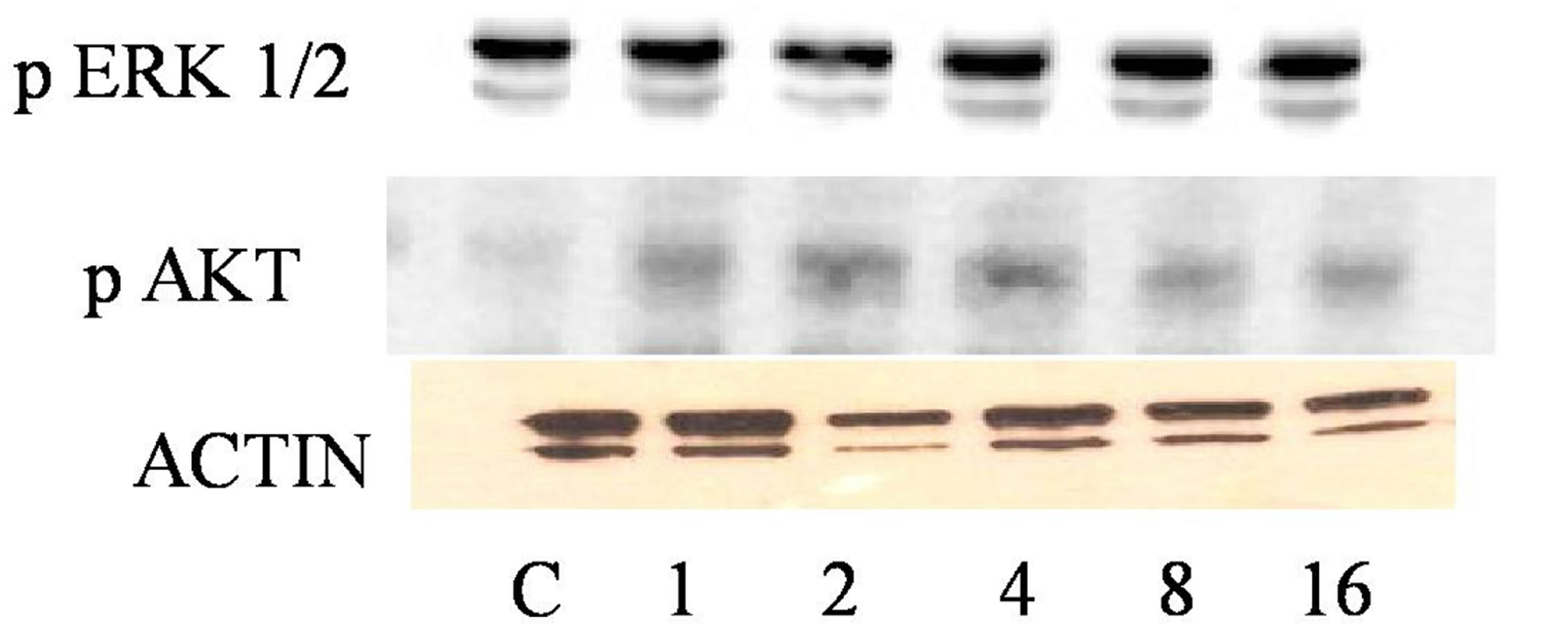
Before we could determine the effect of ETOH on insulin signaling, we first had to determine the maximum concentration of insulin it would take to stimulate phosphorylation of ERK 1/2 and AKT via Western Blotting analysis. HVSMCs were stimulated with a concentration range of 1 - 16 µM of insulin for 30 min. Figure 1 denotes that stimulating HVSMCs with 1 - 16 µM of insulin, increased ERK 1/2 phosphorylation by approximately 23%. In addition, this insulin stimulation increased in AKT expression 33% above basal with a maximum expression detected with 8 µM of insulin (Figure 2). This data suggests that AKT activation in HVSMCs is more sensitive than ERK 1/2 to insulin signaling.
3.2. Insulin Induction Increases ERK 1/2 and AKT Expression in Normal VSMCs
In order to determine the effect of insulin signaling in normal cells, VSMCs were stimulated with a concentration range of 1 - 16 µM of insulin. Maximal expression of phosphorylated ERK 1/2 occurred with 8 µM of insu-
 (a)
(a)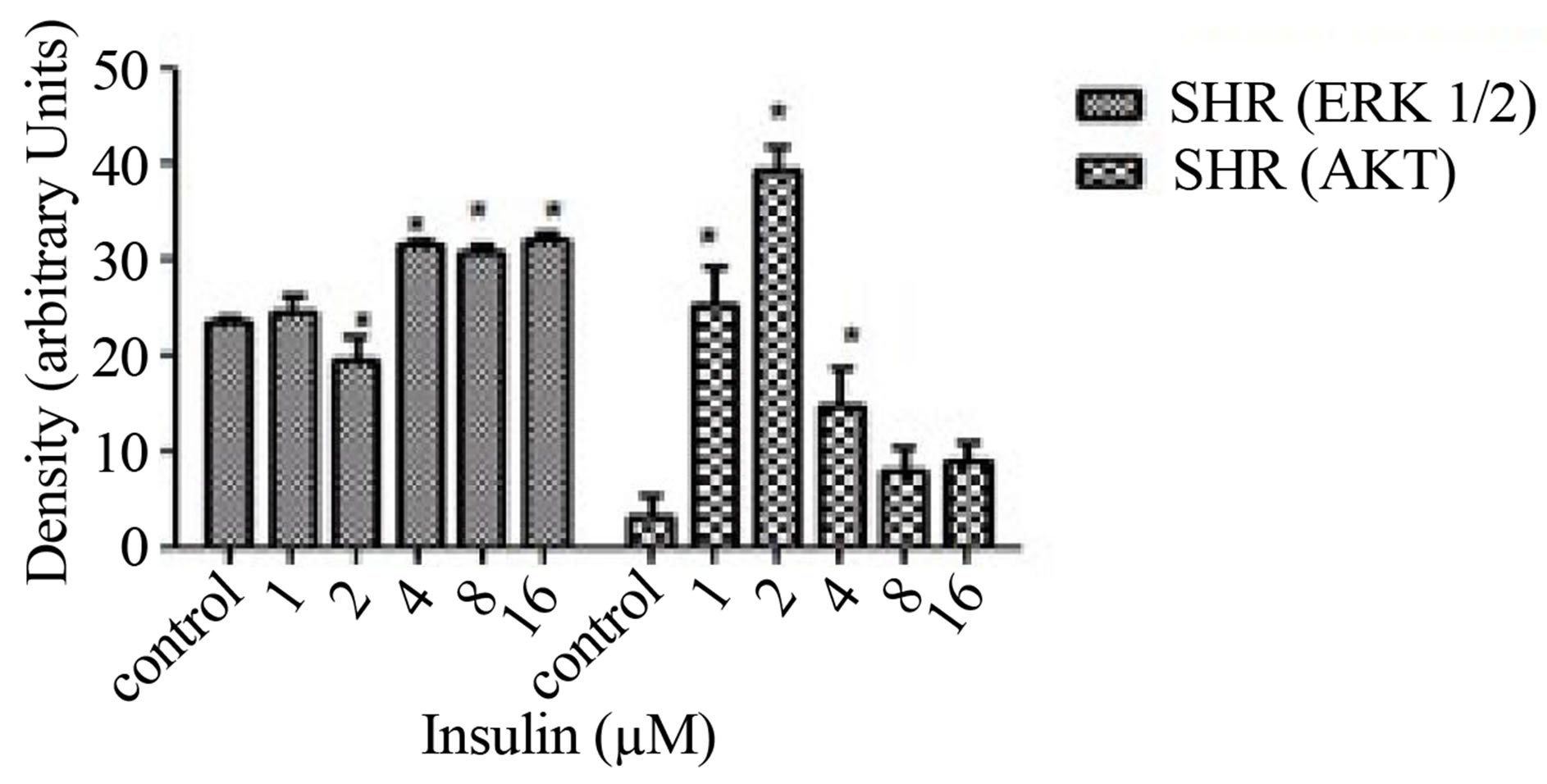 (b)
(b)
Figure 1. Dose response curve for insulin on ERK 1/2 and AKT expression in HVSMCs. Cells were stimulated with 1, 2, 4, 8, and 16 µM insulin for 30 min and lysate harvested. (a) Western Blot Analysis of SHR (Spontaneously Hypertensive) lysate probed with antibodies for ERK 1/2 and AKT expression; (b) Graphical representation of data by densitometry analysis software was taken from a mean of three experiments p < 0.05 compared to control.
 (a)
(a) (b)
(b)
Figure 2. Dose response curve for insulin on ERK 1/2 and AKT expression in normal VSMCs. Cells were stimulated with 1, 2, 4, 8, and 16 µM insulin for 30 min and lysate harvested. (a) Western Blot Analysis of WKY (Wistar Kyoto) lysate probed with antibodies for ERK 1/2 and AKT expression; (b) Graphical representation of data by densitometry analysis software was taken from a mean of three experiments p < 0.05 compared to control.
lin, which was significantly different from basal (Figure 2). No significant increases in ERK 1/2 were observed between 1 - 4 mM (Figure 2). Similar experiments showed that AKT expression was significantly increased throughout all concentrations except 1 µM with maximal expression at 8 µM. Insulin seemingly, also induced increases in AKT with 1 - 8 µM in normal VSMCs (Figure 2). This increase in expression of AKT was not observed when cells were stimulated with 16 µM insulin which resulted in complete inhibition.
3.3. Chronic ETOH Impairs Insulin Signaling in Hypertensive VSMCs
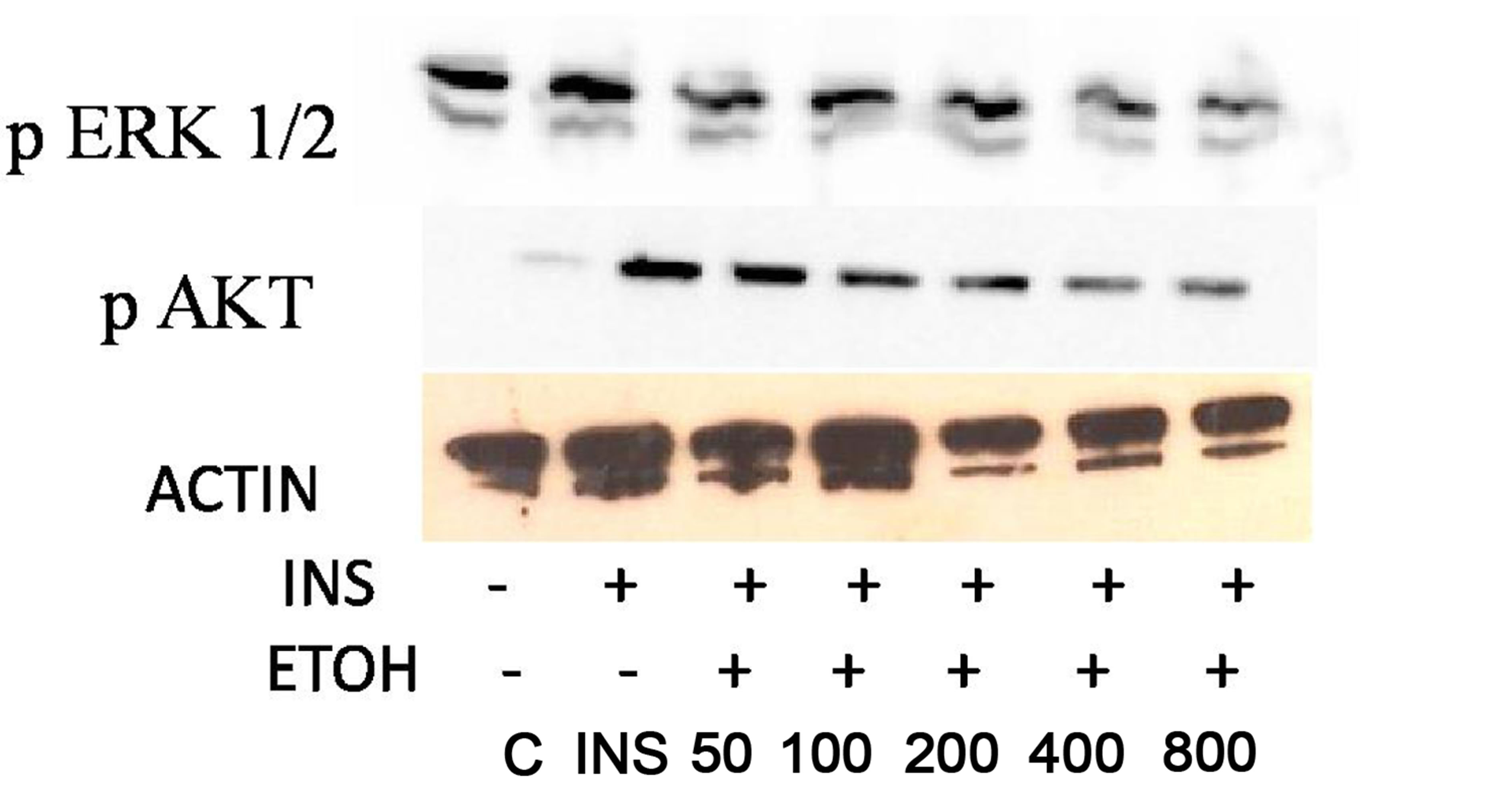
In order to investigate whether ETOH alters insulin signaling in HVSMCs, cells were treated chronically (24 hrs) with 50 - 800 mM of ETOH. After ETOH treatment HVSMCs were stimulated with 8 µM insulin for 30 min and Western Blotting was performed. As a result, ERK 1/2 expression significantly decreased with 50 - 800 mM ETOH treatment by 10% compared to insulin stimulation only (Figure 3). Using the same treatment range (50 - 800 mM) AKT expression was evaluated by chronic ETOH treatment. After 24 hours ETOH treatment, AKT expression HVSMCs significantly decreased (Figure 3). This decrease was gradual as the concentration increased.
 (a)
(a) (b)
(b)
Figure 3. Twenty-four hour ETOH treatment of HVSMCs. Cells were treated with specified concentrations of ETOH (50 - 800 mM) and stimulated with 8 µM insulin for 30 min and lysate collected. (a) Western Blot Analysis of SHR (Spontaneously Hypertensive) lysate probed with antibodies for ERK 1/2 and AKT expression; (b) Graphical representation of data by densitometry analysis software was taken from a mean of three experiments p < 0.05 compared to control.
3.4. Chronic ETOH Impairs Insulin Signaling in Normal VSMCs
In order to investigate whether ETOH alters insulin signaling in normal VSMCs, we evaluated chronic ETOH treatment in them as well. Cells were treated chronically with 50 - 800 mM of ETOH for 24 hr. After ETOH treatment, VSMCs were stimulated with 8 µM insulin for 30 min. Fifty and 100 mM of ETOH had no significant effect on insulin-induced AKT expression when compared to insulin stimulation alone. However, 200 - 800 mM reduced insulin-induced AKT expression approximately 20%. Chronic ETOH treatment of normal VSMCs seems to cause an increase in signal at 50 mM and a decrease at higher concentrations when compared to the insulin induced increase alone (Figure 4).
4. Discussion
The research findings in this paper provide evidence for changes in mitogen-activated protein kinases possibly contributing to the onset of cardiovascular disease. The focus of this study is to show the association of chronic ethanol-induced changes in ERK 1/2 and AKT expression in HVSMCs. Recent reports show that hypertensive
 (a)
(a)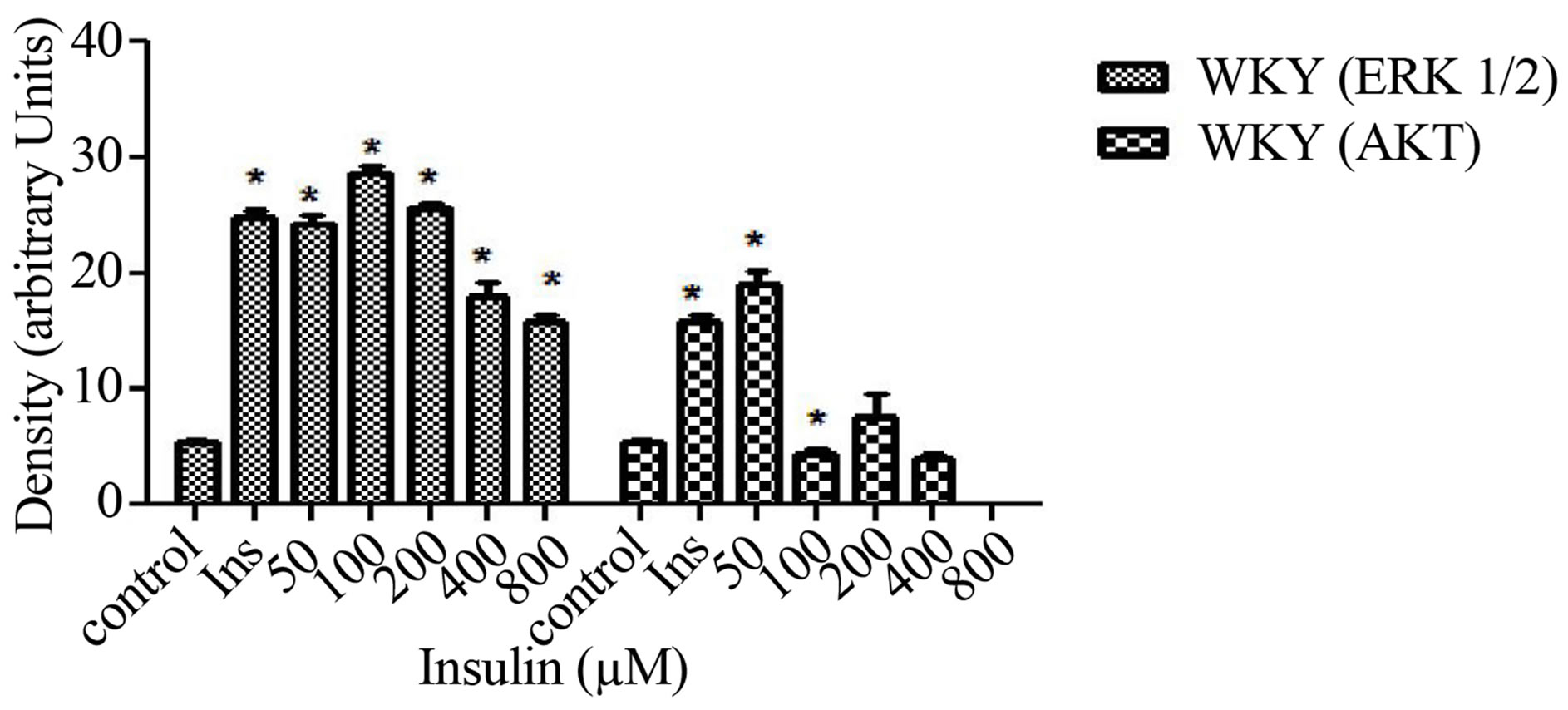 (b)
(b)
Figure 4. Twenty-four hour ETOH treatment of normal VSMCs. Cells were treated with specified concentrations of ETOH (50 - 800 mM) and stimulated with 8 µM insulin for 30 min and lysate collected. (a) Western Blot Analysis of WKY (Wistar Kyoto) lysate probed with antibodies for ERK 1/2 and AKT expression; (b) Graphical representation of data by densitometry analysis software was taken from a mean of three experiments p < 0.05 compared to control.
persons are predisposed to the development of diabetes. [19]. Seventy-five percent of cardiovascular diseases are attributed to diabetes, hypertension, and alcohol, indicating that this is a contributing factor to the onset of hypertension.
In Figure 1, ERK 1/2 phosphorylation was increased in hypertensive cells with increasing concentrations of insulin. This confirms that cells are able to survive with higher concentrations of insulin indicated by ERK 1/2. On the other hand, when AKT stimulation occurs, it has been reported that cellular expression decreases. This could be due to the alteration of insulin signaling. In Figure 2, normal VSMCs with ERK 1/2 and AKT decreased with 1 - 8 µM insulin. With 16 µM of insulin, both ERK 1/2 and AKT expression was inhibited. In Figure 3, hypertensive cells treated with ethanol ERK 1/2 and AKT both decrease in expression as the concentrations of ETOH increase which could mean that ethanol is affecting insulin signaling. Figure 4 depicts a biphasic effect of ETOH on insulin signaling in normal cells which suggest that there may be different effect in normal cells.
Cardiovascular diseases include atherosclerosis and hypertension. Risk factors for such diseases of the cardiovascular system include family history, diabetes, obesity, smoking, excessive alcohol intake, and a diet high in salt and/or low in antioxidant nutrients. Individuals with hypertension are at increased risk for atherosclerotic diseases such as stroke, heart, and kidney disease which can be exacerbated by diabetes and alcohol [20]. The adverse effects of long-term excessive use of alcohol are similar to those seen with other sedative-hypnotics drugs (apart from organ toxicity which is much more problematic with alcohol).
Though the underlying mechanisms remain undefined, accumulating evidence strongly suggests that ETOH interferes with insulin’s action by altering mitogen activated protein kinases. The major signaling molecules, MAPKs, implicated in the biological actions of insulin, and the expression of the insulin receptor may be major factors leading to cardiovascular diseases.
5. Acknowledgements
The authors would like to acknowledge The Research and Engineering Training Program (REAP) and Title III Program for providing support for this work.
REFERENCES
- P. De Meyts, “The Structural Basis of Insulin and Insulin-Like Growth Factor-I Receptor Binding and Negative Cooperativity, and Its Relevance to Mitogenic versus Metabolic Signaling,” Diabetologia, Vol. 37, Suppl. 2, 1994, pp. S135-S148. http://dx.doi.org/10.1007/BF00400837
- E. Conti, F. Andreotti, A. Sciahbasi, P. Riccardi, G. Marra, E. Menini, et al., “Markedly Reduced Insulin-Like Growth Factor-1 in the Acute Phase of Myocardial Infarction,” Journal of the American College of Cardiology, Vol. 38, No. 1, 2001, pp. 26-32. http://dx.doi.org/10.1016/S0735-1097(01)01367-5
- A. Juul, T. Scheike, M. Davidsen, J. Gyllenborg and T. Jorgenson, “Low Serum Insulin-Like Growth Factor 1 Is Associated with Increased Risk of Ischemic Heart Disease: A Population-Based Case Control Study,” Circulation, Vol. 106, No. 8, 2002, pp. 939-944. http://dx.doi.org/10.1161/01.CIR.0000027563.44593.CC
- L. Macho, S. Zorad, Z. Radikova, P. Patterson-Buckedahl and R. Kvetnansky, “Ethanol Consumption Affects Stress Response and Insulin Binding Tissue of Rats Endocrine Regulations,” Endocrine Regulation, Vol. 37, No. 4, 2003, pp. 195-202.
- A. Natali, S. Vichi, P. Landi, S. Severi, A. L’Abbate and E. Ferrannini, “Coronary Atherosclerosis in Type II Diabetes: Angiographic Findings and Clinical Outcome,” Diabetologia, Vol. 43, No. 5, 2000, pp. 632-641. http://dx.doi.org/10.1007/s001250051352
- J. E. P. Brown, D. Onyango, and S. J. Dunmore, “Resistin Down-Regulates Insulin Receptor Expression, and Modulates Cell Viability in Rodent Pancreatic Beta-Cells,” FEBS Letters, Vol. 581, No. 17, 2007, pp. 3273-3276. http://dx.doi.org/10.1016/j.febslet.2007.06.031
- L. He, J. C. Marecki, G. Serrero, F. A. Simmen, M. Ronis and T. Badger, “Dose-Dependent Effects of Alcohol on Insulin Signaling: Partial Explanation for Biphasic Alcohol Impact on Human Health,” Molecular Endocrinology, Vol. 21, No. 21, 2007, pp. 2541-2550. http://dx.doi.org/10.1210/me.2007-0036
- T. Limin, X. Hou, J. Liu, X. Zhang, N. Sun, L. Gao, et al., “Chronic Ethanol Consumption Resulting in the Downregulation of Insulin Receptor-Subunit, Insulin Receptor Substrate-1, and Glucose Transporter 4 Expression in Rat Cardiac Muscles,” Alcohol, Vol. 43, 2009, pp. 51-58. http://dx.doi.org/10.1016/j.alcohol.2008.11.001
- B. Washington, C. Mtshali, S. Williams, H. Smith, J. D. Li, B. Shaw, et al., “Ethanol-Induced Mitogen Activated Protein Kinase Activity Mediated through Protein Kinase C,” Cell and Molecular Biology, Vol. 49, No. 8, 2003, pp. 1351-1356.
- A. L. Johnson, G. D. Goode, C. Mtshali, E. L. Myles and B. Washington, “Protein Kinase C-Alpha/BetaII, Delta, and Zeta/Lambda Involvement in Ethanol-Induced MAPK Expression in Vascular Smooth Muscle Cells,” Cell and Molecular Biology, Vol. 53, No. 4, 2007, pp. 38-44.
- H. Wang, S. Doronin and C. C. Malbon, “Insulin Activation of Mitogen-Activated Protein Kinases Erk1,2 Is Amplified via Beta-Adrenergic Receptor Expression and Requires the Integrity of the Tyr350 of the Receptor,” The Journal of Biological Chemistry, Vol. 275, No. 46, 2000, pp. 36086-26093. http://dx.doi.org/10.1074/jbc.M004404200
- S. Vasdev, V. Gill and P. Singal, “Beneficial Effect of Low Ethanol Intake on the Cardiovascular System: Possible Biochemical Mechanisms,” Journal of Vascular Health and Risk Management, Vol. 2, No. 3, 2006, pp. 263-276. http://dx.doi.org/10.2147/vhrm.2006.2.3.263
- S. Gitlow, “Substance Use Disorders: A Practical Guide,” 2nd Edition, Lippincott Williams and Wilkins, Philadelphia, 2006, pp. 52, 103-121.
- B. Taylor, J. Rehm and G. Gmel, “Moderate Alcohol Consumption and the Gastrointestinal Tract,” Digital Distribution, Vol. 23, No. 3-4, 2005, pp. 170-176.
- A. E. Seiler, B. N. Ross and R. Rubin, “Inhibition of Insulin-Like Growth Factor-1 Receptor and IRS Signaling by Ethanol in SH-SY5Y Neuroblastoma Cells,” Journal of Neurochemistry, Vol. 76, No. 2, 2001, pp. 573-581. http://dx.doi.org/10.1046/j.1471-4159.2001.00025.x
- D. T. Furuya, R. Binsack and U. F. Machado, “Low Ethanol Consumption Increases Insulin Sensitivity in Wistar Rats,” Brazilian Journal of Medical and Biological Research, Vol. 36, No. 1, 2003, pp. 125-130. http://dx.doi.org/10.1590/S0100-879X2003000100017
- J. R. Sowers, M. Epstein, E. D. Frohlich and N. R. Campbell, “Management of Hypertension for People with Diabetes,” Clinical Summary, 2011, pp. 1-52.
- L. Hansen, Y. Ikeda, G. Olsen, A. Busch and L. Mosthaf, “Insulin Signaling Is Inhibited by Micromolar Concentrations of H2O2,” The Journal of Biological Chemistry, Vol. 274, No. 35, 1999, pp. 25078-25084. http://dx.doi.org/10.1074/jbc.274.35.25078
- E. Motley, K. Eguchi, C. Gardner, A. Hicks, C. Reynolds, G. Frank, et al., “Insulin-Induced Akt Activation Is Inhibited by Angiotensin II in the Vasculature through Protein Kinase C,” Hypertension, Vol. 41, No. 3, 2002, pp. 775-780. http://dx.doi.org/10.1161/01.HYP.0000051891.90321.12
- Y. Sasaki, N. Hayashi, T. Ito, H. Fusamoto, T. Kamada and J. R. Wands, “Influence of Ethanol on Insulin Receptor Substrate-1-Mediated Signal Transduction during Rat Liver Regeneration,” Alcoholism Supplements, Vol. 29, No. 1, 1994, pp. 99-106.