Shkurat TP et al. / Open Journal of Genetics 2 (2012) 1-4
Copyright © 2012 SciRes. OJGen
2. MATERIALS AND METHODS
The object of the study were white outbreed rats
Rattus norvegicus treated with high oxygen pressure (0.2
MPa, 1 h). Animals were sacrificed in 3 h after the
treatment and gene expression was studied in the frontal
lobe. The results of full genomic transcriptome study
were previously described in [5].
To perform bioinformatic analysis two groups of
genes were formed. The first group involved genes
up-regulated in response to oxidative stress. The second
group consisted of genes with unchanged expression.
The first grouped was formed by genes
NM_017138:Lamr1, NM_053440:Stmn2,
NM_057207:Sv2b, NM_053339:Acox3, Scn7a, Trpc3,
Nid2:NM_213627, Herc1, Ssc1:BC085795,
Golph2:NM_023977, Actr1a, Crebzf, Pdk4:NM_053551,
Mrpl3, Api5, Zfhx1b, Snrpb:BC083694, Snrpb. The
second group included all genes of the chromosome 20
of R. norvegicus.
Regulatory regions of 264 genes available in
KnownGene with exception of the first and the last gene
were studied.
Two types of regulatory elements were studied in both
groups. These included the promoter regions (1000, 800
b. p. upstream the start point and 200 b. p. downstream
the termination point) and the sequences, overlapping
the potential cys-regulatory elements (5000, 4000 b. p.
upstream the start point and 1000 b. p. downstream the
termination point)
The bioinformatic screening was seeking DNA motifs
located near the first group genes. Here, the “recognition
motif” implies the way to describe a set of similar
oligonucleotides, which can be specifically recognized
and bound by a certain regulatory protein.
To search the motifs we created an integrative
collection of the binding transcription sites on the basis
of JASPAR, TRANSFAC, UCSC ENCODE, Нocomoco
TF Homo sapiens and Uniprobe TF Mus musculus
databases. The integrative collection Uniprobe included
272 motifs for transcription factors of M. musculus. The
collection Нocomoco TF H. sapiens, which was
obtained by means of integration of the data from
different sources, included 332 motives for 321
transcription factor. To perform bioinformatic analysis
we chose the release of R. norvegicus genome rn4
(Baylor 3.4/rn4)
[http :// genome. uc sc.e du/ c gi-bi n/h gGat e wa y].
The conception of homotypic transcription factor
binding site clusters, which were represented by a group
of transcription factor binding site, was used as a model
of a binding region. To assess the statistical confidence
of a binding site cluster the “r-scan” model with fixed
motive acceptance threshold was used (Papatsenko D.,
2007). The statistical confidence of the “r-scan” was
assessed as a probability to find at least the expected
number of transcription factor binding site clusters in a
sequence of certain length on account that at least 1
transcription factor binding site was already found, i.e.
one cluster contains at least one binding site.
In order to perform reliable assessment of the
presence of homotypic clusters they were counted in the
first and second group. We assessed the chance (p) to
choose such a gene, the regulatory region of which
would contain at least one cluster (for promoters) and a
cluster with a confidence of at least 0.0005 for
cis-regulatory elements.
The p value can be assessed using the binomial
distribution as a confidence of the fact that in the studied
sample the transcription factor binding site clusters
contain at least k number of regulatory regions, where k
corresponds to experimentally found number of
sequences which contain the transcription factor binding
site clusters (for promoters) / at least 0.0005 confident
transcription factor binding site clusters for
cis-regulatory elements.
3. RESULTS AND DISCUSSION
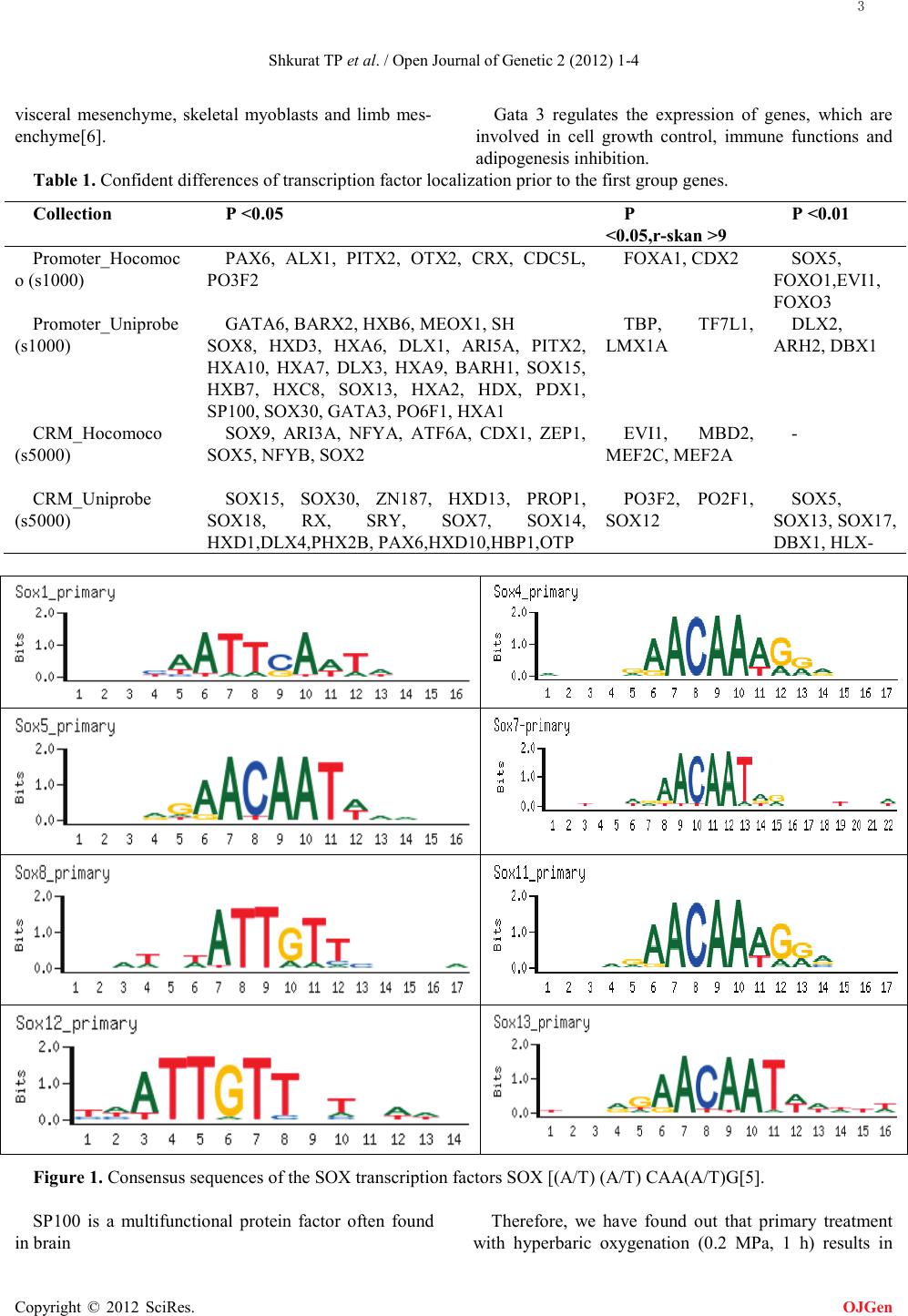
It was shown that transcription factor binding sites of
the SOX subfamily more often preceded the first group
of genes (Table 1). The SOX subfamily involves 30
transcription factors, which contain heptamerous
sequence (A/T) (A/T) CAA(A/T)G (Fig. 1) [5]. It is
considered to be one of the most important subfamilies,
which regulate development of both vertebrate and
invertebrate animals. Biological functions of these
proteins were investigated in a variety of mammalian
tissues and cells during embryogenesis and
postembryonic development. It is suggested that
differentiation of the proteins for groups is typically due
to specificity of their functioning in different tissues.
They initiate differentiation program and activate tissue
specific expression. The SOX subfamily is represented
by multifunctional proteins. Among multiple roles that
SOX proteins play in cells the transcription factors can
either induce or suppress a variety of cellular processes.
Moreover, it was shown that SOX proteins are involved
in DNA reparation processes.
HXA3, hepoxilin A3; in the absence of cytosol glu-
tathione peroxidase is transformed into 12S-hydroper-
oxyeicosatetraenoic acid on biologically active epoxides.
The process is catalyzed by lipoxygenase.
HLX is expressed in mesodermal tissues in embryo-
genesis. Especially high expression was observed in