Y. Ji et al. / Journal of Biosciences and Medicines 1 (2013) 1-5
Copyright © 2013 SciRes. JBM
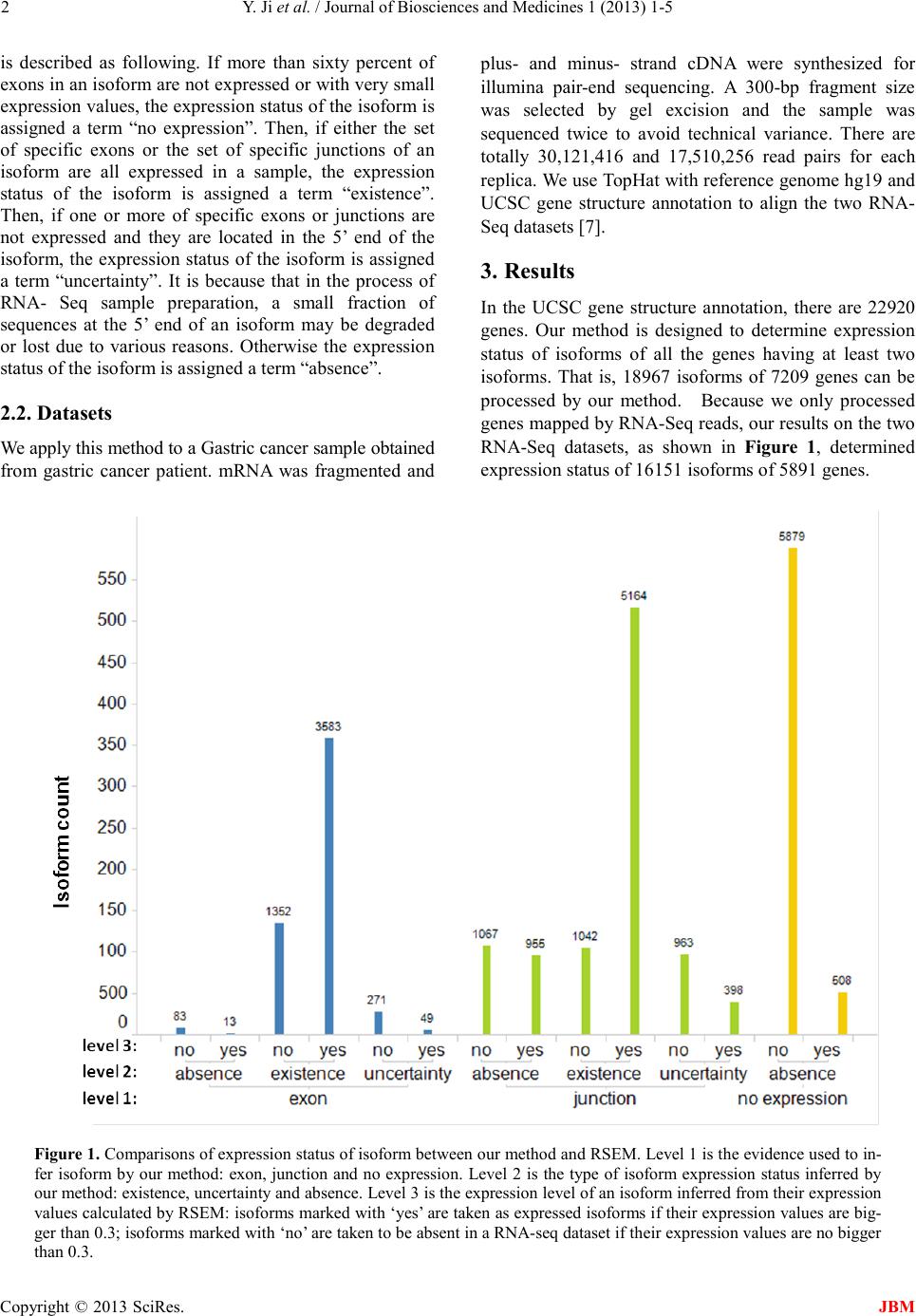
Table 1. Networks constructed from cancer related iso fo r ms
Index Key Network Objects GO Process es
ACACA, eIF4 G1, VEG F -A, MNK1, ABC C1
integrin-mediated signaling pathway, cell-mat r ix adhesi on,
prote in tr ans po r t w ith in lip id bilaye r , calcium -independent cell-ma trix adhesion,
cell mi gr ation involved in spro uting angiogenesis
CDC25B, VAV-2, al ph a-1/beta-1 integrin, ZO-2
posi tive regulati on of cell prolifera tion
Dyn a m in -2, TFIID, MALT1, PDK (PDPK1), Al-
pha-synuclein
protein phosphorylation, cell-cell signaling, synaptic transmission,
phosp horylation, signal transduction
MAP3K3, FANCA, KLF1 1 (TIEG2) , AP-1, WAS P
adenylate cyclase-modulating G-protein coupled receptor signaling pathway
phosphatase activity and G2/M phase transition of cell
cycle. Blocking CDC25B expression leads to inhibition
of cell proliferation, migration and invasion. CDC25B
may be a potential marker and therapeutic target for
hepatocellular carcinoma and pancreatic cancer [9]. In
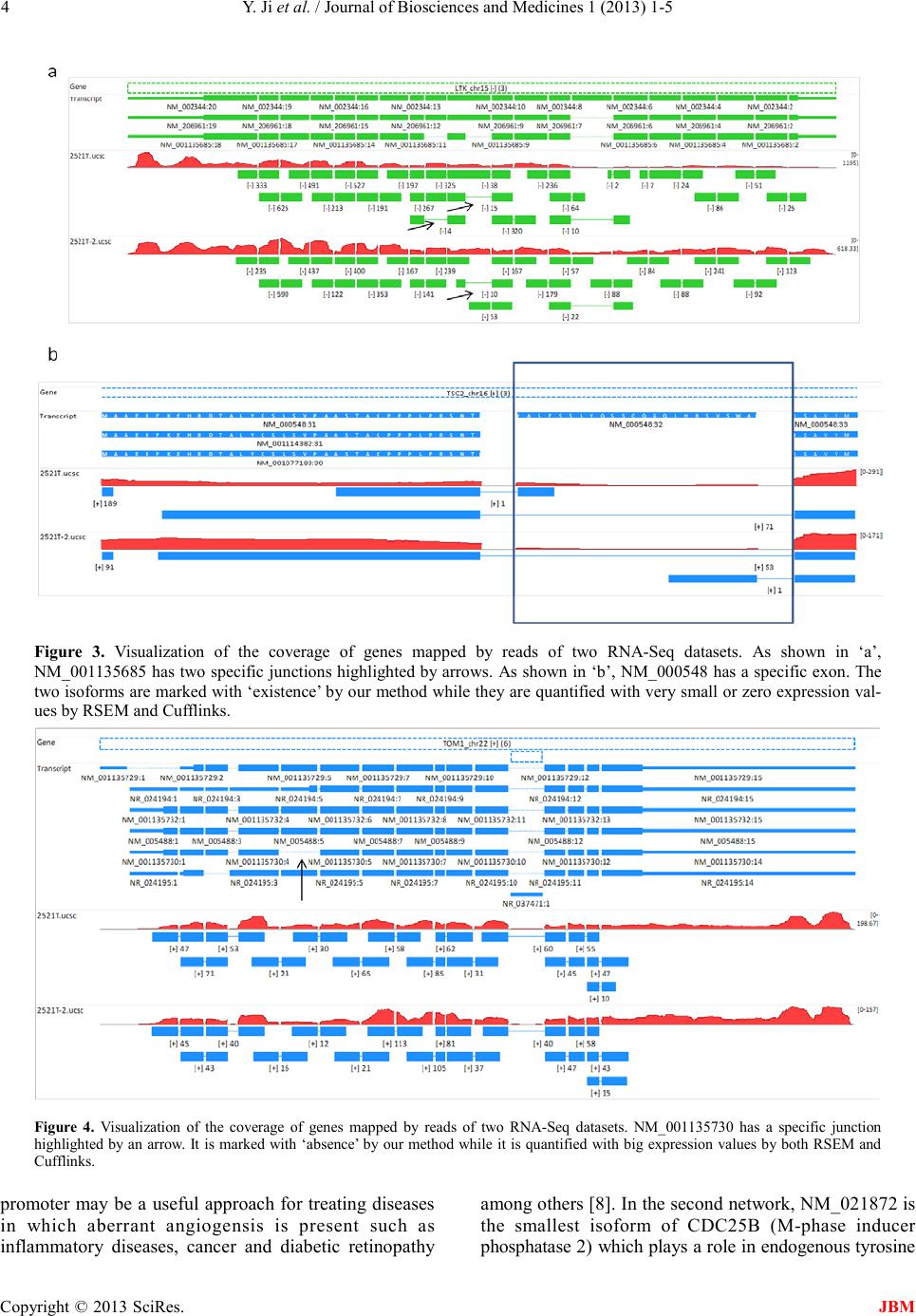
the third network, NM_002613 is the longer isoform of
PDPK1 (3-phosphoinositide-dependent protein kinase 1)
which is involved in the regulation of cell proliferation,
cell survival and signal transduction. PDK1-expressing
MCF-7 cells showed up-regulation of genes of the Wnt
signaling pathway and down-regula tion of p utati ve t umor
suppressor genes. In the fourth network, NM_203351 is
the longest isoform of MAP3K3 (mitogen-activated
protein kinase kinase kinase 3) which is involved in
regulating interleukin 1 receptor (IL-1R), toll-like receptor
4 (TLR4), SAPK and ERK signaling pathways. MEKK3
may be a therapeutic target in controlling the apoptosis
resistance of some cancers [10].
Therefore, accurately inferring isoforms from RNA-
Seq datasets of tumor samples and identifying tumor
driven isoforms have important implications in the field
of cancer drug res earch and development.
4. CONCLUSIONS
We have developed a method for inferring isoforms from
RNA-Seq samples. Its advantages over current isoform
inference tools have been illustrated in previous sections.
Its limitations are that complete sequences and gene
structure annotation of transcriptome of targeted species
have to be available. With advances of sequencing
technologies and genome biology, our method can be
applied to more species. Furthermor e , our method can
detect isoform switching if multiple disease and no rmal
samp l e s are available. If expression status of isoforms
are different between disease and normal samples,
isoform switching can be identi fied . If expression status
of isofo r ms ar e both marked with ‘e xis tenc e’, expr essio n
of specific exons or junctions between samples can be
used to determine whether isoforms are differentially
expressed.
5. ACKNOWLEDGEMENTS
This work was supported in part by a project from
AstraZeneca.
REFERENCES
[1] Met zker, M.L. (2010) Sequencing technologies - the ne xt
generation. Nat R ev Genet, 11(1), 31-46.
[2] Mortazavi, A. Williams, B. A. McCue, K. Schaeffer, L.
Wold, B. (2008) Mapping and quantifying mammalian
transcri pt omes b y RNA-Seq . Nat M ethods, 2008. 5(7): p.
621-8.
[3] Wang, E.T., et al. (2008) Alternative isoform regulation
in human tissue transcriptomes. Nature, 456(7221),
470-6.
[4] Guttman, M., et al. (2010) Ab initio reconstruction of cell
type-specific transcriptomes in mouse reveals the con-
served multi-exonic structure of lincRNAs. Nat Biotech-
nol, 28(5): p. 503-10.
[5] Li, B. and C.N. Dewey (2011) RSEM: accu rate t rans crip t
quantification from RNA-Seq data with or without a ref-
erence genome. BMC Bioinformatics, 12: p. 323.
[6] Trapnel l, C., et al. (2010) Transcript assembly and quan-
tification by RNA-Seq reveals unannotated transcripts
and isoform switching during cell differentiation. Nat
Biotechnol, 28(5): p. 511-5.
[7] Trapnell, C., L. Pachter, and S.L. Salzberg (2009) TopHat:
discovering splice junctions with RNA-Seq. Bioinfor-
matics, 25(9): p. 1105-11.
[8] Snowden, A.W., et al. (2003) Repression of vascular
endothelial growth factor A in glioblastoma cells using
engineered zinc finger transcription factors. Cancer Res,
63(24): p. 8968-76.
[9] Ngan, E.S., et al. (2003) Overexpression of Cdc25B, an
androgen receptor coactivator, in prostate cancer. On co-
gene, 22(5): p. 734-9.
[10] Samanta, A.K., et al.(2004) Overexpression of MEKK3
confers resistance to apoptosis through activation of
NFkappaB. J Biol Chem, 27 9( 9): p. 757 6-83.