Open Journal of Endocrine and Metabolic Diseases
Vol.3 No.5A(2013), Article ID:35964,8 pages DOI:10.4236/ojemd.2013.35A003
The Effects of Insulin Resistance and Inflammation on Renal Proximal Tubule Sodium Transport and Hypertension
Department of Internal Medicine, The University of Tokyo Hospital, Tokyo, Japan
Email: *georgeseki-tky@umin.ac.jp
Copyright © 2013 Shoko Horita et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 18, 2013; revised July 18, 2013; accepted August 10, 2013
Keywords: Insulin; Inflammation; TNF-α; Proximal Tubule; Insulin Resistance; PPARγ; Thiazolidinediones
ABSTRACT
Insulin resistance, closely linked to inflammation, is recognized as a key factor in the onset and aggravation of diabetes, cardio-renal syndrome, hypertension, and obesity. In the renal proximal tubule, insulin resistance may increase renal sodium reabsorption, leading to hypertension, edema and sometimes heart failure. Recently some anti-diabetic agents have been shown to have effects on the transporters in renal proximal tubule. Because renal proximal tubule mediates about 70% of sodium reabsorption, it is quite important to clarify the function of renal proximal tubule under insulin resistance and inflammation.
1. Introduction
The relationship between inflammation and diabetes had been described long ago by the fact that anti-inflammatory drugs such as salicylates decrease blood glucose level [1]. About more than a century ago sodium salicylate was known to reduce or totally eliminate the diabetic symptoms [2,3]. Only recently the relationship between inflammation and insulin resistance began to be investigated vigorously, when Hotamisligil et al. [4] found that a proinflammatory factor tumor necrosis factor (TNF)-α could induce insulin resistance. Karasik et al. [5] reported that TNF-α suppressed insulin-induced tyrosine phosphorylation of insulin receptor and its substrates, resulting in attenuated insulin effect and insulin resistance.
To date there seems to be mainly two ways from inflammation to insulin resistance [1]. One is c-Jun Nterminal protein kinases (JNK) pathway [6,7], which, through the activator protein 1 (AP-1) transcription factor in the nucleus, enhances the transcription of inflammatory genes. JNK pathway also induces serine phosphorylation of insulin-related substrate (IRS-1) [8-11]. Phosphorylated IRS-1 itself induces insulin resistance by direct blocking of insulin signaling pathway through IRS-1 [10,11]. Recently Davis et al. showed that obesity-induced insulin resistance and inflammation are promoted by JNK in macrophage [12]. They produced mice with selective JNK deficiency in macrophages, which showed improved insulin sensitivity under high-fat diet.
The other is via IκB kinase-β (IKKβ)/NFκB pathway [13]. The signals from proinflammatory factors such as TNF-α and interleukin (IL)-1, via IKKβ complex, activate NFκB complex. Then NFκB works as a transcription factor [1]. NFκB is a primary regulator of inflammatory response [14] and requires IKKβ for its activation [15]. IKKβ has been shown to link inflammation to obesityinduced insulin resistance [16]. IKKβ/NFκB pathway is triggered by PI3K/AKT signaling [17-19].
These pathways inducing insulin resistance are triggered by such factors as TNF-α, IL-1, endoplasmic reticulum (ER) stress, oxidative stress, and lipids [1]. The details of the pathways from these factors to the emergence and aggravation of insulin resistance still remain to be clarified. Figure 1 summarizes the main pathways from inflammation to insulin resistance.
2. TNF-α and Sodium Reabsorption in Nephron
TNF-α was originally discovered as an anti-tumor factor [20,21]. At first TNF-α was thought to be produced only by immune cells [22,23]. Now epithelial cells and endo-
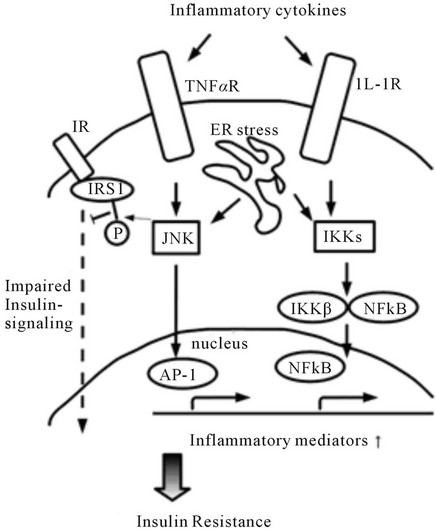
Figure 1. The major pathways from inflammation to insulin resistance. This figure focuses on the two main pathways dependent on JNK or IKK.
thelial cells have been also found to produce TNF-α [24]. TNF-α is generated by tubular epithelial cells and mesangial cells in the kidney [25-27].
The receptors of TNF-α consist of two major subtypes; TNFαR1 (p55) and TNFRα2 (p75) [28,29]. In the kidney TNFαR1 is predominantly expressed in renal vessels and glomeruli, while TNFαR2 is expressed mainly in renal tubules [30].
The role of these two receptors is not clear, although TNFαR1 may mediate the cytotoxic and inflammatory responses of TNF-α, while TNFαR2 may mediate protective effects of this cytokine [31,32]. For example, in cardiomyocytes TNFαR1 mediates ischemic injury, whereas TNFαR2 mediates cardioprotective effects of TNF-α [33, 34]. In the kidney TNFαR1 is supposed to mediate acute renal vasoconstriction and natriuresis in response to high dose of TNF-α [35].
Using TNFαR1 or TNFαR2 knockout (KO) mice Majid and colleagues showed that these receptors play different roles in renal hemodynamic and natriuretic responses to high dose of TNF-α infusion [35]. After TNF- α infusion, the mean arterial pressure slightly decreased from the baseline but renal vascular resistance increased in WT mice. In both KO mice the blood pressure also decreased, while urinary Na+ excretion increased in WT and TNFαR2 KO mice but did not change in TNFαR1 KO mice. These results indicate that TNFaR1 mediates the natriuretic response to high-dose TNF-α.
By contrast, the chronic inflammatory effects of lower-dose TNF-α in the kidney was shown to be mediated by TNFαR2 [36]. Gesek and colleagues showed that chronic TNF-α exposure induced sodium retention by activating ENaC in diabetic rats [37]. They also reported that chronic exposure to low-dose TNF-α stimulates Na+ uptake in isolated rat distal tubule cells [38]. In LLCPK1 cells, the model of renal proximal tubule (PT) cells, TNF-α stimulates Na+-K+-ATPase [39]. TNFαR1−/− mice showed enhanced tubular Na+ reabsorption in response to chronic angotensin II infusion, suggesting that upregulation of TNFαR2 could contribute to this renal response [40]. TNF-α might be one of the therapeutic targets in hypertension and its complications.
3. IRS-1, Insulin Resistance and Renal Proximal Transport
PT plays important roles in the regulation of acid-base and electrolytes homeostasis [41-43]. As for acid-base homeostasis, the Na+-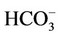 cotransporter NBCe1 in the basolateral side and the Na+/H+ exchanger NHE3 in the apical side of the PT are mainly involved in Na+ coupled
cotransporter NBCe1 in the basolateral side and the Na+/H+ exchanger NHE3 in the apical side of the PT are mainly involved in Na+ coupled  reabsorption. Na+-K+-ATPase gives a driving force for Na+ reabsorption. Insulin is known to be uptaken into proximal tubule [44-46]. Insulin enhances sodium reabsorption from PT [47,48] by stimulating NHE3 [49], Na+-K+-ATPase [50-52], and NBCe1 [53].
reabsorption. Na+-K+-ATPase gives a driving force for Na+ reabsorption. Insulin is known to be uptaken into proximal tubule [44-46]. Insulin enhances sodium reabsorption from PT [47,48] by stimulating NHE3 [49], Na+-K+-ATPase [50-52], and NBCe1 [53].
By using IRS-1 and IRS-2 knockout mice we have clarified [54] that the stimulation of proximal transport by insulin is mediated by IRS-2, not by IRS-1. Signal transduction via Akt, which is thought to mediate the effect of insulin in proximal tubule, is preserved in IRS-1 KO mice but attenuated in IRS-2 KO mice. In insulin resistance signal transduction via IRS-1 is frequently attenuated [55-58]. IRS-2 dependent stimulation of sodium absorption from PT may play an important role in the occurrence of hypertension in insulin resistant status. We recently confirmed that the stimulatory effect of insulin on Na+ absorption from PT is preserved in rats and human species with insulin resistance (“Stimulatory Effect of Insulin on Renal Proximal Na Transport Is Preserved in Insulin Resistance” 2012 Annual Meeting of American Society of Nephrology).
These results strongly suggest that in insulin resistance the stimulatory effect of insulin on PT transport is preserved via IRS-2. While defects in IRS-1 dependent signaling in insulin resistance may induce impaired vasodilation [59], IRS-2 dependent sodium retention from the kidney may play an important role in the onset and aggravation of hypertension in diabetes.
4. Insulin Resistance and Distal Tubules Transporters and Kinases
Insulin also acts on distal tubule, where Na+-Cl− cotransporter (NCC) is located in the luminal side. NCC is regulated by with-no-lysine (WNK) kinase oxidative stress-responsive kinase-1 (OSR1)/STE20/SPS1-related proline-alanine-rich kinase (SPAK) system [60].
WNK was originally discovered as a serine-threonine kinase with an atypical lysine alignment [61]. WNK has five subtypes, WNK1, WNK2, WNK3, WNK4 and transcript variant of WNK1, KS-WNK [62]. The mutations in WNK kinases cause Gordon syndrome (also known as familial hyperkalemic hypertension, FHH or pseudohypoaldosteronism type II, PHAII) [63].
At first WNK4 was thought to suppress the NCC activity by reducing its expression in the plasma membrane [63-67]. WNK1 was shown to suppress WNK4 but not directly inhibit NCC [65,67]. On the other hand, disruption of WNK4 gene was shown to decrease phosphorylation of NCC, inducing enhanced Na+ excretion and lower blood pressure [68]. Furthermore, overexpression of WNK4 was recently shown to induce PHAII phenotypes, suggesting that WNK4 might actually stimulate the NCC activity [69].
NCC was recently shown to be regulated by insulin via WNKs. In cultured mouse distal tubule cells (mpkDCT cells), insulin increased SPAK and NCC phosphorylation. In mpkDCT cultured cells and mouse kidney, insulin induced OSR1/SPAK phosphorylation and consequent NCC phosphorylation. The insulin-induced NCC phosphorylation was lost in SPAK knockout mice [70]. In hypertensive and hyperinsulinemic db/db mice, WNKOSR1/SPAK-NCC is phosphorylated by PI3K/Akt signaling pathway [71]. Moreover, NCC is functionally activated by insulin [72].
These results suggest that the insulin/WNK-OSR1/ SPAK-NCC system may play an important role in Na+ retention and hypertension, and may be another therapeutic target in hypertension associated with insulin resistance.
5. PPARγ, Thiazolidinediones and Kidney
Peroxisome-proliferator-activated receptors (PPARs) were first discovered in 1990s. PPARs belong to the nuclearreceptor superfamily and regulate gene expression in response to binding of the ligands such as fatty acids and oxysterols [73]. PPAR variants include PPARα, β, γ, and δ, among them PPARγ is implicated in the onset of insulin resistance. It is expressed most abundantly in adipose tissue and liver but also exists in pancreatic β cells, vascular endothelial cells and macrophages. PPARγ is a target of thiazolidinediones (TZDs), an insulin sensitizing drug.
Although TZDs are quite effective in improving insulin resistance, they have notorious side effects such as fluid retention leading to heart failure [74,75], and an elevated risk of bladder cancer [76]. The side effect of fluid retention is quite important because diabetic patients are under the risk of heart failure. The mechanism of TZDs-induced fluid retention has been in dispute for a while. The fluid retention effect of TZDs was shown to be dependent on PPARγ in collecting ducts. In particular, the epithelial Na+ channel (ENaC) in collecting ducts was first thought to be the main cause of TZDs-induced fluid retention [77,78]. In addition, TZDs were shown to enhance ENaCα subunit expression through the glucocorticoid inducible kinase SGK1 [79]. On the contrary, the other studies showed that TZDs did not alter ENaC expression and activity [80,81]. Though amiloride, an ENaC inhibitor, prevented TZDs-induced volume expansion in mice [78], it failed to prevent volume expansion induced by GI262570, a non-TZD PPARγ agonist in rats [82]. In mice lacking ENaCα subunit selectively in collecting ducts, TZDs-induced fluid retention was not attenuated [83]. This strongly suggests that ENaC stimulation is not responsible for TZDs-induced fluid retention. On the other hand, some reports suggested that PT transport is stimulated by TZDs both in human [84] and rabbit [85].
We hypothesized that PT may be another important target of TZDs and have proved that TZDs significantly stimulate PT transport within several minutes [86]. This rapid stimulation is dependent on PPARγ-Src-EGFRERK pathway in rabbit, rat and human PTs. However, this rapid stimulation was not observed in mouse PTs, because of constitutive activation of Src/EGFR. The truncated construct representing the ligand-binding domain of PPARγ was also able to mediate the similar signaling, suggesting that the TZDs-induced PT transport stimulation is independent of transcriptional activity of PPARγ. Our work may help to develop new TZDs with fewer side effects. Recently another study showed that TZDs do not increase the ENaC activity and its mRNA expression in the kidney [87], confirming that ENaC is not a main target of TZDs. The actual target of TZDs on distal Na+ transport remains to be determined. Figure 2 shows the proposed mechanisms by which TZDs induce Na+ and fluid retention.
6. PPARγ, TZDs, Insulin Resistance and Inflammation
As described above, insulin resistance is closely related to inflammation. Expansion of adipocyte induces inflammatory reactions such as proliferation of macrophage and release of inflammatory cytokines like TNF-α and IL-1, triggering the onset of insulin resistance [88].
Patients with dominant negative mutations of PPARγ have severe insulin resistance, diabetes and even hypertension [89]. On the other hand, human PPARγ polymorphism Pro12Ala leads to improved insulin sensitivity and glucose tolerance [90]. Moreover, in mice, increased PPARγ activity prevented insulin resistance due to obesity
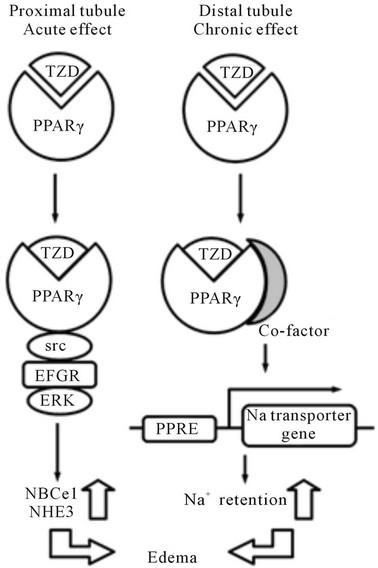
Figure 2. The proposed mechanisms by which TZDs induce Na+ and fluid retention. TZDs may induce acute stimulation of PT transport by non-genomic mechanism, while they induce chronic stimulation of distal transport by activation of undefined Na transporter(s).
obesity [91], while lack of PPARγ in fat, muscle, or liver predispose mice to developing insulin resistance [92-95]. Therefore, PPARγ plays important roles in glucose homeostasis [96].
TZDs can reduce blood glucose by improving insulin resistance [97]. Upon binding to TZDs, PPARγ makes heterodimers with retinoid-X receptors (RXRs). This complex binds to specific DNA sequence known as peroxisome proliferator response elements (PPREs) on the promoters of PPARγ target genes. These genes are transactivated after binding of PPARγ/RXR complex. Some of these genes are involved in metabolic homeostasis [98]. Moreover, PPARγ also regulates inflammatory response genes [99]. TZDs might prevent the onset of insulin resistance and improve glucose tolerance, by either improving metabolic homeostasis or preventing inflammation.
Several studies showed that TZDs exert renoprotective effects [100] and anti-inflammatory effects [101] in the kidney. Moreover, Troglitazone and other PPARγ agonists inhibit inflammation-related chemokine expression in human tubular epithelial cells HK2 [102]. TZDs might have direct anti-inflammatory effects and ameliorate insulin resistance in PTs. Future studies are required to clarify the detailed mechanisms underlying the renoprotective effects of TZDs.
7. Conclusions and Prospectives
We have discussed the relationship between insulin resistance and inflammation in the context of the onset of hypertension. Insulin resistance, caused by inflammation mainly in adipose tissue, is implicated in unfavorable effects on various organs including kidney, resulting in increasing mortality and morbidity in all over the world.
In the kidney, insulin-induced Na+ retention may be causally linked to hypertension. In PT, IRS-2 dependent Na+ reabsorption may be enhanced, even in insulin resistance with defective IRS-1 signaling. In the distal tubule, a WNK-SPAK/OSR1-NCC system may be also activated by insulin.
TZDs can significantly improve insulin sensitivity. However, TZDs-induced fluid retention is a serious problem in diabetes treatment. The development of medications that improve insulin resistance without inducing the serious side effects is expected.
REFERENCES
- S. E. Shoelson, J. Lee and A. B. Goldfine, “Inflammation and Insulin Resistance,” Journal of Clinical Investigation, Vol. 116, No. 7, 2006, pp. 1793-1801. doi:10.1172/JCI29069
- W. Ebstein, “Invited Comment on W. Ebstein: On the Therapy of Diabetes Mellitus, in Particular on the Application of Sodium Salicylate,” Journal of Molecular Medicine (Berlin), Vol. 80, No. 10, 2002, pp. 618-619.
- R. T. Williamson, “On the Treatment of Glycosuria and Diabetes Mellitus with Sodium Salicylate,” British Medical Journal, Vol. 1, No. 2100, 1901, pp. 760-762. doi:10.1136/bmj.1.2100.760
- G. S. Hotamisligil, N. S. Shargill and B. M. Spiegelman, “Adipose Expression of Tumor Necrosis Factor-α: Direct Role in Obesity-Linked Insulin Resistance,” Science, Vol. 259, No. 5091, 1993, pp. 87-91. doi:10.1126/science.7678183
- R. Feinstein, H. Kanety, M. Z. Papa, B. Lunenfeld and A. Karasik, “Tumor Necrosis Factor-α Suppresses InsulinInduced Tyrosine phosphorylation of Insulin Receptor and Its Substrates,” The Journal of Biological Chemistry, Vol. 268, 1993, pp. 26055-26058
- R. Yang and J. M. Trevillyan, “c-Jun N-Terminal Kinase Pathways in Diabetes,” The International Journal of Biochemistry & Cell Biology, Vol. 40, No. 12, 2008, pp. 2702-2706. doi:10.1016/j.biocel.2008.06.012
- R. J. Davis, “Signal Transduction by the JNK Group of MAP Kinases,” Cell, Vol. 103, No. 2, 2000, pp. 239-252. doi:10.1016/S0092-8674(00)00116-1
- V. Aguirre, T. Uchida, L. Yenush, R. Davis and M. F. White, “The c-Jun NH2-Terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser(307),” The Journal of Biological Chemistry, Vol. 275, No. 12, 2000, pp. 9047-9054. doi:10.1074/jbc.275.12.9047
- U. Ozcan, Q. Cao, E. Yilmaz, et al., “Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes,” Science, Vol. 306, No. 5695, 2004, pp. 457- 461. doi:10.1126/science.1103160
- V. Aguirre, E. D. Werner, J. Giraud, Y. H. Lee, S. E. Shoelson and M. F. White, “Phosphorylation of Ser307 in Insulin Receptor Substrate-1 Blocks Interactions with the Insulin Receptor and Inhibits Insulin Action,” The Journal of Biological Chemistry, Vol. 277, No. 2, 2002, pp. 1531-1537. doi:10.1074/jbc.M101521200
- E. D. Werner, J. Lee, L. Hansen, M. Yuan and S. E. Shoelson, “Insulin Resistance Due to Phosphorylation of Insulin Receptor Substrate-1 at Serine 302,” The Journal of Biological Chemistry, Vol. 279, No. 34, 2004, pp. 35298-35305. doi:10.1074/jbc.M405203200
- M. S. Han, D. Y. Jung, C. Morel, et al., “ JNK Expression by Macrophages Promotes Obesity-Induced Insulin Resistance and Inflammation,” Science, Vol. 339, No. 6116, 2013, pp. 218-222. doi:10.1126/science.1227568
- M. J. Lenardo and D. Baltimore, “NF-κB: A Pleiotropic Mediator of Inducible and Tissue-Specific Gene Control,” Cell, Vol. 58, No. 2, 1989, pp. 227-229. doi:10.1016/0092-8674(89)90833-7
- P. J. Barnes and M. Karin, “Nuclear Factor-κB: A Pivotal Transcription Factor in Chronic Inflammatory Diseases,” The New England Journal of Medicine, Vol. 336, No. 15, 1997, pp. 1066-1071. doi:10.1056/NEJM199704103361506
- L. W. Chen, L. Egan, Z. W. Li, F. R. Greten, M. F. Kagnoff and M. Karin, “The Two Faces of IKK and NFκB Inhibition: Prevention of Systemic Inflammation but Increased Local Injury Following Intestinal IschemiaReperfusion,” Nature Medicine, Vol. 9, No. 5, 2003, pp. 575-581. doi:10.1038/nm849
- M. C. Arkan, A. L. Hevener, F. R. Greten, et al., “ IKK-β Links Inflammation to Obesity-Induced Insulin Resistance,” Nature Medicine, Vol. 11, No. 2, 2005, pp. 191- 198. doi:10.1038/nm1185
- L. P. Kane, V. S. Shapiro, D. Stokoe and A. Weiss, “Induction of NF-κB by the Akt/PKB Kinase,” Current Biology, Vol. 9, No. 11, 1999, pp. 601-604. doi:10.1016/S0960-9822(99)80265-6
- O. N. Ozes, L. D. Mayo, J. A. Gustin, S. R. Pfeffer, L. M. Pfeffer and D. B. Donner, “NF-κB Activation by Tumour Necrosis Factor Requires the Akt Serine-Threonine Kinase,” Nature, Vol. 401, No. 6748, 1999, pp. 82-85. doi:10.1038/43466
- A. Salminen and K. Kaarniranta, “Insulin/IGF-1 Paradox of Aging: Regulation via AKT/IKK/NF-κB Signaling,” Cell Signal, Vol. 22, No. 4, 2010, pp. 573-577. doi:10.1016/j.cellsig.2009.10.006
- E. A. Carswell, L. J. Old, R. L. Kassel, S. Green, N. Fiore and B. Williamson, “An Endotoxin-Induced Serum Factor That Causes Necrosis of Tumors,” Proceedings of the National Academy of Sciences of USA, Vol. 72, No. 9, 1975, pp. 3666-3670. doi:10.1073/pnas.72.9.3666
- Y. Ichinose, J. Y. Tsao and I. J. Fidler, “Destruction of Tumor Cells by Monokines Released from Activated Human Blood Monocytes: Evidence for Parallel and Additive Effects of IL-1 and TNF,” Cancer Immunology, Immunotherapy, Vol. 27, No. 1, 1988, pp. 7-12. doi:10.1007/BF00205751
- B. Beutler, D. Greenwald, J. D. Hulmes, et al., “Identity of Tumour Necrosis Factor and the Macrophage-Secreted Factor Cachectin,” Nature, Vol. 316, No. 6028, 1985, pp. 552-554. doi:10.1038/316552a0
- P. G. Tipping, T. W. Leong and S. R. Holdsworth, “Tumor Necrosis Factor Production by Glomerular Macrophages in Anti-Glomerular Basement Membrane Glomerulonephritis in Rabbits,” Laboratory Investigation, Vol. 65, 1991, pp. 272-279.
- V. D. Ramseyer and J. L. Garvin, “Tumor Necrosis Factor-α: Regulation of Renal Function and Blood Pressure,” American Journal of Physiology. Renal Physiology, Vol. 304, No. 10, 2013, pp. F1231-F1242. doi:10.1152/ajprenal.00557.2012
- X. Dong, S. Swaminathan, L. A. Bachman, A. J. Croatt, K. A. Nath and M. D. Griffin, “Resident Dendritic Cells Are the Predominant TNF-Secreting Cell in Early Renal Ischemia-Reperfusion Injury,” Kidney International, Vol. 71, No. 7, 2007, pp. 619-628. doi:10.1038/sj.ki.5002132
- L. Baud, J. P. Oudinet, M. Bens, et al., “ Production of Tumor Necrosis Factor by Rat Mesangial Cells in Response to Bacterial Lipopolysaccharide,” Kidney International, Vol. 35, No. 5, 1989, pp. 1111-1118. doi:10.1038/ki.1989.98
- M. Ruiz-Ortega, V. Esteban, M. Rupérez, et al., “Renal and Vascular Hypertension-Induced Inflammation: Role of Angiotensin II,” Current Opinion in Nephrology and Hypertension, Vol. 15, No. 2, 2006, pp. 159-166. doi:10.1097/01.mnh.0000203190.34643.d4
- G. I. Botchkina, M. E. Meistrell, I. L. Botchkina and K. J. Tracey, “Expression of TNF and TNF Receptors (p55 and p75) in the Rat Brain after Focal Cerebral Ischemia,” Molecular Medicine, Vol. 3, No. 11, 1997, pp. 765-781.
- X. J. Sun, L. M. Wang, Y. Zhang, et al., “Role of IRS-2 in Insulin and Cytokine Signalling,” Nature, Vol. 377, No. 6545, 1995, pp. 173-177. doi:10.1038/377173a0
- R. S. Al-Lamki, J. Wang, J. N. Skepper, S. Thiru, J. S. Pober and J. R. Bradley, “Expression of Tumor Necrosis Factor Receptors in Normal Kidney and Rejecting Renal Transplants,” Laboratory Investigation, Vol. 81, No. 11, 2001, pp. 1503-1515. doi:10.1038/labinvest.3780364
- L. A. Tartaglia, R. F. Weber, I. S. Figari, C. Reynolds, M. A. Palladino and D. V. Goeddel, “The Two Different Receptors for Tumor Necrosis Factor Mediate Distinct Cellular Responses,” Proceedings of the National Academy of Sciences of USA, Vol. 88, No. 20, 1991, pp. 9292-9296. doi:10.1073/pnas.88.20.9292
- M. Feldmann, “Many Cytokines Are Very Useful Therapeutic Targets in Disease,” Journal of Clinical Investigation, Vol. 118, No. 11, 2008, pp. 3533-3536. doi:10.1172/JCI37346
- N. Defer, A. Azroyan, F. Pecker and C. Pavoine, “TNFR1 and TNFR2 Signaling Interplay in Cardiac Myocytes,” The Journal of Biological Chemistry, Vol. 282, No. 49, 2007, pp. 35564-35573. doi:10.1074/jbc.M704003200
- Y. Zhang, J. Zhao, W. B. Lau, et al., “Tumor Necrosis Factor-α and Lymphotoxin-α Mediate Myocardial Ischemic Injury via TNF Receptor 1, but Are Cardioprotective When Activating TNF Receptor 2,” PLoS One, Vol. 8, 2013, Article ID: e60227. doi:10.1371/journal.pone.0060227
- A. Castillo, M. T. Islam, M. C. Prieto and D. S. Majid, “Tumor Necrosis Factor-α Receptor Type 1, Not Type 2, Mediates Its Acute Responses in the Kidney,” American Journal of Physiology. Renal Physiology, Vol. 302, No. 12, 2012, pp. F1650-F1657. doi:10.1152/ajprenal.00426.2011
- P. Singh, L. Bahrami, A. Castillo and D. S. Majid, “TNF- α Type 2 Receptor Mediates Renal Inflammatory Response to Chronic Angiotensin II Administration with High Salt Intake in Mice,” American Journal of Physiology— Renal Physiology, Vol. 304, No. 7, 2013, pp. F991-F999. doi:10.1152/ajprenal.00525.2012
- K. DiPetrillo, B. Coutermarsh, N. Soucy, J. Hwa and F. Gesek, “Tumor Necrosis Factor Induces Sodium Retention in Diabetic Rats through Sequential Effects on Distal Tubule Cells,” Kidney International, Vol. 65, No. 5, 2004, pp. 1676-1683. doi:10.1111/j.1523-1755.2004.00606.x
- K. DiPetrillo, B. Coutermarsh and F. A. Gesek, “Urinary Tumor Necrosis Factor Contributes to Sodium Retention and Renal Hypertrophy during Diabetes,” American Journal of Physiology—Renal Physiology, Vol. 284, No. 1, 2003, pp. F113-F121.
- N. F. Ramia and S. I. Kreydiyyeh, “TNF-α Modulates the Na+/ K+ ATPase and the Na+-K+-2Cl− Symporter in LLCPK Cells,” European Journal of Clinical Investigation, Vol. 39, No. 4, 2009, pp. 280-288. doi:10.1111/j.1365-2362.2009.02098.x
- C. C. Chen, P. L. Pedraza, S. Hao, C. T. Stier and N. R. Ferreri, “TNFR1-Deficient Mice Display Altered Blood Pressure and Renal Responses to ANG II Infusion,” American Journal of Physiology—Renal Physiology, Vol. 299, No. 5, 2010, pp. F1141-F1150. doi:10.1152/ajprenal.00344.2010
- D. Brown and C. A. Wagner, “Molecular Mechanisms of Acid-Base Sensing by the Kidney,” Journal of the American Society of Nephrology, Vol. 23, No. 5, 2012, pp. 774-780. doi:10.1681/ASN.2012010029
- L. A. Skelton, W. F. Boron and Y. Zhou, “Acid-Base Transport by the Renal Proximal Tubule,” Journal of Nephrology, Vol. 23, Suppl. 16, 2010, pp. S4-S18.
- W. F. Boron, “Acid-Base Transport by the Renal Proximal Tubule,” Journal of the American Society of Nephrology, Vol. 17, No. 9, 2006, pp. 2368-2382. doi:10.1681/ASN.2006060620
- J. E. Bourdeau, E. R. Chen and F. A. Carone, “Insulin Uptake in the Renal Proximal Tubule,” American Journal of Physiology, Vol. 225, No. 6, 1973, pp. 1399-1404.
- M. A. Cortney, L. L. Sawin and D. D. Weiss, “Renal Tubular Protein Absorption in the Rat,” Journal of Clinical Investigation, Vol. 49, No. 1, 1970, pp. 1-4. doi:10.1172/JCI106208
- R. Rabkin, A. H. Rubenstein and J. A. Colwell, “Glomerular Filtration and Proximal Tubular Absorption of Insulin 125 I,” American Journal of Physiology, Vol. 223, No. 5, 1972, pp. 1093-1096.
- R. A. DeFronzo, C. R. Cooke, R. Andres, G. R. Faloona and P. J. Davis, “The Effect of Insulin on Renal Handling of Sodium, Potassium, Calcium, and Phosphate in Man,” Journal of Clinical Investigation, Vol. 55, No. 4, 1975, pp. 845-855. doi:10.1172/JCI107996
- R. A. DeFronzo, M. Goldberg and Z. S. Agus, “The Effects of Glucose and Insulin on Renal Electrolyte Transport,” Journal of Clinical Investigation, Vol. 58, No. 1, 1976, pp. 83-90. doi:10.1172/JCI108463
- F. A. Gesek and A. C. Schoolwerth, “Insulin Increases Na+-H+ Exchange Activity in Proximal Tubules from Normotensive and Hypertensive Rats,” American Journal of Physiology, Vol. 260, No. 5, 1991, pp. F695-F703.
- E. Feraille, M. L. Carranza, M. Rousselot and H. Favre, “Insulin Enhances Sodium Sensitivity of Na-K-ATPase in Isolated Rat Proximal Convoluted Tubule,” American Journal of Physiology, Vol. 267, No. 1, 1994, F55-F62.
- C. Rivera, H. Reyes-Santos and M. Marinez-Maldonado, “Response of Dog Renal Na+, K+-ATPase to Insulin in Vitro,” Renal Physiology, Vol. 1, No. 1, 1978, pp. 74-83.
- Z. Taylor, D. S. Emmanouel and A. I. Katz, “Insulin Stimulates Na-K-ATPase Activity of Basolateral Renal Tubular Membranes (Abstract),” Kidney International, Vol. 21, No. 1, 1982, p. 266.
- O. S. Ruiz, Y. Y. Qiu, L. R. Cardoso and J. A. Arruda, “Regulation of the Renal Na-HCO3 Cotransporter: IX. Modulation by Insulin, Epidermal Growth Factor and Carbachol,” Regulatory Peptides, Vol. 77, No. 1-3, 1998, pp. 155-161. doi:10.1016/S0167-0115(98)00115-3
- Y. Zheng, H. Yamada, K. Sakamoto, et al., “Roles of Insulin Receptor Substrates in Insulin-Induced Stimulation of Renal Proximal Bicarbonate Absorption,” Journal of the American Society of Nephrology, Vol. 16, No. 8, 2005, pp. 2288-2295. doi:10.1681/ASN.2005020193
- E. Carvalho, P. A. Jansson, M. Axelsen, et al., “Low Cellular IRS 1 Gene and Protein Expression Predict Insulin Resistance and NIDDM,” The FASEB Journal, Vol. 13, No. 15, 1999, pp. 2173-2178.
- J. E. Friedman, T. Ishizuka, J. Shao, L. Huston, T. Highman and P. Catalano, “Impaired Glucose Transport and Insulin Receptor Tyrosine Phosphorylation in Skeletal Muscle from Obese Women with Gestational Diabetes,” Diabetes, Vol. 48, No. 9, 1999, pp. 1807-1814. doi:10.2337/diabetes.48.9.1807
- L. J. Goodyear, F. Giorgino, L. A. Sherman, J. Carey, R. J. Smith and G. L. Dohm, “Insulin Receptor Phosphorylation, Insulin Receptor Substrate-1 Phosphorylation, and Phosphatidylinositol 3-Kinase Activity Are Decreased in Intact Skeletal Muscle Strips from Obese Subjects,” Journal of Clinical Investigation, Vol. 95, No. 5, 1995, pp. 2195-2204. doi:10.1172/JCI117909
- C. M. Rondinone, L. M. Wang, P. Lonnroth, C. Wesslau, J. H. Pierce and U. Smith, “Insulin Receptor Substrate (IRS) 1 Is Reduced and IRS-2 is the Main Docking Protein for Phosphatidylinositol 3-Kinase in Adipocytes from Subjects with Non-Insulin-Dependent Diabetes Mellitus,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 94, No. 8, 1997, pp. 4171- 4175. doi:10.1073/pnas.94.8.4171
- H. Abe, N. Yamada, K. Kamata, et al., “Hypertension, Hypertriglyceridemia, and Impaired Endothelium-Dependent Vascular Relaxation in Mice Lacking Insulin Receptor Substrate-1,” Journal of Clinical Investigation, Vol. 101, No. 8, 1998, pp. 1784-1788. doi:10.1172/JCI1594
- [61] H. Dimke, “Exploring the Intricate Regulatory Network Controlling the Thiazide-Sensitive NaCl Cotransporter (NCC),” Pflugers Arch, Vol. 462, No. 6, 2011, pp. 767- 777. doi:10.1007/s00424-011-1027-1
- [62] B. Xu, J. M. English, J. L. Wilsbacher, S. Stippec, E. J. Goldsmith and M. H. Cobb, “WNK1, a Novel Mammalian Serine/Threonine Protein Kinase Lacking the Catalytic Lysine in Subdomain II,” The Journal of Biological Chemistry, Vol. 275, No. 22, 2000, pp. 16795-16801. doi:10.1074/jbc.275.22.16795
- [63] J. A. McCormick, C. L. Yang and D. H. Ellison, “WNK Kinases and Renal Sodium Transport in Health and Disease: An Integrated View,” Hypertension, Vol. 51, No. 3, 2008, pp. 588-596. doi:10.1161/HYPERTENSIONAHA.107.103788
- [64] F. H. Wilson, S. Disse-Nicodème, K. A. Choate, et al., “Human Hypertension Caused by Mutations in WNK Kinases,” Science, 2001; Vol. 293, No. 5532, pp. 1107-1112. doi:10.1126/science.1062844
- [65] H. Cai, V. Cebotaru, Y. H. Wang, et al., “WNK4 Kinase Regulates Surface Expression of the Human Sodium Chloride Cotransporter in Mammalian Cells,” Kidney International, Vol. 69, No. 12, 2006, pp. 2162-2170. doi:10.1038/sj.ki.5000333
- [66] A. P. Golbang, G. Cope, A. Hamad, et al., “Regulation of the Expression of the Na/Cl Cotransporter by WNK4 and WNK1: Evidence That Accelerated Dynamin-Dependent Endocytosis Is Not Involved,” American Journal of Physiology—Renal Physiology, Vol. 291, No. 6, 2006, pp. F1369-F1376. doi:10.1152/ajprenal.00468.2005
- [67] M. E. Safar and H. S. Boudier, “Vascular Development, Pulse Pressure, and the Mechanisms of Hypertension,” Hypertension, Vol. 46, No. 1, 2005, pp. 205-209. doi:10.1161/01.HYP.0000167992.80876.26
- [68] C. L. Yang, J. Angell, R. Mitchell and D. H. Ellison, “WNK kinases regulate thiazide-sensitive Na-Cl cotransport,” Journal of Clinical Investigation, Vol. 111, No. 7, 2003, pp. 1039-1045.
- [69] A. Ohta, T. Rai, N. Yui, et al., “Targeted Disruption of the Wnk4 Gene Decreases Phosphorylation of Na-Cl Cotransporter, Increases Na Excretion and Lowers Blood Pressure,” Human Molecular Genetics, Vol. 18, No. 20, 2009, pp. 3978-3986. doi:10.1093/hmg/ddp344
- [70] M. Wakabayashi, T. Mori, K. Isobe, et al., “Impaired KLHL3-Mediated Ubiquitination of WNK4 Causes Human Hypertension,” Cell Reports, Vol. 3, No. 3, 2013, pp. 858-868. doi:10.1016/j.celrep.2013.02.024
- [71] E. Sohara, T. Rai, S. S. Yang, et al., “Acute Insulin Stimulation Induces Phosphorylation of the Na-Cl Cotransporter in Cultured Distal mpkDCT Cells and Mouse Kidney,” PLoS ONE, Vol. 6, No. 8, 2011, Article ID: e24277. doi:10.1371/journal.pone.0024277
- [72] H. Nishida, E. Sohara, N. Nomura, et al., “Phosphatidylinositol 3-Kinase/Akt Signaling Pathway Activates the WNK-OSR1/SPAK-NCC Phosphorylation Cascade in Hyperinsulinemic db/db Mice,” Hypertension, Vol. 60, No. 4, 2012, pp. 981-990. doi:10.1161/HYPERTENSIONAHA.112.201509
- [73] M. Chávez-Canales, J. P. Arroyo, B. Ko, et al., “Insulin Increases the Functional Activity of the Renal NaCl Cotransporter,” Journal of Hypertension, Vol. 31, No. 2, 2013, pp. 303-311.
- [74] H. Yki-Järvinen, “Thiazolidinediones,” The New England Journal of Medicine, Vol. 351, 2004, pp. 1106-1118. doi:10.1056/NEJMra041001
- [75] R. M. Lago, P. P. Singh and R. W. Nesto, “Congestive Heart Failure and Cardiovascular Death in Patients with Prediabetes and Type 2 Diabetes Given Thiazolidinediones: A Meta-Analysis of Randomised Clinical Trials,” Lancet, Vol. 370, No. 9593, 2007, pp. 1129-1136. doi:10.1016/S0140-6736(07)61514-1
- [76] E. Erdmann, B. Charbonnel, R. G. Wilcox, et al., “Pioglitazone Use and Heart Failure in Patients with Type 2 Diabetes and Preexisting Cardiovascular Disease: Data from the PROactive Study (PROactive 08),” Diabetes Care, Vol. 30, No. 11, 2007, pp. 2773-2778. doi:10.2337/dc07-0717
- [77] J. D. Lewis, A. Ferrara, T. Peng, et al., “Risk of Bladder Cancer among Diabetic Patients Treated with Pioglitazone: Interim Report of a Longitudinal Cohort Study,” Diabetes Care, Vol. 34, No. 4, 2011, pp. 916-922. doi:10.2337/dc10-1068
- [78] H. Zhang, A. Zhang, D. E. Kohan, R. D. Nelson, F. J. Gonzalez and T. Yang, “Collecting Duct-Specific Deletion of Peroxisome Proliferator-Activated Receptor γ Blocks Thiazolidinedione-Induced Fluid Retention,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 102, No. 26, 2005, pp. 9406-9411. doi:10.1073/pnas.0501744102
- [79] Y. Guan, C. Hao, D. R. Cha, et al., “Thiazolidinediones Expand Body Fluid Volume through PPARγ Stimulation of ENaC-Mediated Renal Salt Absorption,” Nature Medicine, Vol. 11, No. 8, 2005, pp. 861-866. doi:10.1038/nm1278
- [80] G. Hong, A. Lockhart, B. Davis, et al., “PPARγ Activation Enhances Cell Surface ENaCalpha via Up-Regulation of SGK1 in Human Collecting Duct Cells,” The FASEB Journal, Vol. 17, No. 13, 2003, pp. 1966-1968.
- [81] C. Nofziger, L. Chen, M. A. Shane, C. D. Smith, K. K. Brown and B. L. Blazer-Yost, “PPARγ Agonists Do Not Directly Enhance Basal or Insulin-Stimulated Na+ Transport via the Epithelial Na+ Channel,” Pflügers Archiv, Vol. 451, No. 3, 2005, pp. 445-453. doi:10.1007/s00424-005-1477-4
- [82] J. Song, M. A. Knepper, X. Hu, J. G. Verbalis and C. A. Ecelbarger, “Rosiglitazone Activates Renal Sodiumand Water-Reabsorptive Pathways and Lowers Blood Pressure in Normal Rats,” Journal of Pharmacology and Experimental Therapeutics, Vol. 308, No. 2, 2004, pp. 426- 433. doi:10.1124/jpet.103.058008
- [83] L. Chen, B. Yang, J. A. McNulty, et al., “GI262570, a Peroxisome Proliferator-Activated Receptor γ Agonist, Changes Electrolytes and Water Reabsorption from the Distal Nephron in Rats,” Journal of Pharmacology and Experimental Therapeutics, Vol. 312, No. 2, 2005, pp. 718-725. doi:10.1124/jpet.104.074088
- [84] V. Vallon, E. Hummler, T. Rieg, et al., “Thiazolidinedione-Induced Fluid Retention Is Independent of Collecting Duct αENaC Activity,” Journal of the American Society of Nephrology, Vol. 20, No. 4, 2009, pp. 721-729. doi:10.1681/ASN.2008040415
- [85] A. Zanchi, A. Chiolero, M. Maillard, J. Nussberger, H. R. Brunner and M. Burnier, “Effects of the Peroxisomal Proliferator-Activated Receptor-γ Agonist Pioglitazone on Renal and Hormonal Responses to Salt in Healthy Men,” The Journal of Clinical Endocrinology & Metabolism, Vol. 89, No. 3, 2004, pp. 1140-1145. doi:10.1210/jc.2003-031526
- [86] S. Muto, Y. Miyata, M. Imai and Y. Asano, “Troglitazone Stimulates Basolateral Rheogenic Na+/
 Cotransport Activity in Rabbit Proximal Straight Tubules,” Experimental Nephrology, Vol. 9, No. 3, 2001, pp. 191-197.
Cotransport Activity in Rabbit Proximal Straight Tubules,” Experimental Nephrology, Vol. 9, No. 3, 2001, pp. 191-197. - [87] Y. Endo, M. Suzuki, H. Yamada, et al., “Thiazolidinediones Enhance Sodium-Coupled Bicarbonate Absorption from Renal Proximal Tubules via PPARγ-Dependent Nongenomic Signaling,” Cell Metabolism, Vol. 13, No. 5, 2011, pp. 550-561. doi:10.1016/j.cmet.2011.02.015
- [88] E. Borsting, V. P. Cheng, C. K. Glass, V. Vallon and R. Cunard, “Peroxisome Proliferator-Activated Receptor-γ Agonists Repress Epithelial Sodium Channel Expression in the Kidney,” American Journal of Physiology—Renal Physiology, Vol. 302, No. 5, 2012, pp. F540-F551. doi:10.1152/ajprenal.00306.2011
- [89] G. S. Hotamisligil, “Inflammation and Metabolic Disorders,” Nature, Vol. 444, No. 7121, 2006, pp. 860-867. doi:10.1038/nature05485
- [90] I. Barroso, M. Gurnell, V. E. Crowley, et al., “Dominant Negative Mutations in Human PPARγ Associated with Severe Insulin Resistance, Diabetes Mellitus and Hypertension,” Nature, Vol. 402, No. 6764, 1999, pp. 880-883
- [91] S. S. Deeb, L. Fajas, M. Nemoto, et al., “A Pro12Ala Substitution in PPARγ2 Associated with Decreased Receptor Activity, Lower Body Mass Index and Improved Insulin Sensitivity,” Nature Genetics, Vol. 20, No. 3, 1998, pp. 284-287. doi:10.1038/3099
- [92] S. M. Rangwala, B. Rhoades, J. S. Shapiro, et al., “Genetic Modulation of PPARγ Phosphorylation Regulates Insulin Sensitivity,” Developmental Cell, Vol. 5, No. 4, 2003, pp. 657-663. doi:10.1016/S1534-5807(03)00274-0
- [93] W. He, Y. Barak, A. Hevener, et al., “Adipose-Specific Peroxisome Proliferator-Activated Receptor γ Knockout Causes Insulin Resistance in Fat and Liver but Not in Muscle,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 26, 2003, pp. 15712-15717.
- [94] K. Matsusue, M. Haluzik, G. Lambert, et al., “LiverSpecific Disruption of PPARγ in Leptin-Deficient mice Improves Fatty Liver but Aggravates Diabetic Phenotypes,” Journal of Clinical Investigation, Vol. 111, No. 5, 2003, pp. 737-747.
- [95] A. W. Norris, L. Chen, S. J. Fisher, et al., “Muscle-Specific PPARγ-Deficient Mice Develop Increased Adiposity and Insulin Resistance but Respond to Thiazolidinediones,” Journal of Clinical Investigation, Vol. 112, No. 4, 2003, pp. 608-618. doi:10.1073/pnas.2536828100
- [96] A. L. Hevener, W. He, Y. Barak, et al., “Muscle-Specific PPARγ Deletion Causes Insulin Resistance,” Nature Medicine, Vol. 9, No. 12, 2003, pp. 1491-1497. doi:10.1038/nm956
- [97] M. Lehrke and M. A. Lazar, “The Many Faces of PPARγ,” Cell, Vol. 123, No. 6, 2005, pp. 993-999. doi:10.1016/j.cell.2005.11.026
- [98] J. J. Nolan, B. Ludvik, P. Beerdsen, M. Joyce and J. Olefsky, “Improvement in Glucose Tolerance and Insulin Resistance in Obese Subjects Treated with Troglitazone,” The New England Journal of Medicine, Vol. 331, No. 18, 1994, pp. 1188-1193. doi:10.1056/NEJM199411033311803
- [99] B. Cariou, B. Charbonnel and B. Staels, “Thiazolidinediones and PPARγ Agonists: Time for a Reassessment,” Trends in Endocrinology & Metabolism, Vol. 23, No. 5, 2012, pp. 205-215. doi:10.1016/j.tem.2012.03.001
- [100] D. S. Straus and C. K. Glass, “Anti-Inflammatory Actions of PPAR ligands: New Insights on Cellular and Molecular Mechanisms,” Trends in Immunology, Vol. 28, No. 12, 2007, pp. 551-558. doi:10.1016/j.it.2007.09.003
- [101] L. Petrica, A. Vlad, M. Petrica, et al., “Pioglitazone Delays Proximal Tubule Dysfunction and Improves Cerebral Vessel Endothelial Dysfunction in Normoalbuminuric People with Type 2 Diabetes Mellitus,” Diabetes Research and Clinical Practice, Vol. 94, No. 1, 2011, pp. 22-32. doi:10.1016/j.diabres.2011.05.032
- [102] K. Pegg, J. Zhang, C. Pollock and S. Saad, “Combined Effects of PPARγ Agonists and Epidermal Growth Factor Receptor Inhibitors in Human Proximal Tubule Cells,” PPAR Research, Vol. 2013, 2013, Article ID: 982462.
- [103] W. M. Wang, H. D. Zhang, Y. M. Jin, B. B. Zhu and N. Chen, “PPARγ Agonists Inhibit TGFβ1-Induced Chemokine Expression in Human Tubular Epithelial cells,” Acta Pharmacologica Sinica, Vol. 30, No. 6586, 2009, pp. 107-112. doi:10.1038/aps.2008.15
NOTES
*Corresponding author.