Open Journal of Endocrine and Metabolic Diseases
Vol.3 No.2A(2013), Article ID:31426,6 pages DOI:10.4236/ojemd.2013.32A003
Mitochondrial Dysfunction and Alzheimer’s Disease
Ball State University, Muncie, USA
Email: mlkellyworde@bsu.edu
Copyright © 2013 Fatimah M. Albrekkan, Marie Kelly-Worden. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received February 28, 2013; revised March 26, 2013; accepted April 26, 2013
Keywords: Alzheimer’s; Mitochondria; Cybrid; Oxidative Stress
ABSTRACT
Alzheimer’s disease (AD) is a neurodegenerative disorder that is characterized by progressive loss of basal forebrain cholinergic neurons, leading to reduction in transmission through cholinergic fibers involved in processes of attention, learning, and memory. Mitochondria provide and regulate cellular energy and are crucial for proper neuronal activity and survival. Mitochondrial dysfunction is evident in early stages of AD and is involved in AD pathogenesis. This review focuses on the evidence supporting a clear association between amyloid-β toxicity, mitochondrial dysfunction, oxidative stress and neuronal damage/death in Alzheimer’s disease. To date, the beta amyloid (Aβ) cascade hypothesis still remains the main pathogenetic model of Alzheimer’s disease (AD), but its role in the majority of sporadic AD cases is uncertain. Furthermore, the “mitochondrial cascade hypothesis” could explain many of the biochemical, genetic, and pathological features of sporadic AD. This hypothesis promotes mutations in mitochondrial DNA (mtDNA) as the basis for Alzheimer’s disease. The mutations could lead to energy failure, increased oxidative stress, and accumulation of Aβ, which in a vicious cycle reinforces the mtDNA damage and oxidative stress.
1. Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. It is an epidemic disease with approximately 27 million people affected worldwide and because it is incurable, this number is only expected to rise (quadrupling by 2050). Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder that is characterized by the appearance of amyloid fibrils and plaques. These plaques are formed by the polymerization of the β-amyloid (Aβ) proteins that are derived from the β-amyloid precursor protein (APP). APP is hydrolyzed by β-secretase and by the γ-secretase complex. The amyloid cascade hypothesis suggests that AD arises when the amount of amyloid or nature of amyloid is altered disrupting normal function. This leads to memory loss and neuronal cell death [1]. This is supported in theory by the main pathological feature of AD which is extensive neurodegeneration of the brain in the regions of the median temporal lobe (the hippocampus in particular), parietal lobe, select regions of the frontal cortex and the cingulate gyrus [2]. The hippocampus is the region of the brain involved in the conversion of short term memory into long term memory; a process known as consolidation.
There are two main forms of Alzheimer’s disease (AD). The first type, sporadic AD or SAD, is the most common form of AD and is characterized by a severe progressive decline in cognition and increased neuronal cell death. The second form, familial AD or FAD, progresses much faster and is initiated by mutations in components of the amyloid pathway such as APP, apolipoprotein E4 (ApoE4), presenilin-1 and presenilin-2 (PS1 and PS2) and sortilin-related receptor 1 (SORL1) [1].
The pathogenesis of Alzheimer’s disease is very complex, and involves many molecular, cellular and physiological pathologies. Researchers have implicated soluble Aβ oligomers (also referred to as ADDLs) as the toxic component responsible for the development of ADassociated pathology including tau hyperphosphorylation, spine loss and abnormal spine morphology, formation of reactive oxygen species, prolonged long-term depression and inhibition of long-term potentiation as well as cell death [3].
Although the amyloid-β cascade hypothesis remains the main pathogenetic model of AD, this cascade is possibly viable only for familial AD cases with mutations in AβPP and PS genes and its role in SAD is still uncertain. As a result, research has been directed to the role of mitochondria and oxidative phosphorylation defects in AD [4]. Mitochondrial dysfunction is believed to play a key role in neurodegenerative diseases especially AD. Mitochondria are known to produce the majority of ATP in cells and also function to maintain Ca+2 homeostasis. Since mitochondria are regulators for both cellular metabolism and apoptosis, any oxidation damage to the mitochondria may be relevant to the pathogenesis of AD [5]. This review focuses on the role of mitochondria dysfunction in AD pathophysiology as well as reviewing recent relevant genetic and biochemical data.
2. Review of Literature
2.1. The “Mitochondrial Cascade Hypothesis”
The mitochondrial cascade hypothesis which was first explained by Swerdlow and Kan states that mitochondrial dysfunction is the primary event that causes Aβ deposition, synaptic degeneration and neurofibrillary tangle (NFT) formation in AD (Figure 1). There is much support for this hypothesis since extensive evidence from in vitro, in vivo and human studies has found that mitochondrial abnormalities are a common pathological event in AD [6]. Moreover, the only difference between the sporadic and familial AD is that Aβ seems to be the primary pathological event, causing a secondary mitochondrial dysfunction; though no specific mtDNA gene mutation has been identified [7]. Brain tissue obtained from AD patients displays increased oxidative damage and a significant increase in mtDNA and cytochrome oxidase (COX).
2.2. The “Cybrid Model” of AD
Peripheral tissues are suitable for providing useful biological markers for diagnosis of AD. These tissues include skin, fibroblasts, platelets and lymphocytes. The use of peripheral cells is based on the hypothesis that AD is a systemic disease that affects several tissues in the body. Moreira and colleagues demonstrated that mitochondria isolated from AD platelets have a 15% decrease in COX activity, they also found that platelet ATP levels are diminished in AD while reactive oxygen species (ROS) are increased which leads to energetic failure [6]. The Cytoplasmic Hybrid (“Cybrid”) technique was first described in 1989 and used to determine the origin of mitochondrial oxidative phosphorylation (OXPHOS) defects in SAD. In this technique, mitochondria/mtDNA from human AD and control platelets are transferred to culturable cells depleted of endogenous mtDNA [4].
Many bioenergetic deficits were found in SAD platelet mitochondrial genes expressed in cybrids, such as reduced COX activity, reduced ATP levels and increased oxidative stress, while the citrate synthase activity remained un-changed. In addition, long-term culture showed many abnormal and damaged mitochondria in
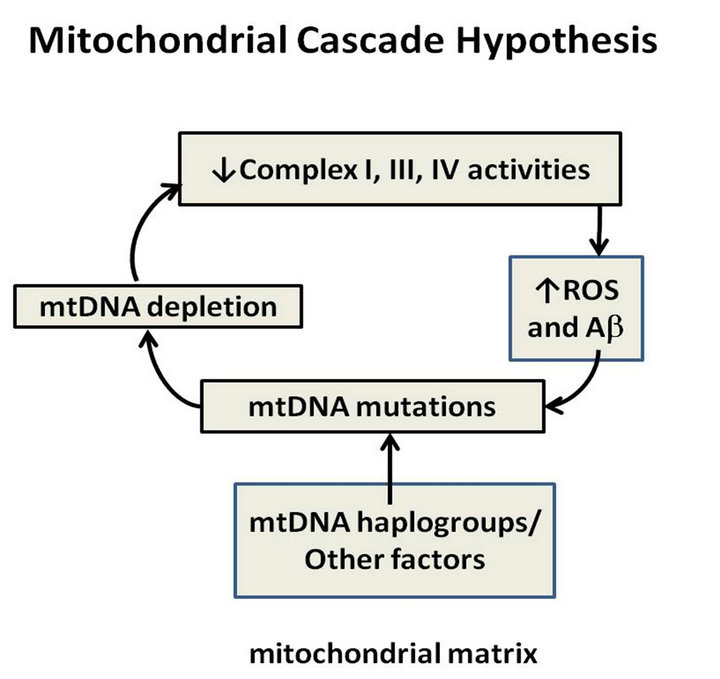
Figure 1. Mitochondrial cascade hypothesis.
SAD cybrids. SAD cybrids also overproduce both of the major amyloidogenic Aβ peptides (1 - 40, 1 - 42) in a caspasedependent manner [4].
Furthermore, the SAD cybrids mimic the amyloid plaques observed in AD brain with the accumulation of Congo red amyloid deposits. These cells manifest an excessive decrease in the mitochondrial membrane potential (Ψ), excessive mitochondrial release of citochrome c and caspase enzyme activation and this reduces the cell’s vulnerability to Aβ 1 - 40 cell death. Continuous culture of AD cybrids lowers Ψ relative to controls, worsening the bioenergetic deficit simultaneously with increased AD mtDNA replication, producing defective mitochondria over time [4].
Finally, a study done by Trimmer and Borland in 2005, demonstrated that the mitochondria found in AD cybrid neurites are elongated compare to short and punctate mitochondria found in controls. Also, the mean velocity of mitochondrial and lysosomal movement, as well as the percentage of moving mitochondria, is significantly reduced in AD cybrids [8-10].
All previous studies demonstrate that mtDNA changes could be responsible for the mitochondrial features found in SAD; since the SAD cybrid cell lines express the same morphological and biochemical phenotype detected in vivo in SAD. These findings together reinforce that mtDNA changes could be the origin of the increased oxidative stress and Aβ deposition found in SAD brains [4].
2.3. TCA Cycle in AD Brains
Since mitochondria are the energy producing cells, any damage to mitochondria will inevitably impair energy metabolism. Brain energy metabolism is found to be significantly decreased in AD patients and this reduction in metabolism can precede the clinical symptoms of AD by many years. The main pathway for oxidation of glucose in brain is the tricarboxylic acid (TCA) cycle (the Krebs’ cycle), which takes place in the mitochondria. Many studies that have been done on AD brains showed significant decrease in the activity of two key enzymes of TCA cycle which are PDHC and KGDHC enzymes. Also, studies have found a significant decrease in the isocitrate dehydrogenase, whereas the activities of succinate dehydrogenase and malate dehydrogenase were increased and the activities of the remaining four TCA cycle enzymes were unchanged. Furthermore, all the changes observed in TCA cycle activities correlated with clinical state, suggesting that the imbalance in AD could lead to diminished brain metabolism resulting in the decline in brain function [4].
2.4. Mitochondrial Genetics and AD
Although mitochondrial dysfunction and the subsequent increased oxidative stress are found in AD brain, the cause of this mitochondrial dysfunction, its relationship to oxidative stress and their roles in synaptic dysfunction and neuronal death are not clear. The discovery of the “classic” mitochondrial diseases where bioenergetic deficiencies were related to the mutations or deletions in mitochondrial DNA (mtDNA) created a search for similar abnormalities in AD samples [7].
Direct metabolic analyses of AD brain mitochondrial function showed some TCA cycle changes that lead to markedly impaired bioenergetics and impaired ability to both metabolize pyruvate and respire properly from the NADH generated by pyruvate oxidation in the TCA cycle. However, the origins of these metabolic problems have not been determined. To date, we know that all TCA enzyme subunits and all but 13 ETC proteins are encoded by the nuclear genome, yet the most common form of AD which is SAD has no definable autosomal genetic inheritance. Therefore, mutations or deletions in mtDNA might explain the impaired COX activity, but still does not provide a direct explanation for TCA cycle deficiencies [7].
Research on mtDNA deletions has revealed an interesting fact that each mtDNA deletion is unique and likely represented by clonal expansion of that deletion [7]. Recent studies also found that polymorphisms in mtDNA may cause refined differences in the encoded proteins which translate to minimum changes in OXPHOS activity resulting in free radical overproduction. This finding leads us to believe that an individual or a population sharing the same mtDNA genotype may have an earlier onset of the apoptotic processes. However, different polymorphism(s) can be beneficial and cause an increase in OXPHOS activity and/or reduction in ROS production. Classes of continent-specific genotypes, haplogroups, which can be detected by RFLP analysis, can be determined by common mtDNA polymorphisms. In Europe, nine different mitochondrial haplogroups have been identified (H, I, J, K, T, U, V, W and X). This lead to a suggestion that inherited European mitochondrial haplogroups may be related to longevity, as well as AD risk in Caucasians [4, 11,12].
Several studies tried to define the specific mtDNA mutation(s) responsible for the mitochondrial dysfunction found in SAD cybrids. Findings suggested that the mtDNA appears to be more vulnerable than nuclear DNA to accumulating oxidative and nucleotide damage due to its proximity to ROS production as well as lack of efficient systems of DNA repair. An increase of somatic mtDNA re-arrangements has been observed in AD brains. The mtDNA “common deletion”, for instance, has been observed to be elevated about 15-fold in AD brains. Moreover, studies comparing AD to control suggest that the A to G transition at nucleotide position 4336 was also noticed more frequently in AD. Recently, somatic mutations in the CR mtDNA region have been associated with aging and with AD [4]. Unfortunately, all the previous studies fail to define the exact role and relevance of these haplogroups and/or mtDNA point mutations.
2.5. Mitochondrial Dysfunction in AD
Mitochondria generate and scavenge free radicals to maintain the viability and functionality of cells. Many studies have shown that mitochondrial function is impaired in AD brains. Findings also suggested that neurons with increased oxidative damage have altered mitochondrial DNA and cytochrome oxidase levels. In addition, evidence from postmortem AD brains show alterations in expression of several proteins involved in mitochondrial fission and fusion, leading to abnormal redistribution of mitochondria [13].
Other than providing the cell with ATP, mitochondria also play a significant role in the regulation of cell death since they harbor several death factors that are released upon apoptotic stimulation. In AD patients, many alterations have been detected; mitochondrial dysfunction, increased oxidative stress, and neurons dying by apoptosis. These findings support the idea that mitochondria may trigger the abnormal onset of neuronal degeneration and death in AD [6].
Findings in transgenic AD mice demonstrate increased levels of H2O2 and decreased levels of cytochrome oxidase at a young age and prior to the appearance of amyloid deposition. Other studies in young transgenic AD mice reported a reduction in mitochondrial membrane potential and of ATP levels. Mitochondrial dysfunction also was found to be associated with increased production of reactive oxygen species (ROS). Data from previous studies also confirmed that amyloid deposition is preceded by deficits in mitochondrial bioenergetics, as evidenced by decreased mitochondrial respiration and pyruvate dehydrogenase levels and activity in transgenic mice with human AβPP, presenilin 1 (PS1), and tau mutations. All these data strongly suggest that mitochondrial dysfunction occurs early in disease pathogenesis [13].
Several mitochondrial key enzymes have been studied in the context of AD. First, in human postmortem AD brain, activity of α-KGDHC was found to be reduced. Additionally, levels of its subunits α-ketoglutarate dehydrogenase (E1; EC 1.2.4.2) and dihydrolipoyl succinyltransferase (E2; EC 2.3.1.61 or DLST) are also diminished in brains of patients bearing the Swedish AβPP mutation KM670/671NL. MnSOD is also an important mitochondrial enzyme that catalyzes the dismutation of superoxide to H2O2, which is then decomposed to water via catalase or glutathione peroxidase. Studies in transgenic AD mice found that partial deficiency of MnSOD increased amyloid plaque deposition and tau phosphorylation. It also accelerated the onset of behavioral abnormalities in transgenic AD mice. On the other hand, MnSOD over expression was found to decrease hippocampal superoxide levels and amyloid plaques as well as improve memory deficit in AD patients [13].
2.6. Aβ Interaction with Mitochondria Aβ has been found to accumulate in mitochondria from AD patients. The amyloid hypothesis of AD states that altered processing of amyloid precursor protein (APP) results in deposition of a neurotoxic form of amyloid-β, and thus plays a central role in AD pathogenesis. Studies which were done on APP transgenic mice supported this finding and showed that immunization against Aβ decreases amyloid levels and improves cognition [14].
Studies also found that Aβ accumulates early on in the disease and mainly in the synaptic mitochondria. These mitochondria where found to be more susceptible to changes in synaptic Ca+2 since they have high levels of cyclophilin D (CypD). CypD is located in the matrix and is a component of the mitochondrial permeability transition pore (mPTP). Cypd translocates from the matrix to the mPTP and interacts with adenine nucleotide translocase in the inner membrane to stimulate pore formation. Opening of mPTP leads to membrane potential collapse that leads to bioenergetic failure and subsequent synaptic failure and ultimately neuronal death. Mitochondrial Aβ has been found to interact with CypD, and CypD deficiency attenuates Aβ induced mitochondrial stress. This suggests that synaptic mitochondria are important in AD pathogenesis since they have high levels of Aβ and CypD [5].
Mitochondrial numbers are decreased and the mitochondria are larger in AD patients, and this explains why fission is more prevalent than fusion in AD. Aβ overproduction leads to APP over expression and that increases the level of proteins associated with fission (Fis1) while decreasing protein levels of those involved in fusion, which include dynamin-like protein and OPA1[5].
Interestingly, Aβ was found to have both antioxidant and pro-oxidant functions. At low concentrations, Aβ remains monomeric and functions as an antioxidant, however, at high concentrations Aβ aggregation will produce some oxidative stress like H2O2 production which can lead to mitochondrial dysfunction [5].
Studies done in APP mutant mice showed that mitochondrial-targeted antioxidants increased Aβ mitochondrial dysfunction and that mitochondrial-induced oxidative stress could initiate the production of Aβ, and thus increases the oxidative stress and mitochondrial dysfunction and provides a feed-forward loop mechanism that increases Aβ levels [15].
2.7. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease
Studies done on human postmortem AD brains and transgenic AD mouse brains showed that oxidative stress and mitochondrial dysfunction are key features in Alzheimer’s disease (AD) and they appear early and contribute to disease progression. Data from these studies suggested that DNA, RNA, lipid and protein oxidation are increased in the cortex and the hippocampus in human AD brains. Moreover, levels of 8-hydroxy-2’-deoxyguanosine (8-OHdG) and nitrotyrosine are found to be significantly augmented in AD patients compared to normal control patients. Oxidative stress markers have been considered as biomarkers for disease progression. Studies done on cerebrospinal fluid (CSF) of AD patients reported that levels of F2-isoprostane were consistently increased compared to normal control patients. All previous studies suggested that increased oxidative stress occurs at early stages of amyloid deposition [13].
In the cortex and hippocampus of transgenic AD mice, increased markers for protein oxidation and lipid peroxidation were noted. This increase was observed prior to the appearance of amyloid plaques and neurofibrillary tangles. Altogether, it suggests that oxidative stress can exacerbate AD pathogenesis [13].
2.8. Role of Amyloid-β Peptide Alcohol Dehydrogenase (ABAD)
Several studies have indicated that intracellular Aβ may be an important contributor to Aβ-mediated cytotoxic events in AD. Identifying specific macromolecular targets is an important means of determining how Aβ affects cells and how this interaction of Aβ with the targets impacts cellular functions. Studies found that a member of the short chain dehydrogenase reductase family which includes amyloid-b alcohol dehydrogenase (ABAD), binds Aβ. Some unique features of ABAD include its presence in endoplasmic reticulum and mitochondria, its broad substrate specificity, and its capacity to bind Aβ and promote Aβ-induced cell stress. Moreover, ABAD is especially enriched in neuronal mitochondria which play an important role in metabolic homeostasis [16].
ABAD has been found to be cytoprotective under normal cell conditions in response to stress. However, these cytoprotective properties of ABAD appear to be negated in an environment rich in Aβ found in AD. Decreased ABAD activity is associated with negative effects on brain energetics and, possibly, other functions. For example, some studies suggest that loss of ABAD’s activity as a hydroxysteroid dehydrogenase might alter metabolism of oestrogenic hormones involved in neuro-protecttion [17].
Taken together, ABAD-Aβ complex (Figure 2) was found to cause cellular dysfunction by several potential mechanisms, such as loss of enzymatic activity, accumulation of upstream toxic metabolites, leakage of mitochondrial oxygen-free radicals, loss of cellular energy charge and enhanced opening of the MPT [16].
2.9. Mitochondrial-Associated Membranes (MAMs)
MAMs originally identified by Vance (1990) are specialized subdomains of the ER that are in close contact with mitochondria. Mitochondria and ER are recognized to have some close connections that are essential for several functions shared by the two organelles, such as transfer of Ca+2, lipid metabolism, morphology of mitochondria and the control of apoptosis. This physical connection is believed to be mediated by phosphofurin acidic cluster sorting protein 2 (PACS2) and mitofusin 2 (MFN2) [18].
Although the awareness of this regions importance in physiology is so crucial, its structural composition remains undetermined [19]. However, studies have shown that PS1 and PS2 are actually highly enriched in only one other compartment of the cell, namely MAM. Moreover, besides identifying MAM as the main subcellular compartment for the presenilins, they also found that γ-secretase activity associated with AβPP processing is essentially confined to the MAM [9].
Since mutations in PS are the major cause of familial AD, this raises the interesting questions whether PS is involved in MAM function and whether this function is disturbed in familial AD. γ-Secretase complex is enriched at the interface between mitochondria and the ER and the communication between the two organelles becomes dysfunctional in models of AD [19].
It has been proposed that the fundamental pathogenetic cause of AD is the alteration of MAM function, which could explain many of the disparate features of the disease, such as elevated cholesterol, altered fatty acid, glu-
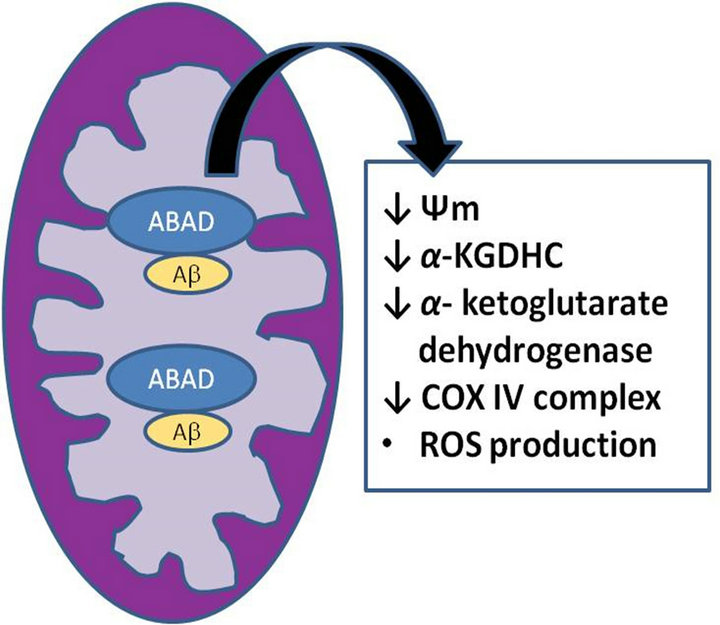
Figure 2. The ABAD-Aβ Complex.
cose, and phospholipids levels, calcium imbalance, and formation of plaques and tangles [10].
3. Conclusion
Mitochondria are involved and play a key role in the progression of several neurodegenerative diseases including AD [20]. There is compelling evidence that AD brains are bioenergetically impaired, that this metabolic deficiency appears early in the clinical evolution of AD, that it worsens with clinical deterioration, and that it is associated with mitochondrial enzymatic impairments in the TCA cycle and ETC. AD cybrid studies over the last decade have shown compelling circumstantial evidence for contributions of systemic mtDNA to AD pathogenesis. However, researchers have been unable to specify how exactly mtDNA may be altered in AD [21,22].
REFERENCES
- C. Lee, P. Linneman and M. J. Berridge, “Calcium Hypothesis of Alzheimer’s Disease,” European Journal of Physiology, Vol. 459, No. 3, 2010, pp. 441-449. doi:10.1007/s00424-009-0736-1
- C. Supnet and I. Bezprozvanny, “Presenilins as Endoplasmic Reticulum Calcium Leak Channels and Alzheimer’s Disease Pathogenesis,” Science China Life Sciences, Vol. 54, No. 8, 2011, pp. 744-751. doi:10.1007/s11427-011-4201-y
- J. Pitt, W. Roth, P. Lacor, A. B. Smith III, M. Blankenship, P. Velasco, F. D. Felice, P. Breslin and W. L. Klein, “Alzheimer’s-Associated Aβ Oligomers Show Altered Structure, Immunoreactivity and Synaptotoxicity with Low Doses of Oleocanthal,” Toxicology and Applied Pharmacology, Vol. 240, No. 2, 2009, pp. 189-197. doi:10.1016/j.taap.2009.07.018
- M. Mancuso, G. Siciliano, M. Filosto and L. Murri, “Mitochondrial Dysfunction and Alzheimer’s Disease: New Developments,” Journal Of Alzheimer’s Disease, Vol. 9, No. 2, 2006, pp. 111-117.
- R. D. Readnower, A. D. Sauerbeck and B. G. Sullivan, “Mitochondria, Amylpoid B, and Alzheimer’s Disease,” International Journal of Alzheimer Disease, Vol. 2011, Article ID: 104545, 2011, pp. 1-5. doi:10.4061/2011/104545
- P. I. Moreira, S. M. Cardoso, M. S. Santos and C. R. Oliveira, “The Key Role of Mitochondria in Alzheimer’s Disease,” Journal of Alzheimer’s Disease, Vol. 9, No. 2, 2006, pp. 101-110.
- I. Onyango, S. Khan, B. Miller, R. Swerdlow, P. Trimmer and J. Bennett Jr., “Mitochondrial Genomic Contribution to Mitochondrial Dysfunction in Alzheimer’s Disease,” Journal of Alzheimer’s Disease, Vol. 9, No. 2, 2006, pp. 183-193.
- P. A. Trimmer and M. K. Borland, “Differentiated Alzheimer’s Disease Transmitochondrial Cybrid Cell Lines Exhibit Reduced Organelle Movements,” Antioxid Redox Signaling, Vol. 7, No. 9-10, 2005, pp. 1101-1109. doi:10.1089/ars.2005.7.1101
- E. Area-Gomez, J. C. de Groof, I. Boldogh, T. D. Bird, G. E. Gibson, C. M. Koehler, W. H. Yu, K. E. Duff, M. P. Yaffe, L. A. Pon and E. A. Schon, “Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria,” The American Journal of Pathology, Vol. 175, No. 5, 2009, pp. 1810-1816. doi:10.2353/ajpath.2009.090219
- M. Dumont, M. T. Lin and M. F. Beal, “Mitochondria and Antioxidant Targeted Therapeutic Strategies for Alzheimer’s Disease,” Journal of Alzheimer’s Disease, Vol. 20, 2010, pp. 633-643.
- J. Zhang, J. Asin-Cayuela, J. Fish, Y. Michikawa, M. Bonafé, F. Olivieri, G. Passarino, G. De Benedictis, C. Franceschi and G. Attardi, “Strikingly Higher Frequency in Centenarians and Twins of mtDNA Mutation Causing Remodeling of Replication Origin in Leukocytes,” Proceedings of National Academy of Science of USA, Vol. 100, No. 3, 2003, pp. 1116-1121. doi:10.1073/pnas.242719399
- A. Eckert, K. L. Schulz, V. Rhein and J. Götz, “Convergence of Amyloid-b and Tau Pathologies on Mitochondria in Vivo,” Molecular Neurobiology, Vol. 41, No. 2-3, 2010, pp. 107-114. doi:10.1007/s12035-010-8109-5
- R. Castellani, K. Hirai, G. Aliev, K. Drew, A. Nunomura, A. Takeda, A. D. Cash, M. E. Obrenovich, G. Perry and M. A. Smith, “Role of Mitochondrial Dysfunction in Alzheimer’s Disease,” Journal of Neuroscience Research, Vol. 70, No. 3, 2002, pp. 357-360. doi:10.1002/jnr.10389
- A. K. Niemi, J. S. Moilanen, M. Tanaka, A. Hervonen, M. Hurme, T. Lehtimäki, Y. Arai, N. Hirose and K. Majamaa. “A Combination of Three Common Inherited Mitochondrial DNA Polymorphisms Promotes Longevity in Finnish and Japanese Subject,” European Journal of Human Genetics, Vol. 13, 2005, pp. 166-170. doi:10.1038/sj.ejhg.5201308
- S. D. Yan and D. M. Stern, “Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Amyloid-β Peptide Alcohol Dehydrogenase (ABAD),” International Journal of Experimental Pathology, Vol. 86, No. 3, 2005, pp. 161-171. doi:10.1111/j.0959-9673.2005.00427.x
- He X.Y., MerZ G., Mehta P., SchulZ H., Yang S.Y. “Human Brain Short Chain L-3-hydroxyacyl-coenzyme A Dehydrogenase Is a Single-Domain Multifunctional Enzyme: Characterization of a Novel 17β-Hydroxysteroid Dehydrogenase,” Journal of Biological Chemistry, Vol. 274, 1999, pp. 15014-15019. doi:10.1074/jbc.274.21.15014
- E. A. Schon and E. Area-Gomez, “Is Alzheimer’s Disease a Disorder of Mitochondria-Associated Membranes?” Journal of Alzheimer’s Disease, Vol. 20, No. 2, 2010, pp. S281-S292.
- V. A. Morais and B. D. Strooper, “Mitochondria Dysfunction and Neurodegenerative Disorders: Cause or Consequence,” Journal of Alzheimer’s Disease, Vol. 20, No. 4, 2010, pp. 255-263.
- J. E.Vance, “Molecular and Cell Biology of Phosphatidylserine and Phosphatidylethanolamine Metabolism,” Progress in Nucleic Acid Research and Molecular Biology, Vol. 75, 2003, pp. 69-111. doi:10.1016/S0079-6603(03)75003-X
- M. Mancuso, V. Calsolaro, D. Orsucci, C. Carlesi, A. Choub, S. Piazza and G. Siciliano, “Mitochondria, Cognitive Impairment, and Alzheimer’s Disease,” International Journal of Alzheimer’s Disease, Vol. 2009, Article ID: 951548, 2009, pp. 1-8. doi:10.4061/2009/951548
- S. M. Cardoso, I. Santana, H. Russell, Swerdlow and C. R. Oliveira, “Mitochondria Dysfunction of Alzheimer’s Disease Cybrids Enhances Aβ Toxicity,” Journal of Neurochemistry, Vol. 89, No. 6, 2004, pp. 1417-1426. doi:10.1111/j.1471-4159.2004.02438.x
- S. M. Cardoso, M. T. Proença, S. Santos, I. Santana and C. R. Oliveira, “Cytochrome C Oxidase is Decreased in Alzheimer’s Disease Platelets,” Neurobiology of Aging, Vol. 25, No. 1, 2004, pp. 105-110. doi:10.1016/S0197-4580(03)00033-2