International Journal of Clinical Medicine
Vol.3 No.4(2012), Article ID:20871,9 pages DOI:10.4236/ijcm.2012.34051
Regulation of 11β-Hydroxysteroid Dehydrogenase Types 1 and 2 in Rheumatoid Arthritis
![]()
1Institute of Medical Education, Department of Rheumatology, Clinical Immunology and Allergology, University Hospital of Berne, Berne, Switzerland; 2Departments of Nephrology and Hypertension and Clinical Research, University Hospital of Berne, Berne, Switzerland; 3Academic Department of Musculoskeletal Medicine, University of Leeds, Leeds, UK.
Email: *brigitte.frey@dkf.unibe.ch
Received March 19th, 2012; revised April 17th, 2012; accepted May 4th, 2012
Keywords: Rheumatoid Arthritis; Basic Clinical Scienc; Hormones; Inflammation; Biomarkers
ABSTRACT
Objective: The sequential activities of the 11β-hydroxysteroid dehydrogenase enzymes type 1 and 2 have not been investigated so far in patients suffering from rheumatoid arthritis before and after starting treatment though the enzymes balancing cortisol and cortisone levels are involved in regulating various inflammatory diseases. Methods: In a retrospective study, a panel of 41 urinary steroid metabolites has been analysed in a group of 18 patients with active rheumatoid arthritis (RA) as they were brought under control with disease modifying drugs. Results: No major changes were found in a variety of androgen, oestrogen and progesterone metabolites, however the ratio of THF + 5αTHF/THE as an index of 11β-HSD1 oxidative activity demonstrated down-regulation with modification correlating significantly with change in acute phase reactants as the disease came under control. These findings were supported by a tendency of a reduced ratio of F/E, an index of 11β-HSD2 oxidative activity, resulting in a significant correlation of the two ratios (p < 0.001). This parallelism of the two enzymes with functions of clinical laboratory parameters during drug-induced improvement of the disease is novel. Conclusions: Urinary steroid metabolites, which alter with disease activity, may provide further insight into the mechanisms by which stress can modify arthritis through hormones.
1. Introduction
11β-hydroxysteroid dehydrogenase (11β-HSD) enzymes type 1 and 2 convert cortisol into receptor inactive cortisone and vice versa (type 1 only). The type 2 isoform, mainly localised in the distal tubule of the kidney, inactivates cortisol to cortisone in a unidirectional manner, while 11β-HSD type 1, mainly localised in the liver, acts bi-directionally and can therefore restore cortisone to active cortisol. The balance between serum cortisol and cortisone is altered during the acute phase response in inflammatory disease, with a shift towards active cortisol [1]. Thus there is evidence that 11β-HSD enzymes play an important role in the modulation of systemic availability of cortisol during acute illness [2,3].
This mechanism may also be relevant for non-inflammatory diseases. Hypothalamic regulation of adiposity is partly mediated through 11β-HSD raising the relevance of this pathway to inflammatory conditions that may be hormonally influenced, such as rheumatoid disease [4]. Moreover, the pathway is also sensitive to influences of various drugs. TNF-α enhances intracellular glucocorticoid availability [5,6] and interleukin-1β activates the cortisone/cortisol shuttle [7].
11β-HSD enzymes play a pivotal role in the cardiovascular system [8] as well as asthma [9] and, in particular, ulcerative colitis, which can be associated with inflammatory arthritis [10]. Cytokines, hormones and drugs such as ACE inhibitors, furosemide and carbenoxolone act on 11β-HSD enzymes in various ways [11,12]. We have earlier studied urinary 6β-hydroxycortisol excretion in rheumatoid arthritis (RA) as part of a larger pharmacogenetic study to determine whether the activity of the cytochrome P450 isoenzyme CYP3A4 was altered by disease activity in RA [13] Urinary 6β-hydroxycortisol excretions however did not correlate with measurements of disease activity such as plasma viscosity (PV) in 21 closely monitored patients with RA. The ratio also appeared to be unaltered by disease modifying drugs being used at this time (sulphasalazine, sodium aurothiomalate and d-penicillamine) [13]. The recent demonstration that RA synovial cells might have a reduced capacity for reactivation of glucocorticoids [14] prompted us to analyze in a retrospective manner (investigation performed before year 2000) the urine samples stored from the original study to determine the ratio of the tetrahydro-metabolites of cortisol, THF + 5αTHF, and of cortisone, THE, as an index of 11β-HSD1 activity and the ratio of F and E as an index of 11β-HSD2 activity. We hypothesized based on own observations investigating inflammatory cytokines on steroid metabolism, that untreated inflammatory changes in rheumatoid disease down-regulate the enzyme activity while effective treatment might normalize the ratio of these metabolites [5-7].
2. Patients and Methods
Patients and disease assessments. Patients with RA (ARA criteria) [15] and disease activity sufficient to justify the use of DMARDs were recruited (PV > 1.72 mPa or Ritchie articular index (AI) > 16 or early morning stiffness [16] > 1 h) [17]. Patients with a history of or with laboratory evidence of hepatitis (excessive alcohol intake, infectious, autoimmune or storage diseases) were excluded.
Blinded to the results of steroid metabolites patients were allocated either to sulphasalazine (SASP), sodium aurothiomalate (gold) or d-penicillamine (DPA) according to clinical criteria by the investigating physician. Standard dosage regimens were used [18]. Dosage adjustments, due to toxicity, and concomitant drug therapy, particularly drugs potentially interfering with 11β-HSD activities, were meticulously recorded. Whenever possible the concomitant drug therapy remained constant throughout the study. No patient received systemic steroid therapy or intra articular steroid injections during the three months before the investigation.
At 0, 12 and 24 weeks disease activity of RA and safety of the DMARD treatments were assessed as reported earlier [13,18]. With each clinic visit a 24 h urine collection was obtained. Urinary aliquots were stored at –20˚C.
Ethical committee approval was obtained from the Ethics Committee of the Leeds Western Health Authority. All patients gave informed consent.
Laboratory tests. Analysis of urinary steroid metabolites was performed by gas chromatography-mass spectrometry as previously described [19]. Briefly, urine samples were pre-extracted, enzymatic hydrolysed, extracted from the hydrolysis mixture, derivatised and gel filtrated. Medroxyprogesterone was added as a recovery standard. The samples extracted on Sep-Pak C18 columns (Waters Corporation, Milford, Massachusetts USA) were hydrolysed with sulfatase (Sigma-Chemical, St. Louis, Missouri USA) and β-glucuronidase/arylsulfatase (Roche Diagnostics, Rotkreuz, Switzerland). The resulting free steroids were extracted once more on a Sep-Pak C18 cartridge and a mixture of 5α-androstane-3α, 17α- diol, stigmasterol, cholesteryl butyrate, and 3β5β-tetrahydroaldosterone added as internal standards. Samples were derivatised to form methyloxime-trimethylsilyl ethers. After adding 100 µl of methoxyamine HCl in pyridine (Pierce Biotechnology, Rockford, Illinois USA) the extracts were heated at 60˚C for one hour. Then pyridine was evaporated and 100 µl of trimethylsilylimidazole (TMSI) (Pierce) added to form trimethylsilyl ethers. The derivatisation was carried out at 100˚C for 16 hours. The derivatives were purified by gel filtration on Lipidex-5000 columns (Perkin Elmer, Boston, Massachusetts USA). Samples were analysed on a gas chromatograph 6890N equipped with an autoinjector 7683a and mass selective detector 5973 (Agilent Technologies, La Jolla, CA USA) by selective ion monitoring (SIM). One characteristic ion was chosen for each compound being measured. Derivatised steroids were analyzed using a temperature-programmed run (210˚C - 265˚C) over 35-min on a HP-1 MS column (15 m, 0.25 mm I.D., 0.25 µm film thickness) (Agilent). Calibration was performed on a regular basis. Each “ion-peak” abundance was quantified using the corresponding ion-peak of the internal standard stigmasterol. 3α5β-tetrahydroaldosterone was quantified using 3β5β-tetrahydroaldosterone.
Statistical analysis. Statistical analyses included median, interquartile range (IQR) and Wilcoxon matched pair test for differences between paired observations. Linear correlation coefficients (r) were calculated where appropriate. The significance level was set at p = 0.05 with two-sided testing. PV was considered as the primary reference parameter of disease activity. The power to detect difference between week 0 and week 24 was calculated to be 1.000 (albumin), 0.9306 (platelets), 0.8286 (PV), 0.5949 [8] 0.5444, (THF + 5αTHF/THE), 0.3062 (CRP), 0.2602 [16] and 0.0606 (γGT), respectively) [20].
3. Results
Eighteen patients, 1 male and 17 females, suffering from RA of at least 6 months duration (median 11 yr; IQR 7 - 20) were studied. Eight patients were positive for rheumatoid factor, 5 for antinuclear factor and 12 presented extra-articular manifestations, in particular rheumatoid nodules and keratoconjunctivitis. The median age was 58 yrs (IQR 53 - 68) and median weight 59 kg (IQR 53 - 72). Median blood pressure was 140/80 mmHg (IQR 120/70 to 150/85 mmHg). The median creatinine clearance was 60 ml/min/1.73 m2 at week 0 with no significant change over the 24 weeks period. Five patients were smoking 5 - 30 cigarettes/day (median 20 cigarettes), while 13 patients were non-smokers. Three patients were drinking alcohol > 70 g/week, five patients ≤ 70 g/week and 10 patients denied alcohol consumption. In the past eleven out of the 18 patients (61%) had been treated with a drug regimen conventional at this time, namely DMARD (DPA (n = 3), gold (n = 2), SASP (n = 2), hydroxychloroquine (HCQ) (n = 2), auranofin (n = 1) and methotrexate (MTX) (n = 1). All 11 patients started a different DMARD allowing a treatment interval of at least three months for gold, one month for MTX and one week for the remainder.
At the beginning of the study all 18 patients were allocated to a new regimen of DMARD treatment. Eight patients were started on gold, 5 on SASP and 5 on DPA. No patient was given furosemide, a known inhibitor of 11β-HSD2 [11]. One patient was taking hydrochlorothiazide 25 mg/day from week 0 to 24, a questionable inhibitor of 11β-HSD2 [21]. All other drugs used, such as non-steroidal anti-inflammatory drugs, analgesics, other diuretics, β-blockers, were not reported to influence 11β-HSD2. One patient was taking quinalbarbitone 100 mg/day from week 0 to 24, a well-established inducer of cytochrome P450. Eight patients were given substrates of the CYP3A4 isoenzyme that might influence the 6β- OHC output, such as nifedipine (one patient taking 80 mg/day from week 0 to 24), codeine (one patient taking 24 mg/day at week 12 and one 48 mg/day and 32 mg/day at week 12 and 24 respectively) and dihydrocodeine (two patients taking between 20 and 40 mg/day from week 0 to 24, one patient with 30 mg/day at week 12, one patient with 120 mg/day at week 0 and, finally, one with 120 mg/day and 60 mg/day at week 12 and 24 respectively). All other drugs used were judged unlikely to influence the CYP3A4 isoenzyme.
Over the 24 weeks period disease activity of RA decreased significantly (median PV week 0 = 1.85 mPa (IQR 1.76 - 1.90), week 12 = 1.77 (IQR 1.71 - 1.89), week 24 = 1.77 (1.66 - 1.84), p < 0.01; median AI week 0 = 24 (IQR 17 - 29), week 12 = 15 (10 - 23), week 24 = 11 (IQR 6 - 23), p < 0.02, respectively). Concurrently, significant changes from baseline were found for platelets (decrease), haemoglobin (increase), albumin (increase), globulins (decrease), IgG, IgA, IgM (decrease), but not for CRP, γGT, leucocytes, lymphocyte count, C3, C4 and EMS (data not shown). No significant changes of urinary steroid metabolite excretion (median values) were detected over the observed time periods (Tables 1 and 2) and all urinary steroid levels were in the normal range. Over the same time period the ratio of the tetrahydro-metabolites THF + 5αTHF/THE, an index of 11β- HSD1 activity, decreased significantly (median week 0 =
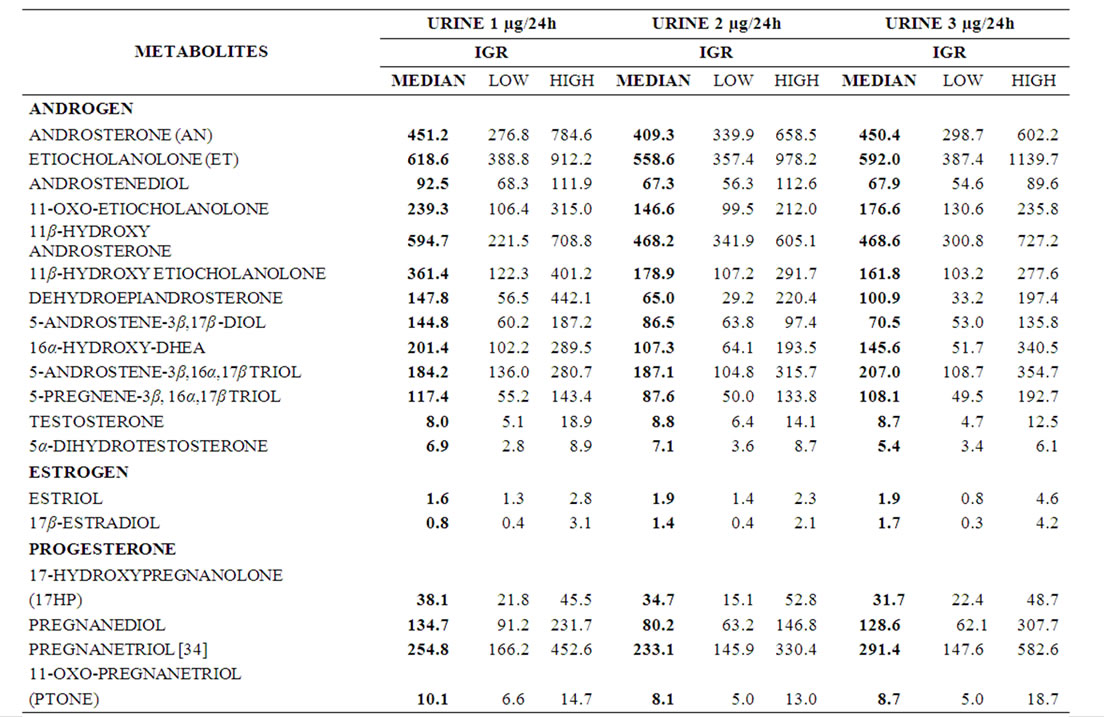
Table 1. Sequential data of urinary sex steroid metabolites (μg/24h). Median and Interquartile range (IQR) of RA patients (n = 18) before (urine 1), 12 (urine 2) and 24 (urine 3) weeks after treatment are given.
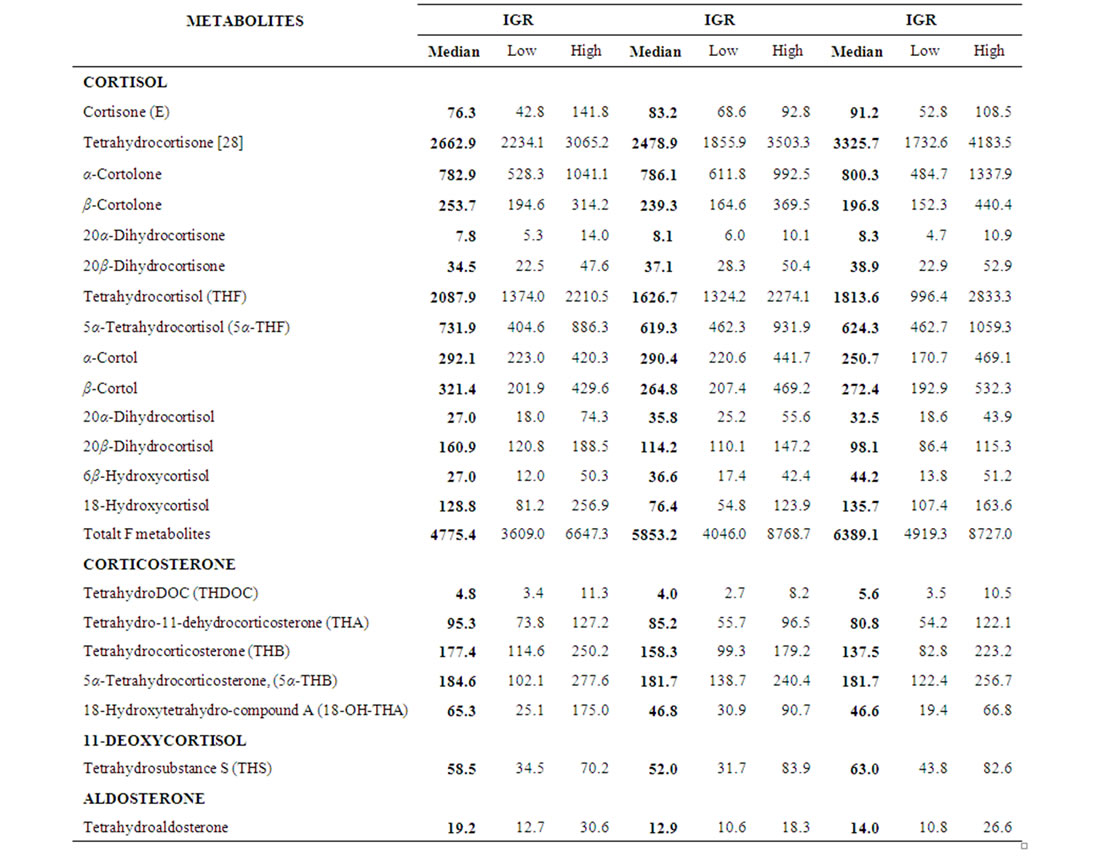
Table 2. Sequential data of urinary glucocorticosteroid and mineralocorticosteroid metabolites (μg/24h). Median and Interquartile range (IQR) of RA patients (n = 18) before (urine 1), 12 (urine 2) and 24 (urine 3) weeks after treatment are given.
1.05 (IQR 0.91-1.25), median week 12 = 0.91 (IQR 0.83 - 1.05), median week 24 = 0.85 (IQR 0.77 - 1.17) (Table 3 and Figure 1, p < 0.05). Similarly, the ratios of cortoles/cortolones and 11OH-andro + 11OH-etio/11oxo-etio indicated a slightly impaired oxidative activity of 11β-HSD1 (Figure 1, p < 0.05). Likewise, a tendency, but not significant changes of the 11β-HSD2 and 5α/5β reductase activities were observed (Figure 1). Overall a significant correlation of the oxidative activities of the two 11β-HSD enzymes was obtained (Figure 2, p < 0.001).
Importantly correlations between the ratios of cortisol to cortisone metabolites, (THF + 5aTHF/THE) and F/E as index markers of the activities of 11β-HSD1 and 2, respectively, [22,23], and various clinical and laboratory parameters of disease activity were found (Figures 3 and 4). These correlations present evidence, that the enzyme activities are regulated by the disease activity (Figures 3 and 4). The ratios are correlating significantly with the PV (r = 0.482 and 0.452, p < 0.001 for both), platelets (r = 0.426 and 0.513, p < 0.01 and p < 0.001) and albumin (r = 0.348 and 0.504, p < 0.05 and p < 0.001) (Figures 3 and 4), CRP (r = 0.560 and 0.606, p < 0.001 for both), γGT (r = 0.496 and 0.564 p < 0.001 for both) (data not given). Correlations however with clinical parameters such as AI (r = 0.010 and 0.040, ns for both) (Figures 3 and 4) and EMS (r = 0.386 and 0.254, p < 0.01 and ns) (data not given) were less prominent. In addition, a weak
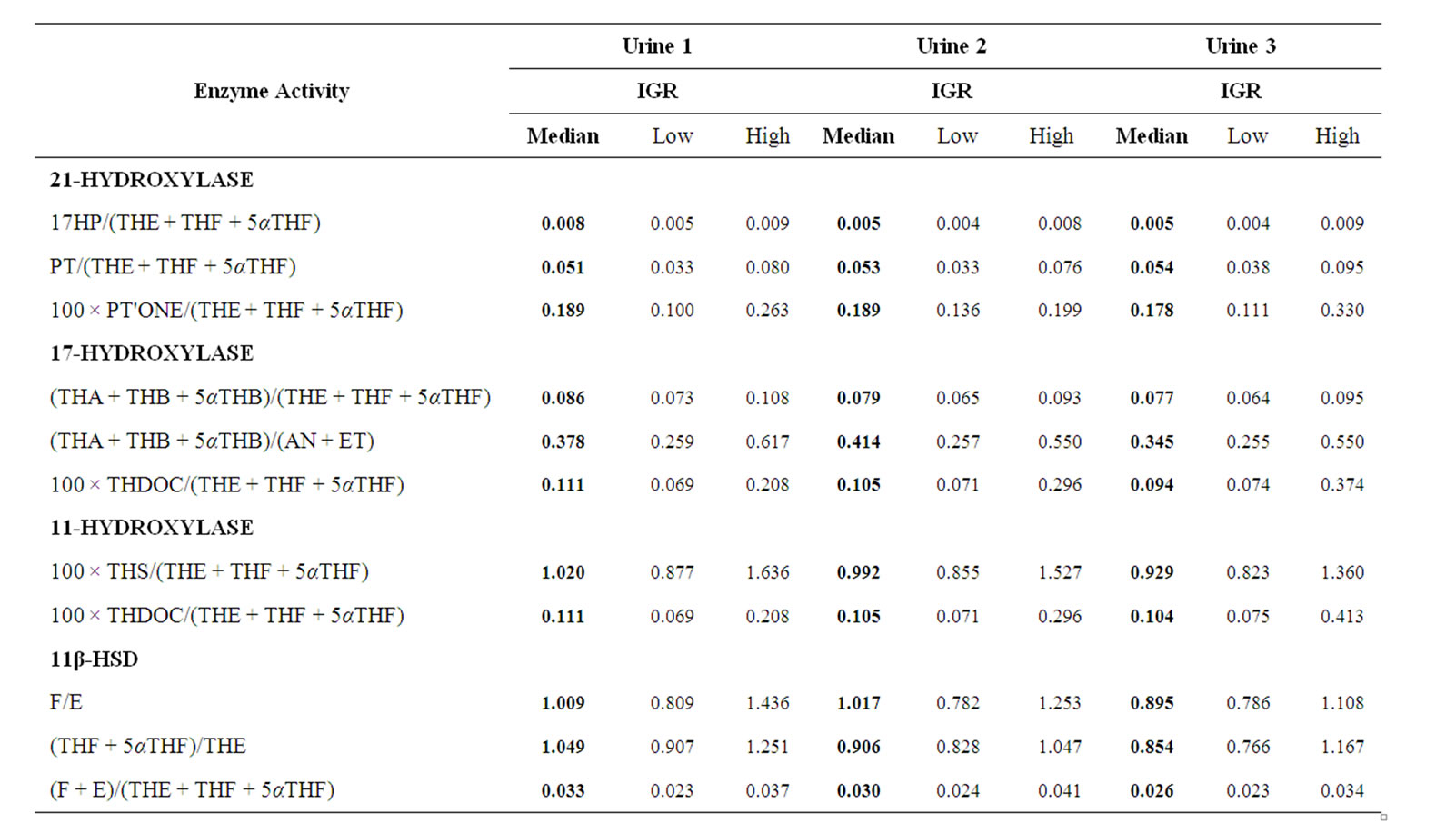
Table 3. Enzyme activities. Median and Interquartile range (IQR) of RA patients (n = 18) before (urine 1), 12 (urine 2) and 24 (urine 3) weeks after treatment are given.
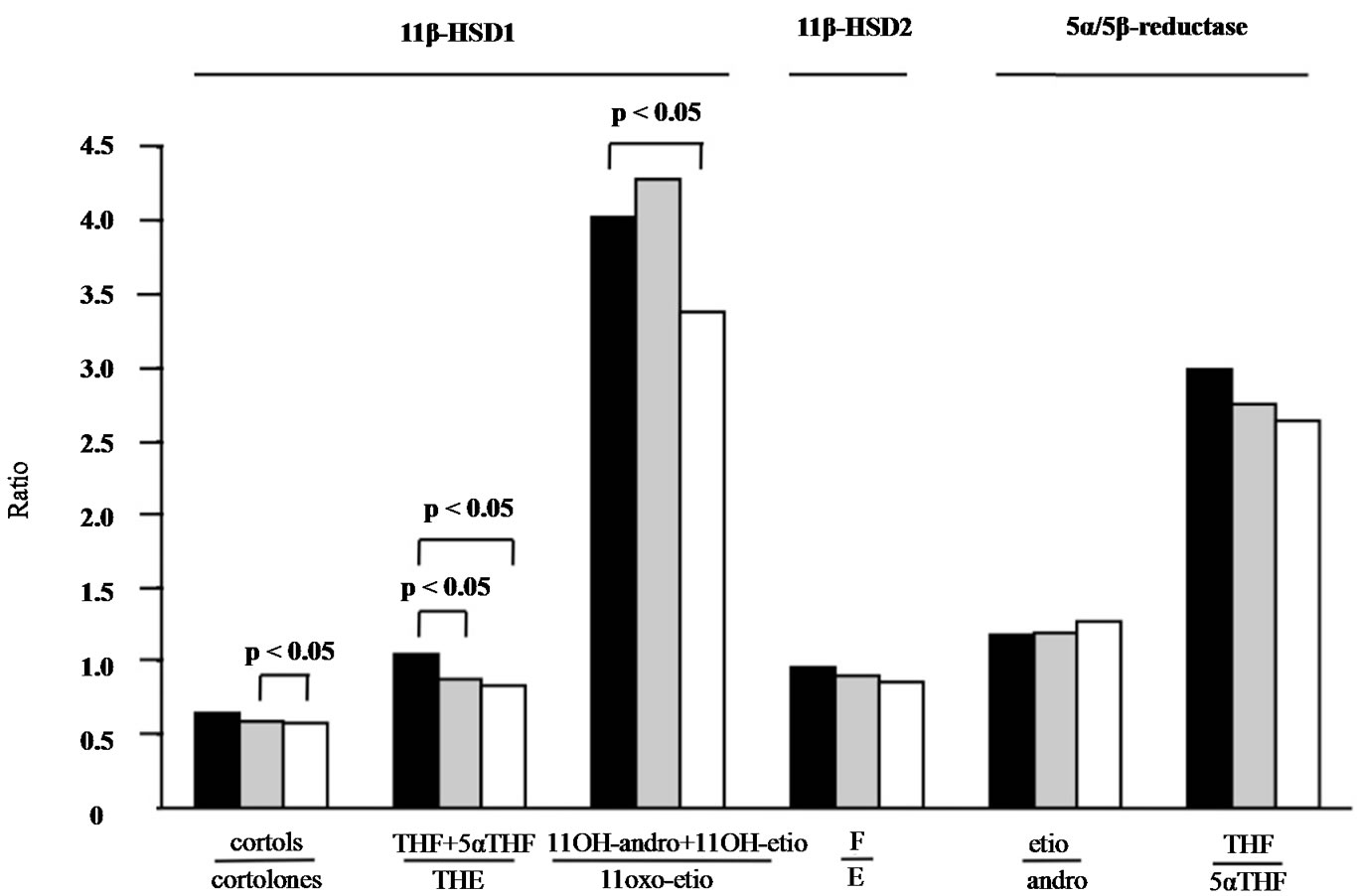
Figure 1. Urinary steroid metabolite ratios from patients with RA before and after treatment for 12 and 24 weeks. Median values of ratios representing oxidative 11β-HSD1 activity decreased significantly (p < 0.05) as a function of treatment time, indicating higher amounts of metabolites of the receptor inactive cortisone formed. A similar tendency was observed for 11β-HSD2 and 5α/5β-reductase activities. The median was calculated from 18 patients at week 0 (black column), week 12 (grey column) and week 24 (white column).
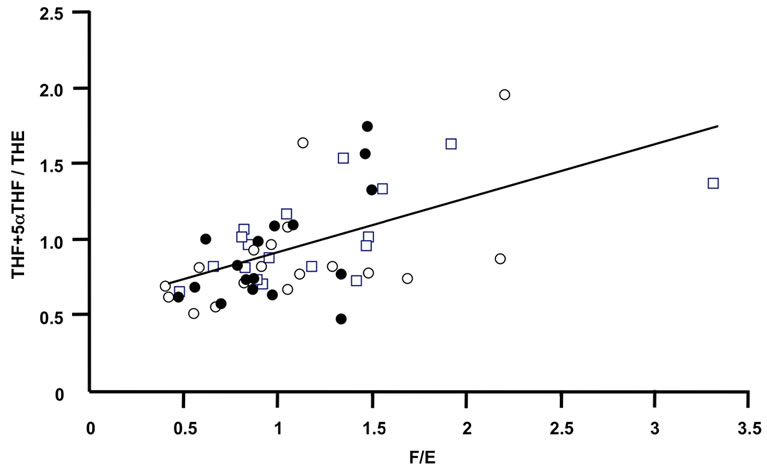
Figure 2. The oxidative activity of 11β-HSD2 correlates with the oxidative activity of 11β-HSD1. The ratios of F/E determined in urine from RA patients after 0 week (closed circle), 12 weeks (open circles) and 24 weeks (open quadrangles) treatment correlated significantly with the ratios of the tetrahydro-metabolites (p < 0.001).
correlation with creatinine clearance was observed, however not reaching significance.
There was no significant correlation with any other laboratory or demographic features studied (haemoglobin, leucocytes, lymphocytes, globulins, IgG, IgA, IgM, C3, C4, age, weight and disease duration, respectively). Notably, the ratio THF + 5αTHF/THE and F/E of the only male studied was within the range of the 17 females.
4. Discussion
To our knowledge this is the first study measuring a large panel of urinary steroid metabolites prospectively in a group of patients with active RA brought under control using a regimen of disease-modifying drugs. Although there was little serial change in the majority of hormones and their metabolites, the correlation of the ratio of cortisol to cortisone tetrahydro-metabolites as an indicator of 11β-HSD1 activity rather than F/E as an indicator of 11β-HSD2 activity was significant for those acute phase reactants proposed to be the most reliable indicators of inflammation in rheumatoid disease (Figures 1, 3 and 4). The two ratios correlated significantly in a positive manner plotting the 3 time points of 0, 12 and 24 weeks of treatment (Figure 2) indicating a parallel oxidative activity of the two enzymes. In this respect we propose that RA could be added to the list of conditions where altered 11β-HSD regulation might be of pathogenetic importance.
That hormonal changes are important in the progression of rheumatoid arthritis has long been recognised. Evidence for this comes from changes occurring in rheumatoid arthritis during pregnancy [24] and circadian variation [25], presumably because of the intricate association between the hypothalamus, the endocrine system and circulating hormones [26,27]. This probably accounts
 (a)
(a)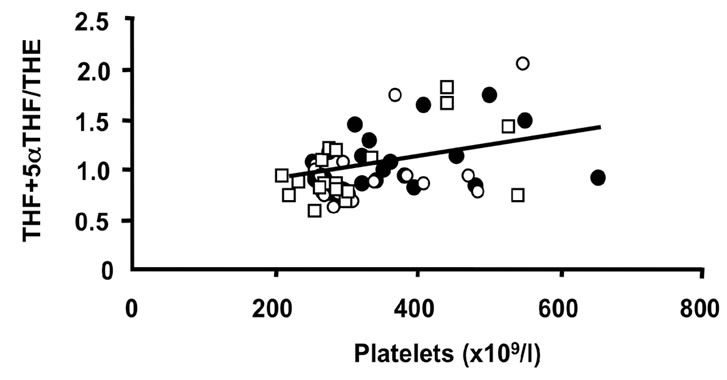 (b)
(b)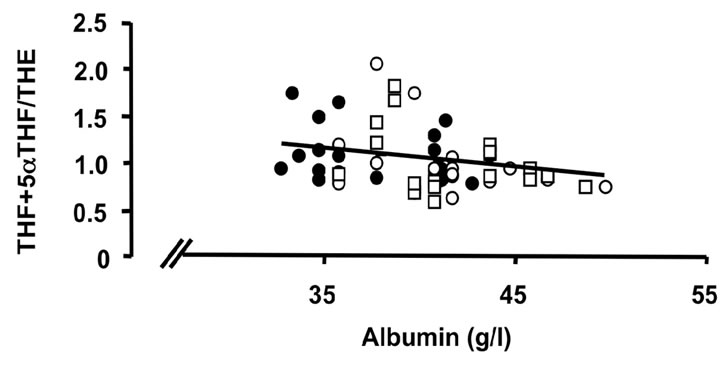 (c)
(c)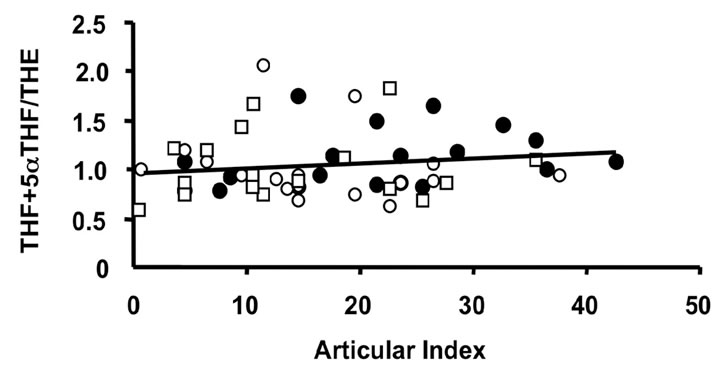 (d)
(d)
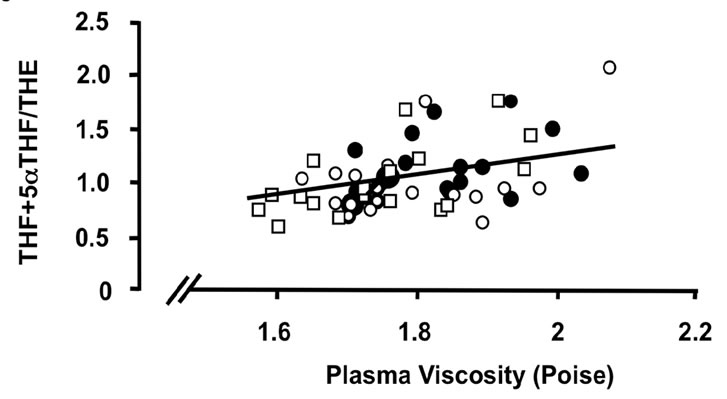
Figure 3. Correlations of the oxidative 11β-HSD1 activity expressed as the ratio THF + 5αTHF/THE and selected parameters of disease activity. Oxidative 11β-HSD1 activity was correlated to plasma viscosity ((a), p < 0.001)), platelet counts ((b), p < 0.01), plasma albumin levels ((c), p < 0.05) and articular index (d) (ns) in all patients at week 0 (closed circles), week 12 (open circles) and week 24 (open quadrangles). The normal range for plasma viscosity is <1.72 poise, for platelets 1.50 - 4.00 × 1011/l, for albumin 37 - 49 g/l and for articular index 0.
 (a)
(a)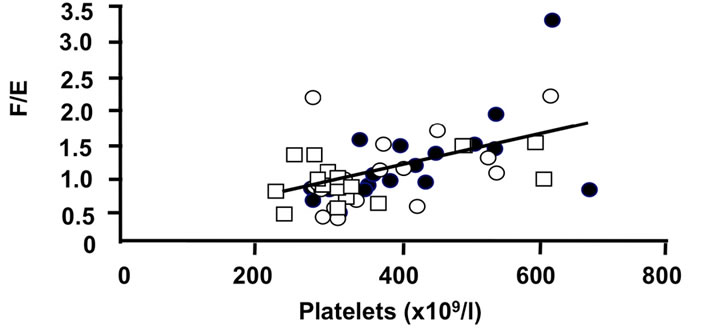 (b)
(b)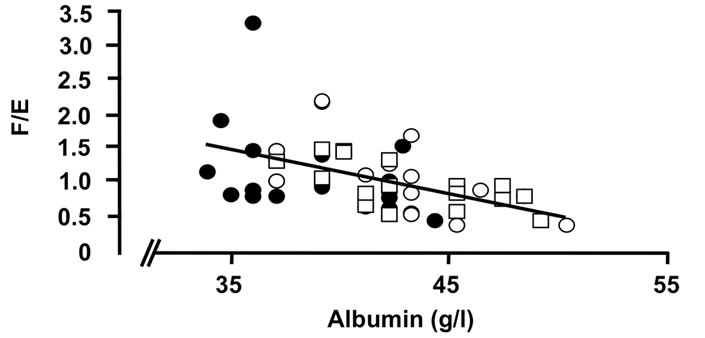 (c)
(c)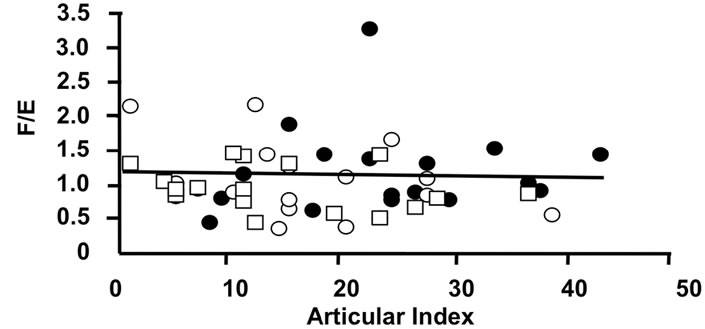 (d)
(d)
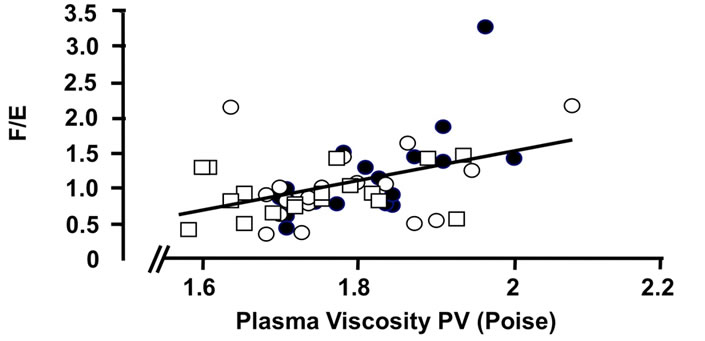
Figure 4. Correlations of the oxidative 11β-HSD2 activity expressed as the ratio F/E and selected parameters of disease activity. Oxidative 11β-HSD2 activity was correlated to plasma viscosity (a) (p < 0.001)), platelet counts; (b) (p < 0.001), plasma albumin levels; (c) (p < 0.001) and articular index; (d) (ns) in all patients at week 0 (closed circles), week 12 (open circles) and week 24 (open quadrangles). The normal range for plasma viscosity is given on Figure 3.
for diurnal variation in symptoms, particularly grip strength and tiredness, as well as disability [28]. This does not just affect rheumatoid arthritis. In polymyositis, there is a circadian variation in serum myoglobin levels [29] confirming end-organ sensitivity to hormonal change.
The last decade has seen heightened interest in diurnal variation not just in hormones but also in cytokines. Of necessity, our results derive from an era before cytokines were manipulated as part of conventional therapy for rheumatoid arthritis. Debatably, amongst cytokines, interleukin-6 is one of those displaying most diurnal change, particularly in polymyalgia rheumatica, a rheumatic disease in which diurnal variation is arguably even more pronounced [30]. It would be of interest to repeat this study in a group of rheumatoid patients brought under control not only with TNF-α blockers but also with particular attention to any sub-group requiring the use of interleukin-6 blockade to relieve symptoms.
Surprisingly, and somehow at variance with the literature, amongst the large panel of steroids studied, androgen [14,31,32], oestrogen [16,33] and progesterone [32] metabolites did not display a change in their level as a function of treatment. In part this may be because the group was selected for need of immediate disease control, not because coming from age or gender groups in which hormones might have displayed greater influence. Neither does hormonal influence necessarily need to be mediated through metabolic pathways of disease activity. There is increasing recognition from studies of inherited abnormalities of collagen that progestogens particularly can make previously asymptomatic hyperlax joints symptomatic. If it is accepted that joint laxity exhibits a Gaussian distribution in a normal population without disease, the soil on which the seed of rheumatoid arthritis is sown may also deserve consideration.
There is also no doubting the close relationship between psychological factors and rheumatoid arthritis. Whether rheumatoid arthritis can be precipitated by a psychological event is not certain but the undoubted tiredness, intriguingly often relieved by TNF-α blockade, the undoubted variation in symptoms in some patients in relation to the menstrual cycle and the influence of stress both psychological and physical on the condition [34,35] all represent areas deserving of attention for which variation in all steroid metabolites, measured by means of a urinary test, may provide the sensitive marker we are needing.
It remains a possibility that urinary hormonal metabolite levels might act as a surrogate for monitoring disease progression. Traditionally this had been done with acute phase reactants and quantitative serology. Cytokine levels might provide more refinement but are expensive and not always available. Serial arthroscopy with synovial histology felt to represent the gold standard is invasive and expensive, even though it offers the possibility of histochemical staining demonstrating differential cytokine expression [36,37]. Complementing these existing methods of disease assessment with urinary steroid metabolite analysis might add interesting insight into the mechanisms and periodicity of this disease.
5. Acknowledgements
We thank Miss Susan Dimmock and Mr Robert Addyman for technical assistance, and Dr. Rainer Hofer for statistical advice.
6. Grant Support
This work was supported by grants from the Arthritis Research Campaign (B0132), United Kingdom; the Swiss National Foundation for Scientific Research (3100A0 102153/2 and 320000-122135/1 to B. Frey); Dr Christine Beyeler was in receipt of a Swiss National Foundation Fellowship.
REFERENCES
- M. Vogeser, R. Zachoval, T. W. Felbinger and K. Jacob, “Increased Ratio of Serum Cortisol to Cortisone in Acute-Phase Response,” Hormone Research, Vol. 58, No. 4, 2002, pp. 172-175. doi:10.1159/000065486
- K. E. Chapman, A. Coutinho, M. Gray, J. S. Gilmour, J. S. Savill and J. R. Seckl, “Local Amplification of Glucocorticoids by 11beta-Hydroxysteroid Dehydrogenase Type 1 and Its Role in the Inflammatory Response,” Annals of the New York Academy of Sciences, Vol. 1088, 2006, pp. 265-273. doi:10.1196/annals.1366.030
- K. Raza, R. Hardy and M. S. Cooper, “The 11beta-Hydroxysteroid Dehydrogenase Enzymes—Arbiters of the Effects of Glucocorticoids in Synovium and Bone,” Rheumatology, Vol. 49, No. 11, 2010, pp. 2016-2023. doi:10.1093/rheumatology/keq212
- Z. Hochberg, M. Friedberg, L. Yaniv, T. Bader and D. Tiosano, “Hypothalamic Regulation of Adiposity: The Role of 11beta-Hydroxysteroid Dehydrogenase Type 1,” Hormone and Metabolic Research, Vol. 36, No. 6, 2004, pp. 365-369. doi:10.1055/s-2004-814570
- C. D. Heiniger, M. K. Rochat, F. J. Frey and B. M. Frey, “TNF-Alpha Enhances Intracellular Glucocorticoid Availability,” Federation of European Biochemical Societies Letters, Vol. 507, No. 3, 2001, pp. 351-356. doi:10.1016/S0014-5793(01)03004-6
- R. M. Kostadinova, A. R. Nawrocki, F. J. Frey and B. M. Frey, “Tumor Necrosis Factor Alpha and Phorbol 12-Myristate-13-Acetate Down-Regulate Human 11betahydroxysteroid Dehydrogenase Type 2 through p50/p50 NF-KappaB Homodimers and Egr-1,” Federation of American Societies for Experimental Biology Journal, Vol. 19, No. 6, 2005, pp. 650-652.
- G. Escher, I. Galli, B. S. Vishwanath, B. M. Frey and F. J. Frey, “Tumor Necrosis Factor Alpha and Interleukin 1beta Enhance the Cortisone/Cortisol Shuttle,” Journal of Experimental Medicine, Vol. 186, No. 2, 1997, pp. 189- 198. doi:10.1084/jem.186.2.189
- Z. Krozowski and Z. Chai, “The Role of 11beta-Hydroxysteroid Dehydrogenases in the Cardiovascular System,” Endocrinology Journal, Vol. 50, No. 5, 2003, pp. 485-489.
- B. E. Orsida, Z. S. Krozowski and E. H. Walters, “Clinical Relevance of Airway 11beta-Hydroxysteroid Dehydrogenase Type II Enzyme in Asthma,” American Journal of Respiratory Critical Care Medicine, Vol. 165, No. 7, 2002, pp. 1010-1014.
- K. I. Takahashi, K. Fukushima and H. Sasano, “Type II 11beta-Hydroxysteroid Dehydrogenase Expression in Human Colonic Epithelial Cells of Inflammatory Bowel Disease,” Digestive Diseases and Sciences, Vol. 44, No. 12, 1999, pp. 2516-2522. doi:10.1023/A:1026699324927
- D. Fuster, G. Escher, B. Vogt and F. J. Frey, “Furosemide Inhibits 11beta-Hydroxysteroid Dehydrogenase Type 2,” Endocrinology, Vol. 139, No. 9, 1998, pp. 3849-3854. doi:10.1210/en.139.9.3849
- J. W. Tomlinson and P. M. Stewart, “Cortisol Metabolism and the Role of 11beta-Hydroxysteroid Dehydrogenase,” Best Practice & Research Clinical Endocrinology & Metabolism, Vol. 15, No. 1, 2001, pp. 61-78. doi:10.1053/beem.2000.0119
- C. Beyeler, B. M. Frey and H. A. Bird, “Urinary 6 Beta-Hydroxycortisol Excretion in Rheumatoid Arthritis,” British Journal of Rheumatology, Vol. 36, No. 1, 1997, pp. 540-558. doi:10.1093/rheumatology/36.1.54
- M. Schmidt, C. Weidler, H. Naumann, S. Anders, J. Scholmerich and R. H. Straub, “Reduced Capacity for the Reactivation of Glucocorticoids in Rheumatoid Arthritis Synovial Cells: Possible Role of the Sympathetic Nervous System?” Arthritis Rheumatism, Vol. 52, No. 6, 2005, pp. 1711-1720. doi:10.1002/art.21091
- F. C. Arnett, S. M. Edworthy and D. A. Bloch, “The American Rheumatism Association 1987 Revised Criteria for the Classification of Rheumatoid Arthritis,” Arthritis Rheumatism, Vol. 31, No. 3, 1988, pp. 315-324. doi:10.1002/art.1780310302
- U. Islander, C. Jochems, M. K. Lagerquist, H. Forsblad-d’Elia and H. Carlsten, “Estrogens in Rheumatoid Arthritis; The Immune System and Bone,” Molecular Cellular Endocrinology, Vol. 335, No. 1, 2011, pp. 14- 29. doi:10.1016/j.mce.2010.05.018
- D. M. Ritchie, J. A. Boyle and J. M. McInnes, “Clinical Studies with an Articular Index for the Assessment of Joint Tenderness in Patients with Rheumatoid Arthritis,” Quaterly Journal of Medicine, Vol. 37, No. 147, 1968, pp. 393-406.
- C. Beyeler, A. K. Daly, M. Armstrong, C. Astbury, H. A. Bird and J. R. Idle, “Phenotype/Genotype Relationships for the Cytochrome P450 Enzyme CYP2D6 in Rheumatoid Arthritis: Influence of Drug Therapy and Disease Activity,” Journal of Rheumatoly, Vol. 21, No. 6, 1994, pp. 1034-1039.
- C. Quattropani, B. Vogt, A. Odermatt, B. Dick, B. M. Frey and F. J. Frey, “Reduced Activity of 11 Beta-Hydroxysteroid Dehydrogenase in Patients with Cholestasis,” Journal Clinical Investigation, Vol. 108, No. 9, 2001, pp. 1299-1305.
- B. Kirkwood, “Essentials of Medical Statistics,” WileyBlackwell, New York, 2003.
- M. L. Licketts and P. M. Stewart, “Regulation of 11BetaHydroxysteroid Dehydrogenase Type 2 by Diuretics and the Renin-Angiotensin-Aldosterone Axis,” Clinical Science, Vol. 96, No. 6, 1999, pp. 669-675.
- M. Quinkler, D. Zehnder and J. Lepenies, “Expression of Renal 11beta-Hydroxysteroid Dehydrogenase Type 2 Is Decreased in Patients with Impaired Renal Function,” European Journal of Endocrinology, Vol. 153, No. 2, 2005, pp. 291-299. doi:10.1530/eje.1.01954
- E. A. Walker, A. Ahmed and G. G. Lavery, “11BetaHydroxysteroid Dehydrogenase Type 1 Regulation by Intracellular Glucose 6-Phosphate Provides Evidence for a Novel Link between Glucose Metabolism and Hypothalamo-Pituitary-Adrenal Axis Function,” Journal of Biological Chemistry, Vol. 282, No. 37, 2007, pp. 27030- 27036. doi:10.1074/jbc.M704144200
- B. H. Scott, “Auto-Immunity and Connective Tissue Disorders,” Oxford Medical Publications, Oxford University Press, Oxford, 1990.
- M. Cutolo, B. Seriolo, C. Craviotto, C. Pizzorni and A. Sulli, “Circadian Rhythms in RA,” Annals of the Rheumatic Diseases, Vol. 62, No. 7, 2003, pp. 593-596. doi:10.1136/ard.62.7.593
- A. K. Bhall, “Hormones and the Immune Response,” Annals of the Rheumatic Diseases, Vol. 48, No. 1, 1989, pp. 1-6. doi:10.1136/ard.48.1.1
- B. W. Kirkham and G. S. Panayi, “Diurnal Periodicity of Cortisol Secretion, Immune Reactivity and Disease Activity in Rheumatoid Arthritis: Implications for Steroid Treatment,” British Journal of Rheumatology, Vol. 28, No. 2, 1989, pp. 154-157. doi:10.1093/rheumatology/28.2.154
- N. Bellamy, R. B. Sothern, J. Campbell and W. W. Buchanan, “Circadian Rhythm in Pain, Stiffness, and Manual Dexterity in Rheumatoid Arthritis: Relation between Discomfort and Disability,” Annals of the Rheumatic Diseases, Vol. 50, No. 4, 1991, pp. 243-248. doi:10.1136/ard.50.4.243
- S. Bombardieri, A. Clerico, L. Riente, M. G. Del Chicca and C. Vitali, “Circadian Variations of Serum Myoglobin Levels in Normal Subjects and Patients with Polymyositis,” Arthritis Rheumatism, Vol. 25, No. 12, 1982, pp. 1419-1424. doi:10.1002/art.1780251205
- M. G. Perry, J. R. Kirwan, D. S. Jessop and L. P. Hunt, “Overnight Variations in Cortisol, Interleukin 6, Tumour Necrosis Factor Alpha and Other Cytokines in People with Rheumatoid Arthritis,” Annals of the Rheumatic Diseases, Vol. 68, No. 1, 2009, pp. 63-68. doi:10.1136/ard.2007.086561
- M. Cutolo, “Androgens in Rheumatoid Arthritis: When Are They Effectors?” Arthritis Research & Therapy, Vol. 11, No. 5, 2009, p. 126. doi:10.1186/ar2804
- A. Martocchia, M. Stefanelli, S. Cola and P. Falaschi, “Sex Steroids in Autoimmune Diseases,” Current Topics in Medicinal Chemistry, Vol. 11, No. 13, 2011, pp. 1668- 1683. doi:10.2174/156802611796117595
- M. Cutolo, B. Villaggio, C. Craviotto, C. Pizzorni, B. Seriolo and A. Sulli, “Sex Hormones and Rheumatoid Arthritis,” Autoimmunity Reviews, Vol. 1, No. 5, 2002, pp. 284-289. doi:10.1016/S1568-9972(02)00064-2
- A. Gupta and A. J. Silman, “Psychological Stress and Fibromyalgia: A Review of the Evidence Suggesting a Neuroendocrine Link,” Arthritis Research & Therapy, Vol. 6, No. 3, 2004, pp. 98-106. doi:10.1186/ar1176
- R. H. Straub, F. S. Dhabhar, J. W. Bijlsma and M. Cutolo, “How Psychological Stress via Hormones and Nerve Fibers May Exacerbate Rheumatoid Arthritis,” Arthritis Rheumatism, Vol. 52, No. 1, 2005, pp. 16-26. doi:10.1002/art.20747
- F. Buttgereit, H. Zhou and M. J. Seibel, “Arthritis and Endogenous Glucocorticoids: The Emerging Role of the 11Beta-HSD Enzymes,” Annals of the Rheumatic Diseases, Vol. 67, No. 9, 2008, pp. 1201-1203. doi:10.1136/ard.2008.092502
- R. Hardy, E. H. Rabbitt and A. Filer, “Local and Systemic Glucocorticoid Metabolism in Inflammatory Arthritis,” Annals of the Rheumatic Diseases, Vol. 67, No. 9, 2008, pp. 1204-1210. doi:10.1136/ard.2008.090662
NOTES
*Corresponding author.