Open Journal of Marine Science
Vol.3 No.4(2013), Article ID:37963,7 pages DOI:10.4236/ojms.2013.34020
A Depth Resolved Insight into Benzoyl CoA Reductase and Benzoate Dioxygenase Gene Copy Numbers within a Marine Sediment Associated with Methane Seepage
1School of Biological Sciences, Queen’s University Belfast, Belfast, UK
2School of Chemical Sciences, Dublin City University, Dublin, Ireland
3Geological Survey of Ireland, Beggar’s Bush, Dublin, Ireland
Email: *c.allen@qub.ac.uk
Copyright © 2013 Paul V. Flanagan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 28,, 2013; revised August 4, 2013; accepted August 21, 2013
Keywords: Aromatic Compounds; qPCR; bcr; bdo
ABSTRACT
A marine sediment core obtained from a methane seepage site off the northern coast of Ireland was analysed, at 3 depths, for catabolic genes associated with the aerobic and anaerobic degradation of aromatic compounds. Catabolic gene copy numbers varied through the core peaking at 2.1 meters below sediment surface (mbsf)-just above the sulphate boundary. Beyond the sulphate boundary gene copy numbers fell considerably, suggesting the boundary may be a critical factor in the degradation of aromatic compounds within marine sediments. At the 2.1 mbsf depth our data also suggest that known benzoyl CoA reductase utilizing bacteria are readily detectable.
1. Introduction
The importance of marine sediments, in terms of bioremediation, has grown significantly in recent times [1]. Various studies show that marine sediments, and the indigenous microorganisms therein, possess the potential to degrade a wide array of aromatic compounds [2]. Benzene, a classical, persistent aromatic compound [3], has been shown to be degraded in laboratory microcosms under anaerobic conditions where marine sediments were used as a source of microorganisms in the presence of nitrate or sulphate as an electron acceptor [3-5]. Both monocyclic and polycyclic aromatic compounds, have been shown to be degraded under a variety of conditions when marine sediment is used as a source of microorganisms in enrichment studies [2].
The discovery of catabolic gene sequences associated with the degradation of aromatic compounds from marine sediments via metagenomic research consolidates the fact that eubacteria associated with these sediments are capable of carrying out diverse aromatic compound degradation under both aerobic and anaerobic conditions [6]. This is of particular relevance as aromatic substrates commonly occur in marine sediments, and are derived from both natural and anthropogenic sources. Hydrophobic aromatic compounds commonly migrate and exhibit recalctrance in marine sediments [7].
However, despite the importance of microbes associated with the degradation of ubiquitous aromatic compounds, little is known about their actual distribution in marine sediments and subsequently their role in the degradation of these substrates.
In recent work we characterised a 6 metre marine sediment core from a methane seepage site [8] and found that chemistry and microbial activity in the core are influenced by the presence of methane and sulphate. In this study we wanted to investigate the quantities of benzoyl CoA reductase (bcr) and benzoate dioxygenase (bdo)- catabolic genes associated with the anaerobic and aerobic degradation of various aromatic compounds—at defined depths—as markers for microbial activity associated with aromatic degradation. We then wanted to consider the significance of these data in conjunction with the nature of the overall eubacterial population from within the methane seepage site. We wanted to see if heterotrophic aromatic substrate-degrading bacteria also played a potential role in the carbon cycle in these methane-rich sites.
2. Methods
2.1. Sample Collection and Nucleic Acid Isolation
Sediment samples were collected, characterised and stored as described in our previous work [8]. Frozen sediment was taken at 0.2, 2.1 and 5.9-metre depths from a single core. Total DNA was extracted using a combination of heat and chemical lysis to disrupt microbial cells [9]. Total DNA was then subjected to purification with chloroform: isoamyl alcohol (Sigma). Finally DNA was precipitated with 0.6 volumes of isopropanol and eluted in 100 µL of sterile TE buffer. Extracts of DNA were quantified spectrophotometrically as described in the literature [9] and stored at −20˚C.
2.2. 16S rRNA Gene Amplification
16S bacterial rRNA polymerase chain reactions were carried out using DNA Engine DYADTM Peltier Thermal Cycler. 16S bacterial rRNA sequences were amplified from genomic DNA using universal 63f (5’-CAGGCCTAACACATGCAAGTC-3’) forward primer and 1387r (5’-GGGCGGWGTGTACAAGGC-3’) reverse primer [10]. PCR was performed as follows: denaturation step of 95˚C for 5 min; followed by 33 cycles of 94˚C for 30 s, 55˚C for 30 s and finally 72˚C for 1 min 30 s.
16S PCR products were visualised by UV transillumination (Biorad) and bands were removed and purified prior to cloning using a DNA purification kit (Fermentas).
2.3. Cloning and Sequence Analysis
Cloning of 16S rRNA genes was carried out as described previously [11] at depths of 2.1 m and 5.9 m as these depths were found on either side of the sulphate boundary [8]. Operational taxonomic units (OTUs) were grouped by restriction analysis using Fastdigest RSaI and HaeIII (Fermentas) restriction enzymes. Restriction profiles were analysed on 1.4% agarose gel and each gel was standardized to allow comparison between gels. Clones showing identical restriction profiles were assigned to the same OTU group using a total of 60 clones from 2.1 mbsf and 52 clones from 5.9 mbsf. The nucleotide sequence data reported in this study were deposited in the GenBank nucleotide sequence database under the accession numbers JQ349446 to JQ349503.
2.4. Phospholipid Fatty Acid (PLFA) Analysis
Lyophilized sediment samples were extracted using the modified Bligh-Dyer method, according to White & Ringelberg [12]. Total lipid extracts (TLEs) were desulphurised by mixing with activated Cu powder overnight. TLEs were fractionated into neutral, glycolipids and polar lipids using solid phase extraction on solvent-cleaned aminopropyl cartridges (Alltech 500 mg Ultra-Clean, USA) according to Pinkart et al. [13]. Phospholipids were transesterified by mild alkaline methanolysis using 0.5 M sodium methoxide (Sigma Aldrich) for 30 min at 50˚C. Fatty acid methyl esters (FAMEs) were analysed using an Agilent 6890 gas chromatograph (GC) coupled to Agilent 5973 N quadruopole mass selective detector (MSD). The GC was fitted with a HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm). Splitless injection mode was used while other GC and MSD operating conditions were as described in Otto et al. [14]. Unsaturation position on monounsaturated FAMEs were identified according to Nichols et al. [15]. Quantification was performed using relative response factors for a model FAME (tetradecanoic acid methyl ester) to 5-α-cholestane internal standard (100 μg∙mL−1).
2.5. qPCR
The abundance of catabolic genes was quantified by qPCR using primer pair bdoF/R [16] for the detection of the bdo gene or BZAQF [17] coupled to PFR1 (5’TCCTGMCCGCCSATGTCSAG’3) for the detection of the bcr gene. The thermal cycling program consisted of an initial hotstart at 95˚C for 10 min, followed by 35 cycles of 95˚C for 20 s, 58˚C for 20 s and 72˚C for 20 s. Plate reads were taken following each extension step at 72˚C. Gel electrophoresis (Biorad) of products was performed to ensure specificity of product and in the construction of standards. At all other times melting curves were performed at the end of a qPCR run to confirm reaction specificity. All Quantitative PCR reactions were performed in an Opticon 3 real-time PCR machine (Biorad) using the Maxima SYBR Green MasterMix (Fermentas). Each 25 µL reaction contained 1 µL of template DNA and a final primer concentration of 0.35 µM each.
Standard curves, for each target gene, were generated using purified PCR products from environmental samples [18]. Briefly, PCR products, specific to each target gene, from environmental samples were excised from an agarose gel and purified using an agarose gel PCR purification kit (Fermentas). DNA concentrations were then measured spectrophotometrically at 260 nm using a microcell cuvette (Hellma). Gene copy numbers were calculated according to the size of the amplicon [19]. The standard curves were linear over 5 orders of magnitude with an r2 value above 0.95. Amplification efficiency ranged from 91% to 105% with samples analyzed in triplicate. Samples of qPCR products were chosen at random, cloned, as described previously [11] and sequenced to confirm product identity.
3. Results
3.1. 16S rRNA Cloning
An overview of the composition of the 2.1 mbsf and 5.9 mbsf bacterial clones libraries is given in Table 1. Clone library analysis indicates that proteobacteria are the dominant phylum within this particular marine sediment, representing 93.4% and 98.1% of the bacterial population at 2.1 mbsf and 5.9 mbsf respectively. Also evident in Table 1 is the presence of Sequences sharing homology with Psychrobacter spp and Sulfitobacter spp which are apparent at both the 2.1 m and 5.9 m depths—both known marine sediment inhabitants [20]. Some minor components of the community at 2.1 m but not 5.9 m were shown to be similar to facultative anaerobes previously shown to utilise the benzoate CoA reductase enzyme for aromatic hydrocarbon biodegradation. A notable example here is the OUT group 8—comprising 1.6% of the community at 2.1 mbsf.
3.2. Distribution of Catabolic Genes
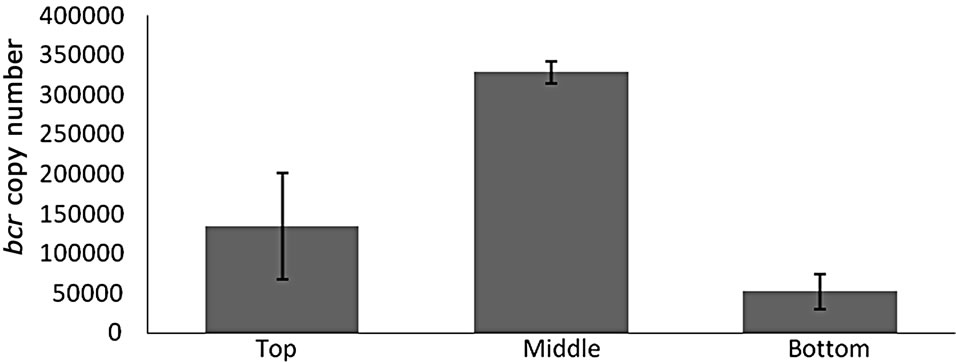
For both catabolic genes tested it is evident that variations in copy numbers of the genes occur throughout the sediment core (Figure 1). The highest abundance of both bcr and bdo genes is noted at the 2.1 m depth of the sediment (Figure 1) with 3.3 × 105 and 1.8 × 105 copies detected respectively. The lowest abundance of both catabolic genes is observed at the 5.9 m depth where 5.2 × 104 and 9.9 × 104 copies of bcr and bdo are detected. Also evident at the 2.1 m depth is an almost 2:1 abundance of bcr relative to bdo. In total 10 clones of each gene were sequenced, to confirm qPCR product identity, with sequences sharing homologies of 85% - 91% and 87% - 92% to known bcr and bdo genes held within the NCBI database respectively.
3.3. Phospholipid Fatty Acid (PLFA) Analysis
PLFA profiles were overall similar down-core, dominated by saturated fatty acids from C14 to C24 (31% to 80% of total PLFA), C16 and C18 monounsaturated fatty acids (10% to 15% of total PLFA) and methyl-branched fatty acids from C14 to C16 (2.5% to 7.5% of total PLFA.) and are likely derived from bacteria [12,21,22]. PLFA abundance has been used to estimate the number of viable cells in environmental samples, based on the estimation that the average bacterium the size of E. Coli contains approximately 1.7 × 10−17 mol PLFA per bacterial cell [23,24]. This corresponds to between 1.9 and 4.7 ×107

Table 1. Operational taxonomic unit (OTU) table of 16S rRNA bacterial clone 2.1 and 5.9 mbsf libraries from the pockmark.
 (a)
(a)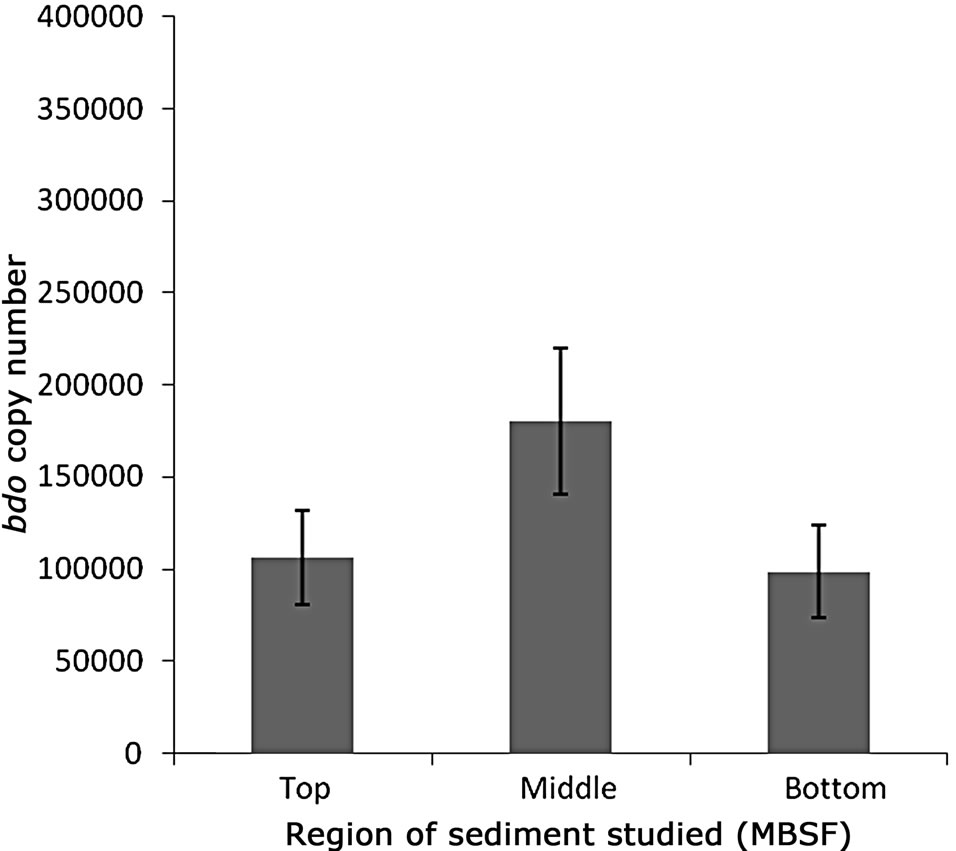 (b)
(b)
Figure 1. Mean catabolic gene copy numbers of bcr (a) and bdo (b)/0.5 g sediment were determined by qPCR on DNA isolated from different regions of the Malin core sample. Analysis was conducted on triplicate DNA extractions. Error bars represent standard deviation from the mean copy number.
cells∙(g∙dry∙wt)−1 at the sediment depths investigated.
4. Discussion
Low growth rates, or intermittent periods of rapid growth interspersed by long periods of non-growth and starvetion is the norm for microbes in the environment [25], with bacterial biomass concentration in marine sediments is usually in the region of 108 to 1010 cells∙g−1 [26,27]. In this study the abundance of prokaryotic biomass appears to be at least an order of magnitude lower than typical at the depths investigated supplementing our previous work which suggested a reduction in microbial activity within the marine sediment examined [8].
The lowest abundance of PLFA’s is noted at the 2.1 mbsf which, incidentally, is where the highest relative abundance of both catabolic genes is observed. This may imply that the viable bacterial population at this depth is lower than other depths examined but of those viable microorganisms, and indeed previous populations at this depth, a high proportion of eubacteria in this region of the sediment may harbor the catabolic genes targeted in this study. However this remains a theory as mRNA studies would need to be performed to link viable eubacteria to expressed genes in this marine environment.
Psychrobacter spp and Sulfitobacter spp are apparent at both the 2.1 m and 5.9 metre depths (Table 1). Both genera have been associated with marine sediments in previous studies [20,28,29]. However as this study focuses on different depths within a marine sediment we observed that sequences from both genera tend to cluster according to the depth at which they are found (Figure 2) indicating subtle differences within relatively similar eubacterial populations potentially being influenced by the chemistry-such as the sulphate boundary-of the sediment region. Also noted in Table 1 is the presence of an OTU group (group 8) whose 16S rRNA sequence shares homology with that of Thauera spp and Azoarcus spp— both facultative, bcr harbouring, β proteobacteria capable of aromatic compound degradation under both aerobic and anaerobic conditions [30]. Another OTU group of interest at the 2.1 m depth is group 6 which has highest known species similarity to a desulfobacterium spp—a known sulphate reducer [30] and aromatic compound degrader via the bcr pathway [31,32].
As stated the highest abundance of both bcr and bdo genes is noted at the 2.1 m depth of the sediment (Figure 1). At this depth aromatic compounds appear to be at the lowest abundance [8]. The aromatic compounds detected within the site are believed to consist of, amongst other groups, benzoate and naturally occurring phenol aromatic derivatives (e.g. vanillic acid). Also evident at the 2.1 m depth is an approximate relevant abundance of 2:1 for bcr relative to bdo suggesting that facultative anaerobes - such as Thauera spp.—may have been metabolic active this region of the sediment. This concept is further enhanced by the detection of sequences sharing homology with known facultative aromatic degraders through clone studies. Intriguingly at 5.9 mbsf more bdo copies are detected relative to bcr. This could be attributed to the presence of bdo harbouring Psychrobacter spp at this depth and the potential redundancy of the energy expensive bcr beyond the sulphate boundary [30].
Previous studies suggest that microbial populations in marine sediments are shaped in their bioremediation potential by an abundance of sulphate [33]. However this is the first study to show that the distribution of catabolic genes, associated with bioremediation of aromatic compounds, fluctuates through a marine sediment core within a methane seepage site, and varies in response to chemical factors, such as the proximity of a sulphate boundary, within the sediment (3). The highest abundance of catabolic genes associated with the aerobic and anaerobic degradation of aromatic compounds was found above the sulphate boundary at a depth of 2.1 mbsf suggesting that
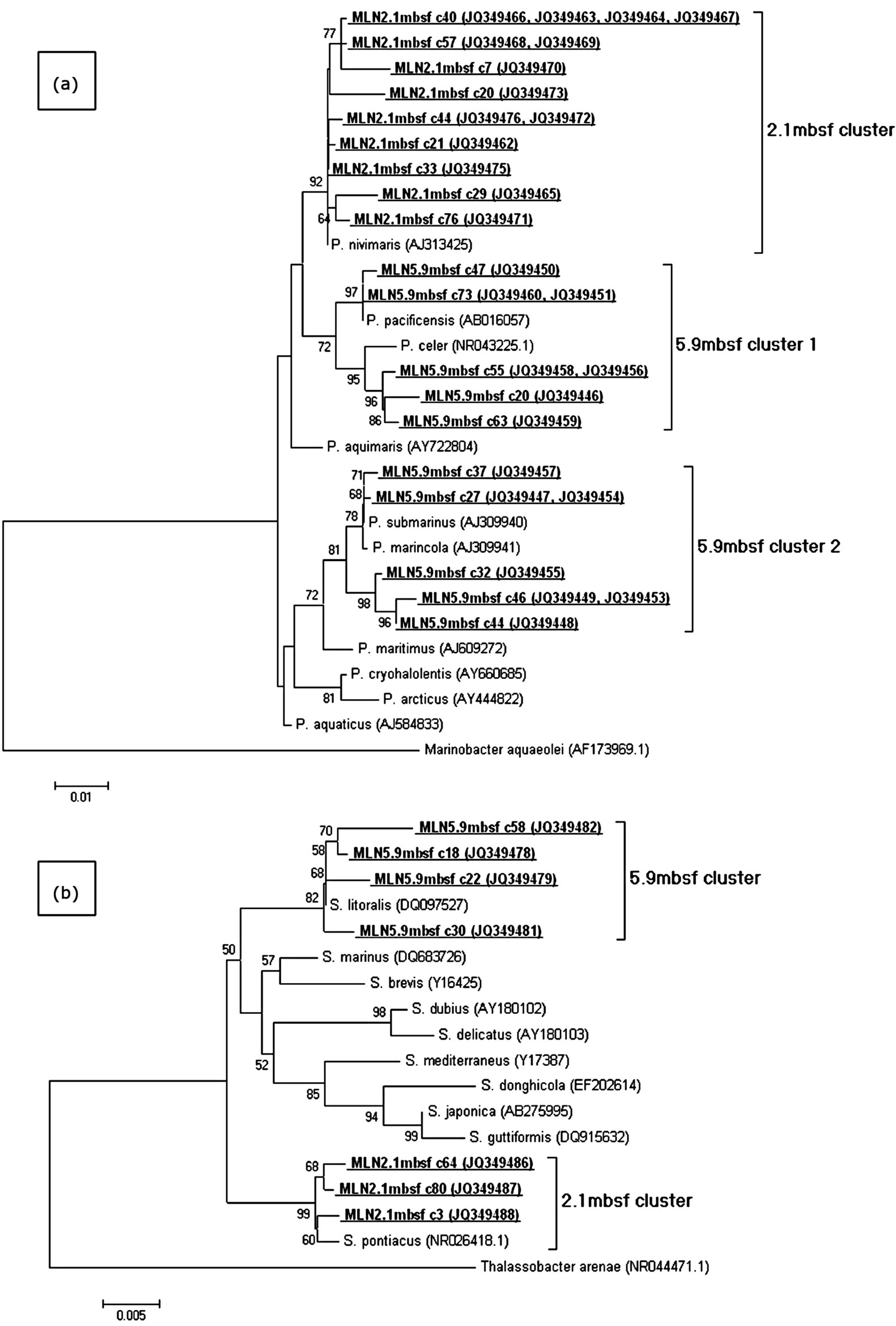
Figure 2. Maximum likelihood phylogenetic tree for selected clones from the Psychrobacter (a) and Sulfitobacter (b) OTU’s from the 2.2 mbsf and 5.9 mbsf clone libraries. Nucleotide sequences from this study are in underlined bold. Accession numbers are in brackets. The scale bars represent the number of base substitutions per site. The tree was subjected to bootstrap analysis (n = 1000) to assess confidence intervals. Bootstrap values > 50 are reported. This shows distinct clustering of both bacterial populations at the two depths. At 2.1 mbsf Psychrobacter clones (n = 14) are phylogenetically most closely related to P. nivimaris, while at 5.9 mbsf (n = 14) are more diverse and cluster into groups related to P. celer, P. marincola and P. submarinus (Romanenko et al., 2002). Sulfitobacter clones at 2.1 mbsf (n = 3) are phylogenetically most closely related to S. pontiacus, while at 5.9 mbsf (n = 6) are most related to S. litoralis.
facultative microorganisms, possibly utilizing sulphate as an electron acceptor, play a vital role in the breakdown of these ubiquitous aromatic compounds within marine sediments.
5. Acknowledgements
We wish to thank the Geological Survey of Ireland, the INtegrated Mapping FOr the Sustainable Development of Ireland’s MArine Resource (INFOMAR) programme, the Irish Environmental Protection Agency, Science Foundation of Ireland, QUESTOR (Queens University Belfast) and the Irish Council for Science, engineering & technology (IRCSET) for funding this research. The survey data utilized in the research has been co-funded by the Geological Survey of Ireland and the Offshore Irish Petroleum Infrastructure Programme (PIP; Ref. No: IS05/16 Malin Basin EM).
REFERENCES
- D. R. Lovley, “Cleaning up with Genomics: Applying Molecular Biology to Bioremediation,” Nature Reviews Microbiology, Vol. 1, No. 1, 2003, pp. 35-44. http://dx.doi.org/10.1038/nrmicro731
- F. Widdel and R. Rabus, “Anaerobic Biodegradation of Saturated and Aromatic Hydrocarbons,” Current Opinion in Biotechnology, Vol. 12, No. 3, 2001, pp. 259-276. http://dx.doi.org/10.1016/S0958-1669(00)00209-3
- C. D. Phelps, J. Kazumi and L. Y. Young, “Anaerobic Degradation of Benzene in BTX Mixtures Dependant on Sulphate Reduction,” FEMS Microbiology Letters, Vol. 145, No. 3, 1996, pp. 433-437. http://dx.doi.org/10.1111/j.1574-6968.1996.tb08612.x
- D. Canfield, B. Thamdrup and J. W. Hansen, “The Anaerobic Degradation of Organic Matter in Danish Coastal Sediments: FE reduction, Mn Reduction and Sulfate Reduction,” Geochimica et Cosmochimica Acta, Vol. 57, No. 16, 1993, pp. 3867-3883. http://dx.doi.org/10.1016/0016-7037(93)90340-3
- F. Musat and F. Widdel, “Anaerobic Degradation of Benzene by a Marine Sulphate Reducing Enrichment Culture, and Cell Hybridization of the Dominant Pylotype,” Environmental Microbiology, Vol. 10, No. 1, 2008, pp. 10-19.
- M. Kube, A. Beck, A. Meyerdierks, R. Amann, R. Reinhardt and R. Rabus, “A Catabolic Gene Cluster for Anaerobic Benzoate Degradation in Methanotrophic Microbial Black Sea Mats,” Systematic and Applied Microbiology, Vol. 28, No. 4, 2005, pp. 287-294. http://dx.doi.org/10.1016/j.syapm.2005.02.006
- S. Dahle, V. M. Savinov, G. G. Matishov, A. Evenset and K. Naes, “Polycyclic Aromatic Hydrocarbons (PAHs) in Bottom Sediments of the Kara Sea Shelf, Gulf of Ob and Yenisei Bay,” The Science of the Total Environment, Vol. 306, No. 1-3, 2003, pp. 57-71. http://dx.doi.org/10.1016/S0048-9697(02)00484-9
- M. T. Szpak, X. Monteys, S. O’Reilly, A. J. Simpson, X. Garcia, R. L. Evans, C. C. R. Allen, D. J. McNally, D. Courtier-Murias and B. P. Kelleher, “Geophysical and Geochemical Survey of a Large Marine Pockmark on the Malin Shelf, Ireland,” Geochemistry Geophysics Geosystems, Vol. 13, No. 1, 2012, p. Q01011. http://dx.doi.org/10.1029/2011GC003787
- J. Zhou, M. Bruns and J. M. Tiedje, “DNA Recovery from Soils of Diverse Composition,” Applied and Environmental Microbiology, Vol. 62, No. 2, 1996, pp. 316- 322.
- J. R. Marchesi, T. Sato, A. J. Weightman, T. A. Martin, J. C. Fry, S. J. Hiom and W. G. Wade, “Design and Evaluation of Useful Bacterium-Specific PCR Primers That Amplify Genes Coding for 16S rRNA,” Applied and Environmental Microbiology, Vol. 64, No. 2, 1998, pp. 795- 799.
- A. Del Casale, P. V. Flanagan, M. J. Larkin, C. C. R. Allen and L. A. Kulakov, “Extent and Variation of PhageBorne Bacterial 16S rRNA Gene Sequences in Wastewater Environments,” Applied and Environmental Microbiology, Vol. 77, No. 15, 2011, pp. 5529-5532. http://dx.doi.org/10.1128/AEM.00457-11
- D. C. White and D. B. Ringelberg, “Signature Lipid Biomarker Analysis,” In: R. S. Burlage, R. Atlas, D. Stahl, G. Geesey and G. Sayler, Eds., Techniques in Microbial Ecology, Oxford University Press, Oxford, 1998, pp. 255- 288.
- H. C. Pinkart, R. Devereux and P. J. Chapman, “Rapid Separation of Microbial Lipids Using Solid Phase Extraction Columns,” Journal of Microbiological Methods, Vol. 34, No. 1, 1998, pp. 9-15. http://dx.doi.org/10.1016/S0167-7012(98)00060-8
- A. Otto, C. Shunthirasingham and M. J. Simpson, “A Comparison of Plant and Microbial Biomarkers in Grassland Soils from the Prairie Ecozone of Canada,” Organic Geochemistry, Vol. 36, No. 3, 2005, pp. 425-448. http://dx.doi.org/10.1016/j.orggeochem.2004.09.008
- P. D. Nichols, J. B. Guckert and D. C. White, “Determination of Monounsaturated Fatty Acid Double-Bond Position and Geometry for Microbial Monocultures and Complex Consortia by Capillary GC-MS of Their Dimethyl Disulphide Adducts,” Journal of Microbiological Methods, Vol. 5, No. 1, 1986, pp. 49-55. http://dx.doi.org/10.1016/0167-7012(86)90023-0
- D. Kim, S. W. Kim, K. Y. Choi, J. S. Lee and E. Kim, “Molecular Cloning and Functional Characterization of the Genes Encoding Benzoate and P-Hydroxybenzoate Degradation by the Halophilic Chromohalobacter sp. Strain HS-2,” FEMS Microbiology Letters, Vol. 280, No. 2, 2008, pp. 235-241. http://dx.doi.org/10.1111/j.1574-6968.2008.01067.x
- B. Song and B. B.Ward, “Genetic Diversity of Benzoyl Coenzyme A Reductase Genes Detected in Denitrifying Isolates and Estuarine Sediment Communities,” Applied and Environmental Microbiology, Vol. 71, No. 4, 2005, pp. 2036-2045. http://dx.doi.org/10.1128/AEM.71.4.2036-2045.2005
- S. Dhanasekaran, T. M. Doherty and J. Kenneth, “Comparison of Different Standards for Real-Time PCR-Based Absolute Quantification,” Journal of Immunological Methods, Vol. 354, No. 1-2, 2010, pp. 34-39.
- J. A. Whelan, N. B. Russell and M. A. Whelan, “A Method for the Absolute Quantification of cDNA Using Real-Time PCR,” Journal of Immunological Methods, Vol. 278, No. 1-2, 2003, pp. 261-269. http://dx.doi.org/10.1016/S0022-1759(03)00223-0
- B. McKew, A. J. Dumbrell, S. D. Daud, L. Hepburn, E. Thorpe, L. Morgensen and C. Whitby, “Characterization of Geographically Distinct Bacterial Communities Associated with the Coral Mucus from Acropora spp. and Porites spp,” Applied and Environmental Microbiology, Vol. 78, No. 15, 2012, pp. 5229-5237. http://dx.doi.org/10.1128/AEM.07764-11
- N. Rajendran, O. Matsuda, Y. Urushigawa and U. Simidu, “Characterization of Microbial Community Structure in the Surface Sediment of Osaka Bay, Japan, by Phospholipid Fatty Acid Analysis,” Applied and Environmental Microbiology, Vol. 60, No. 1, 1994, pp. 248-257.
- T. Stoeck, I. Kröncke, C. A. Duineveld and Palojärvi, “Phospholipid Fatty Acid Profiles at Depositional and Non-Depositional Sites in the North Sea,” Marine Ecology Progress Series, Vol. 241, 2002, pp. 57-70. http://dx.doi.org/10.3354/meps241057
- D. L. Balkwill, F. Leach, J. T. Wilson, J. F. Mcnabb and D. C. White, “Equivalence of Microbial Biomass Measures Based on Membrane Lipid and Cell Wall Components, Adenosine Triphosphate and Direct Counts in Subsurface Aquifer Sediments,” Microbial Ecology, Vol. 16, No. 1, 1988, pp. 73-83. http://dx.doi.org/10.1007/BF02097406
- J. Guezennec and A. Fiala-Medioni, “Bacterial Abundance and Diversity in the Barbadose Trench Determined by Phospholipid Analysis,” FEMS Microbiology Ecology, Vol. 19, No. 2, 1996, pp. 83-93. http://dx.doi.org/10.1111/j.1574-6941.1996.tb00201.x
- S. D’Hondt and S. Rutherford, “Metabolic Activity of Subsurface Life in Deep Sea Sediments,” Science, Vol. 295, No. 5562, 2002, pp. 2067-2070. http://dx.doi.org/10.1126/science.1064878
- D. M. Alongi, “Vertical Profiles of Bacterial Abundance, Productivity and Growth Rates in Coastal Sediments of the Central Great Barrier Reef Lagoon,” Marine Biology, Vol. 112, No. 4, 1992, pp. 657-663. http://dx.doi.org/10.1007/BF00346184
- G. M. Luna, E. Manini and R. Danovaro, “Large Fraction of Dead and Inactive Bacteria in Coastal Marine Sediments: Comparison of Protocols for Determination and Ecological Significance,” Applied and Environmental Microbiology, Vol. 68, No. 7, 2002, pp. 3509-3513. http://dx.doi.org/10.1128/AEM.68.7.3509-3513.2002
- F. Inagaki, M. Suzuki, K. Takai, H. Oida, T. Sakamoto, K. Aoki, K. H. Nealson and K. Horikoshi, “Microbial Communities Associated with Geological Horizons in Coastal Subseafloor Sediments from the Sea of Okhotsk,” Applied and Environmental Microbiology, Vol. 69, No. 12, 2003, pp. 7224-7235. http://dx.doi.org/10.1128/AEM.69.12.7224-7235.2003
- S. R. Prabagaran, R. Manorama, D. Delille and S. Shivaji, “Predominance of Roseobacter, Sulfitobacter, Glaciecola and Psychrobacter in Seawater Collected off Ushuaia, Argentina, Sub-Antarctica,” FEMS Microbiology Ecology, Vol. 59, No. 2, 2007, pp. 342-355. http://dx.doi.org/10.1111/j.1574-6941.2006.00213.x
- J. Heider, A. M. Spormann, H. R. Beller and F. Widdel, “Anaerobic Bacterial Metabolism of Hydrocarbons,” FEMS Microbiology Reviews, Vol. 22, No. 5, 1999, pp. 459-473. http://dx.doi.org/10.1111/j.1574-6976.1998.tb00381.x
- M. Boll, G. Fuchs and J. Heider, “Anaerobic Oxidation of Aromatic Compounds and Hydrocarbons,” Current Opinion in Biotechnology, Vol. 6, No. 5, 2002, pp. 604-611.
- S. Sehnell and B. Sehink, “Anaerobic Aniline Degradation via Reductive Deamination of 4-Aminobenzoyl-CoA in Desulfobacterium anilini,” Archives of Microbiology, Vol. 155, No. 2, 1991, pp. 183-190.
- J. D. Coates, J. Woodward, J. Allen, P. Philip and D. R. Lovley, “Anaerobic Degradation of Polycyclic Aromatic Hydrocarbons and Alkanes in Petroleum-Contaminated Marine Harbour Sediments,” Applied and Environmental Microbiology, Vol. 63, No. 9, 1997, p. 3589.
NOTES
*Corresponding author.