Paper Menu >>
Journal Menu >>
 Advances in Ma teri als Physics and Chemistry, 2012, 2, 1-4 doi:10.4236/ampc.2012.24B001 Published Online December 2012 (http://www.Sci RP.org/journal/ampc) Copyright © 2012 SciRes. A MPC A First-Principles Study of Structure-Property Correlation and the Origin of Ferrimagnetism in Gallium Ferrite Amritendu Roy1, Ashish Garg1, Rajendra Prasad2, Sushil Auluck3 1Department of M ater ial s Science an d Engi neering, Indian Institute of Technology Kanpur, Kanpur, India 2Department of Physics, Indian Institute of Technology Kanpur, Kanpur, India 3National Physical Laboratory, Dr. K. S. Krishnan Marg, New Delhi, India Email: rprasad@iitk.ac.in Received 2012 ABSTRACT A fir st-prin ciples study of structure property correlation and the origin of ferrimagnetism is presented based on LSDA+U method. In particu lar, the resu lts for the grou nd state stru cture, electron ic band structure, den sity of states, Born effecti ve charges, spontaneous pol arization and cationic disorder are discussed. The calculations were done using Vienna ab-initio simulation package (VASP) with projector augmented wave method. We find that the ground state structure is orthorhombic and insulating having A-type antiferro- magnetic sp in configuration. The cationic disorder is found to play an important role. Although the cationic site disorder is not spon- taneous in the ground state, interchange of octahedrally coordinated Ga2 and Fe2 sites is most favored. We find that ferrimagnetism in gallium ferrite is p rimaril y due to this exch ange between Ga-Fe si tes such th at Fe spins at Ga1 and Ga2 si tes are antiferro magneti- cally aligned while maintaining ferromagnetic coupling between Fe spins at Ga1 and Fe1 sites as well as between Fe spins at Ga2 and Fe2 sites. Further, the partial density of states shows noticeable hybridization of Fe 3d, Ga 4s, Ga 4p and O 2p states indicating some covalent character of Ga/Fe-O bonds. However, the charge density and electron localization functions show largely the ionic character of these bonds. Our calculation predicts spontaneous polarization of ~59 μC/cm2 along b-axis. Keywords: Gallium Ferrite; LSDA+U; Spontaneous Polarization; Ferrimagnetism; Cation Site Disorder 1. Introduction Gallium ferrite (GaFeO3 or GFO) is a room temperature pie- zoelectric an d a ferrimagnet who se magnetic transit ion temper- ature (TC) is slightly lower than room temperature [1] but tuna- ble to room temperature and beyond by tailoring the composi- tion [1,2] and processing conditions.[1-3] Thus, composition- ally modulated GFO is a promising candidate for room temper- ature magn etoelectric applications. Early studies on GFO [2] predicted simultaneous piezoelec- tricity and ferromagnetism. Structural characterization using x-ray [1,4,5] and neutron [1,4,6] diffraction techniques con- cluded an orthorhombic structure (Space Group: Pc21n) with eight formula units per unit cell, is stable over a wide tempera- ture domain (4K-700K)[1,7]. The unit cell comprises of two types of Ga (Ga1 and Ga2) and Fe (Fe1 and Fe2) ions and six types of O (O1, O2,….O6) ions.[1] The above studies also suggest substantial cationic site disorder i.e. some of the Ga sites are always occupied by Fe ions and vice-versa.[1] How- ever, magnetic behavior of GFO had been a matter of uncer- tainty for a long time. Initial prediction of ferromagnetic order- ing [2] was challen ged by Franke l et al.[8] who using high field Mössbauer spectroscopy and macroscopic magnetic measure- ments proved collinear ferrimagnetism in GFO. Ferrimagnetic ordering has been further demonstrated by almost all subse- quent studies using neutron diffraction technique.[1,4] Piezoe- lectricity in GFO, on the other hand, has been hardly studied with few exceptions showing that the piezoelectric constants are almost double to that of quartz [9] which is attributed to the asymmetric Ga1-O tetrahedron in the GFO unit cell. [10] In spite of extensive experimental studies, first-principles cal- culations on GFO, have not been carried out much, partly due to the co mplex cr ystal st ru ctu re with substantially large number of ions in the unit cell having partial site occupancies of the cations. However, such type of studies have been quite successful in predicting and analyzing structure-property correlations in complex mat erial s ystems an d in th is regard, a detailed study on GFO would be particularly interesting to probe the hitherto disputed issues such as the ground state structure, magnetic structure and the piezoelectric response of the material. Our study, using first-principles calculati ons, shows that the ground state structure of GFO is A-type antiferromagnetic. The calcu- lations indicate the presence of large spontaneous polarization (Ps) of ~ 59 µC/cm2 along crystallographic b-axis. Finally, we predi ct that th e observed ferri magnetis m is solely due the inhe- rent cation site disorder in the material. 2. Calculation Methodolo gy We used Vienna ab -initio simulation package (VASP ) [11] with projector augmented wave method (PAW) [12] in our work. The Kohn-Sham equation [13] was solved using local spin density approximation (LSDA+U) [14] with Hubbard parame- ter, U = 5 eV, and the exchange interacti on, J = 1 eV. Our cal- culations for the determination of the ground states structure are based on the stoichiometric GFO assuming no partial occupan- cies of the co nstituent ions with startin g parameters taken fro m previo us literatur e.[1] We con sidered 3 valence elect rons of Ga 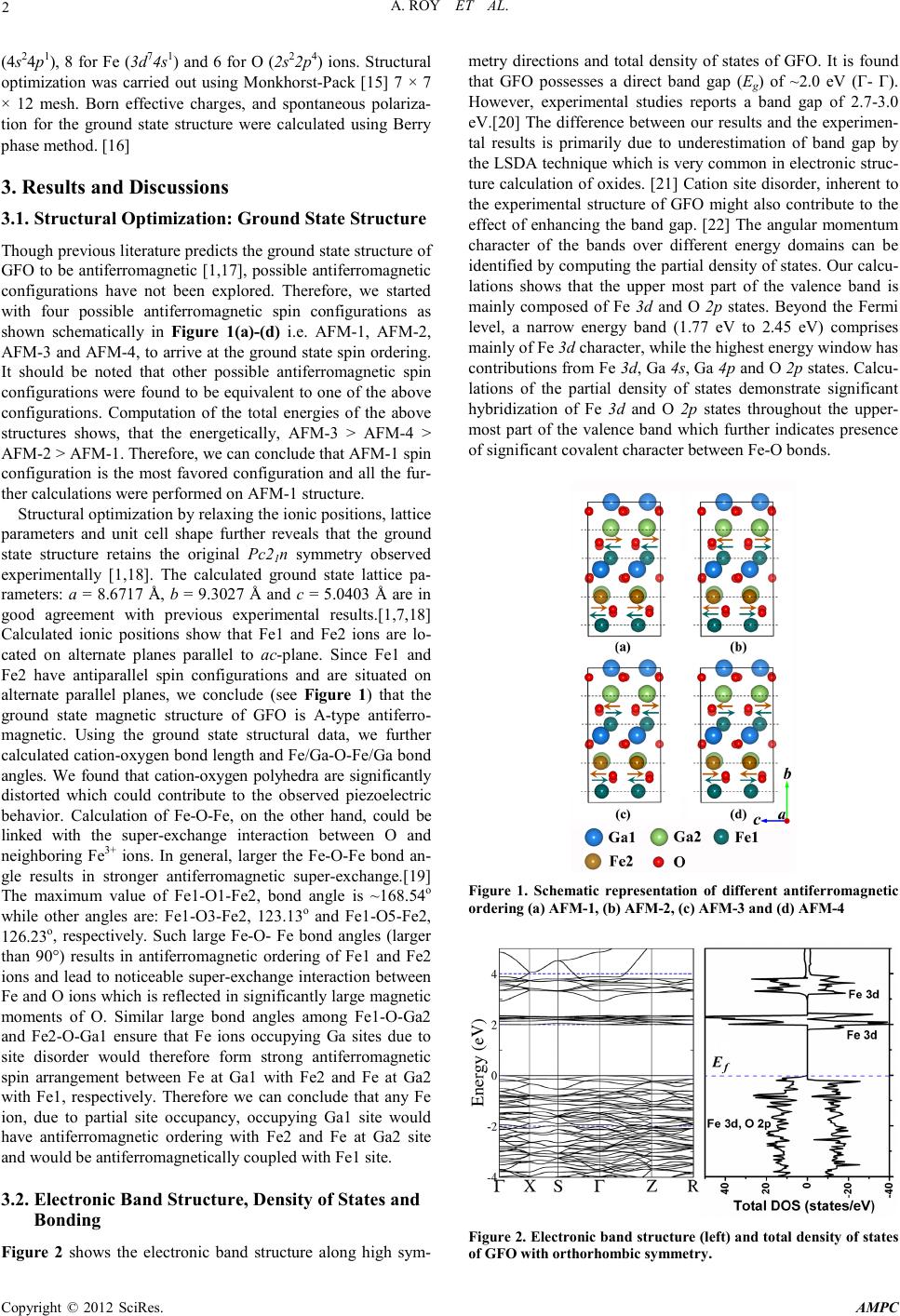 A. ROY ET AL. Copyright © 2012 SciRes. AMPC 2 (4s24p1), 8 for F e (3d74s1) and 6 for O (2s22p4) ions. Structural optimization was carried out using Monkhorst-Pack [15] 7 × 7 × 12 mesh. Born effective charges, and spontaneous polariza- tion for the ground state structure were calculated using Berry phase m e t hod. [ 16 ] 3. Results and Discussions 3.1. Structural Optimization: Ground State Structure Though previous literature predicts the ground state structure of GFO to be antiferromagnetic [1,17], possible antiferromagnetic configurations have not been explored. Therefore, we started with four possible antiferromagnetic spin configurations as shown schematically in Figure 1(a)-(d) i.e. AFM-1, AFM-2, AFM-3 and AFM-4, to arrive at the ground state spin ordering. It should be noted that other possible antiferromagnetic spin configurations were found to be equivalent to one of the above configurations. Computation of the total energies of the above structures shows, that the energetically, AFM-3 > AFM-4 > AFM-2 > AFM -1 . Therefore, we can co n clude th at AFM-1 spi n configuration is the most favored configuration and all the fur- ther calculations were performed o n AFM-1 structure. Structural optimization by relaxing the ionic positions, lattice parameters and unit cell shape further reveals that the ground state structure retains the original Pc21n symmetry observed experimentally [1,18]. The calculated ground state lattice pa- rameters: a = 8.6717 Å, b = 9.3027 Å and c = 5.0403 Å are in good agreement with previous experimental results.[1,7,18] Calculated ionic positions show that Fe1 and Fe2 ions are lo- cated on alternate planes parallel to ac-plane. Since Fe1 and Fe2 have antiparallel spin configurations and are situated on alternate parallel planes, we conclude (see Figure 1) that the ground state magnetic structure of GFO is A-type antiferro- magnetic. Using the ground state structural data, we further calculat ed catio n-oxygen bond length and Fe/Ga-O-Fe/Ga bond angles. We found that cation-oxygen polyhedra are significantly distorted which could contribute to the observed piezoelectric behavior. Calculation of Fe-O-Fe, on the other hand, could be linked with the super-exchange interaction between O and neighboring Fe3+ ions. In general, larger the Fe-O-Fe bond an- gle results in stronger antiferromagnetic super-exchange.[19] The maximum value of Fe1-O1 -Fe2, bond angle is ~168.54o while other angles are: Fe1-O3-Fe2, 123.13o and Fe1-O5-Fe2, 12 6.23o, respectively. Such large Fe-O- Fe bond angles (larger than 90°) results in antiferromagnetic ordering of Fe1 and Fe2 ions and lead to not iceable super-exchange i nteraction b etween Fe and O ions which is reflected in significantly large magnetic moments of O. Similar large bond angles among Fe1-O-Ga2 and Fe2-O-Ga1 ensure that Fe ions occupying Ga sites due to site disorder would therefore form strong antiferromagnetic spin arrangement between Fe at Ga1 with Fe2 and Fe at Ga2 with Fe1, respectively. Therefore we can conclude that any Fe ion, due to partial site occupancy, occupying Ga1 site would have antiferromagnetic ordering with Fe2 and Fe at Ga2 site and would be antiferromagnetically coupled with Fe1 site. 3.2. Electronic Band Struc ture, Density of States and Bonding Figure 2 shows the electronic band structure along high sym- metry directions and total density of states of GFO. It is found that GFO possesses a direct band gap (Eg) of ~2.0 eV (Γ- Γ). However, experimental studies reports a band gap of 2.7-3.0 eV.[20] The difference bet ween ou r results and the exper imen- tal results is primarily due to underestimation of band gap by the LSDA techn ique which is very co mmon in electron ic str uc- ture calculation of oxides. [21] Cation site disorder, inherent to the experimental structure of GFO might also contribute to the effect of enhancing the band gap. [22] The angular momentum character of the bands over different energy domains can be identified by computing the partial density of states. Our calcu- lations shows that the upper most part of the valence band is mainly composed of Fe 3d and O 2p states. Beyond the Fermi level, a narrow energy band (1.77 eV to 2.45 eV) comprises mainly o f Fe 3d character, while the h ighest en ergy window has contributions from Fe 3d, Ga 4s, Ga 4p and O 2p states. Calcu- lations of the partial density of states demonstrate significant hybridization of Fe 3d and O 2p states throughout the upper- most part of the valence band which further indicates presence of significant covalent character between Fe-O bonds. Figure 1. Schematic representation of different antiferromagnetic ordering (a) AFM-1, (b) AFM-2, (c) AFM-3 and (d) AFM-4 Figure 2 . Electronic band structure (lef t) and total densi ty of states of GFO with orthorhom b ic sym metry.  A. ROY ET AL. Copyright © 2012 SciRes. AMPC 3 Analysis of the chemical bonding can further be carried out by plotting electron localization function (ELF) which gives a measure of the local influence of the Pauli repulsion on the behavior of the electrons. A large value of ELF function indi- cates space with anti-parallel spin configuration .[23] Our cal- culation of ELF distribution in GFO unit cell (not shown here) depicts maximum ELF value at O sites and small values at Fe and Ga sites i nd icati ng ch arge tra n sfer fro m Fe/ Ga to O sites . A complete charge transfer was found between Fe2 and O3 ions. Similar charge transfer was also noticed between Fe1 and O1, O2 ions. Thus, we can predict that Fe-O bonds in GFO are mostly ionic. In comparison, finite value of ELF between O and Ga1 and Ga2 i ndicat e s ome degree of covalent characteris tics. 3.3. Cation Site Disorder and Ferrimagnetism Calculation of the magnetic moments of the constituent ions in the ground state shows that Fe1 and Fe2 ions have magnetic moments of + 4.05 µB and - 4.04 µB, resp ectively. We find that the magnitude of moments agrees reasonably well with the experimental data. [1] It was also found that the oxygen ions surrounding the Fe ions manifest small but finite magnetic moments attributed to super-exchange interactions with the surrounding Fe ions. Till now, our calculations have been based on the ground state structure containing no partial site occupancies of the cations which is however, in contrast with the experimental repor ts showing significant Fe occu pancies at the Ga sites. Thus, to probe the effect of cation site disorder on the magnetic cha- racteristi cs of GFO, we selectively interchanged F e and Ga sites and computed total energy of the system. Since, GFO unit-cell contains four ions of each type of cation, such an interchange would result in ¼th site occupancy of Fe ions at Ga sites and vice-versa. Calculations of total energy of these disordered structures show that partial site occupancy is not favored in the ground state, also observed previously by Han et al. [17] How- ever, it was found that among various possible cases of site disorders, Fe2 ions preferentially occupying Ga2 sites is most probable since ∆E, the energy difference with respect to the ground state in that case is minimum. Although these energy differences may be affected by the computational methodology, the magnitude of the available thermal energy at room temper- ature (kT ~25 meV) is of the order of the energy difference for Fe2-Ga2 site disorder indicating towards the role of thermally originated defects. An important observation of the inclusion of cation site disorder in the calculation would be on the modifica- tion of the local magnetic moments. It was observed that upon interch angin g Fe1 and Ga1 s ites, the avera ge magneti c moment of Fe ion at Ga1 site becomes 3.99 µB. On the other hand, the magnetic momen t of Fe ion at G a2 site becomes 4 .11 µB when Fe2 and Ga2 sites are interchanged. In order to analyze the observed ferrimagnetism as against the antiferromagnetic or- dering in the ground state, we further calculated the total mag- netic moment of the disordered structure, using the partial site occupancies from the Rietveld refinement data [24] and taking the magnetic moments for different cation sites, from our cal- culation with site disorder. We estimated net magnetic mo ment of 0.24 µB/ Fe site which agrees quite well with experimental results. [25] Therefore, we conclude that ferrimagnetism in GFO is solely due to site disorder in the structure. 3.4. Born Effectiv e C harge and Spontaneous Polariza t ion Born effective charges (BEC or Z*), are important quantities in characterizing the piezoelectrics, ferroelectrics and multifer- roics since they relate the lattice displacements and electric field and therefo re give a measure long range Coulomb interac- tion, whose competition with the short range forces leads to the ferroelectric transition. Recent first-principles calculations show anomalously large BECs for some ions in common fer- roelectrics [21] which are often explained as manifestation of strong covalent char acter of bonds bet ween the sp ecific ion s. In GFO, fro m the E LF pl ots, we find th at charge sharin g between the Ga/Fe and O ions in cation-oxygen bonds is insignificant while the structural data indicates large distortion of the ca- tion-oxygen polyhedra. Since ferroelectric and/ piezoelectric responses are combined manifestations of structural distortions and effecti ve charges of const ituent ions [26] it is imperati ve to calculate the Born effective charges of ions in GFO. Here we calculate the BEC tensors of nonequivalent ions of GFO by slightly displacing each ion, one at a time, along three axes of Cartesian co-ordinates and then calculating the resulting dif- ference in p ol arization, using Berry phase method. Table 1 lists the diagonal elements of BEC tensor for each ion. It is noticed that the principal elements of BEC tensor for Ga1 are close to the nomin al ionic ch arge of Ga i.e., +3 . Thus we pred ict that all the Ga1-O bonds are mainly ionic in nature. On the other hand, Ga2 develops a maximum effective charge of 3.53, ~ 18% higher with respect to its nominal charge. In contrast, both Fe1 and Fe2 ions show much higher increase in the effective charges, 36% and 27% respectively. Oxygen ions show a maximum reduction of 39.5% with respect to the nominal ionic charge. Structural analysis of GFO shows that while the constituent ion s possess inversi on symmetry i n the crystallographic a and c directions, the inversion symmetry is lost in crystallographic b-direction. Since existence of spontaneous polarization (Ps) is manifestation of the lack of inversion symmetry, we argue that the direction of spontaneous polarization in GFO is only along the b-direction. Using the Born effective charges from Tabl e 1, we further calculated the magnitude of Ps of GFO as ~ 58.63 µC/cm2 along the b-direction which is an order of magnitude larger than that predicted by Arima et al.[1] who neglected the role of effective charges and other ions in determining the magnitude of Ps. A calculation on the relative contribution of Table 1. Principal elements of Born effective charge tensors of constituent ions along with their nominal charges, in GFO. Ion Nominal ionic charge (e) Z* (e) Zxx Zyy Zzz Ga1 +3 3.01 3.11 2.99 Ga2 +3 3.5 7 3.16 3.53 Fe1 +3 3.66 3.78 4.08 Fe2 +3 3.68 3 .3 8 3.82 O1 -2 -2.29 -2.58 -2.79 O2 -2 -2.45 -2.29 -2.41 O3 -2 -2.54 -2.30 -2.75 O4 -2 -2.27 -2.85 -2.17 O5 -2 -2.50 -2.16 -2.79 O6 -2 -2.32 -2.08 -2.40  A. ROY ET AL. Copyright © 2012 SciRes. AMPC 4 individual ions shows that the contribution of Ga1 is largest. However, it is balanced by opposite contributions from Fe1, O1, O2 and O6. Structural data further substantiate that these ions are the most as ymmetrical ly placed around the i nversion center whereas Ga2 and Fe2 cations maintain nearly centrosymmetric configuration and contribute minimum to the total polarization. Therefore, we predict that Ps in GFO is primarily contributed from Ga1, Fe1, O1, O2 and O6 ions. 4. Summary To summarize, we show, using first-principles calculations using LSDA+U that orthorhombic Pc21n symmetry with A-type antiferromagnetic spin configuration is the ground state structure of gallium ferrite. The calculated ground state lattice parameters, bond strength and bond angles agree well with the reported as well as our experimental results. Electronic density of states showed hybridization among Fe 3d, Ga 4s, Ga 4p and O 2p states. From the electron localization function (ELF) cal- culation, we find almost complete charge transfer between Fe2 and O3 and F e1 and O1 , O2 ions suggesti ng th at F e-O bonds in GFO have mostly ionic character. Calculations also showed a spontaneous polarization of ~ 59 µC/cm2 along b-direction i.e. [010]-axis of GFO, attributed to the non-centrosymmetry and effective charges of Ga1, Fe1, O1, O2 and O6 ions. We find that the cation site disorder, although not preferred in the ground state, is the most favored configuration in the disordered state. An examination of the role of cation site disorder on magnetic structure of GFO shows modification of the local magnetic str uctur e with alter ed magnetic moments of Fe ions at Ga site. This suggests that ferrimagnetism in GFO is solely due to the site disorder. 5. Acknowledgements The work was supported by Department of Science and Tech- nology, Govt. of India through project number SR/S2/CMP- 0098/2010. SA thanks NPL for the J C Bose Fellowship. REFERENCES [1] T. Arima, D. Higashiyama, Y. Kaneko, J. P. He, T. Goto, S. Miyasaka, T. Kimura, K. Oikawa, T. Kamiyama, R. Kumai, and Y. Tokura, Physical Review B 70 (6), 064426 (2004). [2] J. P. Remeika, Journal of Applied Physics 31 (5), S263 (1960). [3] C. H. Nowlin and R. V. Jones, Journal of Applied Physics 34 (4), 1262 (1963). [4] M. B. Mohamed, A. Senyshyn, H. Ehrenberg, and H. Fuess, Journal of Alloys and Compounds 492 (1-2), L20. [5] W. Kim, J. H. We, S. J. Kim, and C. S. Kim, Journal of Applied Physic s 101 (9), 09M515 (2007) . [6] Y. Kaneko, T. Arima, J. P. He, R. Kumai, and Y. Tokura, Jour- nal of Magnetism and Magnetic Materials 272-276 (Part 1), 555 (2004). [7] S. Mukherjee, A. Garg, and R. Gupta, Journal of Physics: Con- densed Matter 23 (44), 445403 (2011). [8] R. B. Frankel, N. A. Blum, S. Foner, A. J. Freeman, and M. Schieber, Physical Review Letters 15 (25), 958 (1965). [9] D. L. White, Bull. Am. Phys. Soc. 5, 189 (1960). [10] S. C. Abrahams and J. M. Reddy, Physical Review Letters 13 (23), 688 (1964). [11] G. Kresse and D. Joubert, Physical Review B 59 (3), 1758 (1999). [12] P. E. Blöchl, Physical Review B 50 ( 24), 1795 3 (1994). [13] W. Kohn and L. J. Sham, Physical Review 140 (4A), A1133 (1965). [14] V. I. Anisimov, F. Aryasetiawan, and A. I. Lichtenstein, Journal of Physics: Condensed Matter 9 (4), 767 (1997). [15] H. J. Monkhorst and J. D. Pack, Physical Review B 13 (12), 5188 (1976). [16] R. D. King-Smith and D. Vanderbilt, Physical Review B 47 (3), 1651 (1993). [17] M. J. Han, T. Ozaki, and J. Yu, Physical Review B 75 (6), 060404 (2007). [18] S. C. Abrahams, J. M. Reddy, and J. L. Bernstein, The Journal of Chemical Physics 42 (11), 3957 (1965). [19] J. B. Goodenough, Physical Review 100 (2), 564 (1955). [20] Z. H. Sun, S. Dai, Y. L. Zhou, L. Z. Cao, and Z. H. Chen, Thin Solid Films 516 (21), 7433 (2008). [21] A. Roy and et al., Journal of Physics: Condensed Matter 22 (16), 165902 (2010). [22] S. Chen, X. G. Gong, A. Walsh, and S.-H. Wei, Applied Physics Letters 94 (4), 041903 (2009). [23] A. Savin, R. Nesper, S. Wengert, and T. F. Fässler, Angewandte Chemie International Edition in English 36 (17), 1808 (1997). [24] A. Roy, S. Mukherjee, R. Gupta, S. Auluck, R. Prasad, and A. Garg, Journal of Physics: Condensed Matter 23 (32), 325902 (2011). [25] A. Roy, R. Prasad, S. Auluck, and A. Garg, Journal of Applied Physic s 111 (4), 043915 (2012 ). [26] R. E. Cohen, in Piezoelectricity (Springer Berlin Heidelberg, 2008), Vol. 114, pp. 471. |