 Open Journal of Synthesis Theory and Applications, 2012, 1, 44-57 http://dx.doi.org/10.4236/ojsta.2012.13008 Published Online October 2012 (http://www.SciRP.org/journal/ojsta) A Simple and Convenient Synthesis of Isolated-Fused Heterocycles Based on: 2-Imino-N-phenyl-2H-chromene-3-carboxamide Islam H. El Azab*, Fawi M. Abd El Latif Chemistry Department, Faculty of Science, Aswan University, Aswan, Egypt Email: *ihelmy2003@yahoo.com Received June 23, 2012; revised July 26, 2012; accepted August 29, 2012 ABSTRACT Starting from 2-imino-N-phenyl-2H-chromene-3-carbox-amide, (1) a series of functionalized chromenes were achieved; such as, 2-ethoxy-2,3-dihydro-3-phenylchromeno[2,3-d]pyrimidin-4-one (2), and 2-hydrazinyl-2,3-dihydro-3-phenyl- chromeno-[2,3-d]pyrimidin-4-one (3). Furthermore, reactions of (3) with some of laboratory available compounds gave pyrazoles (4 - 9, 12, 13a, 13b), tetrazoles (11), 2-(2-benzylidenehydrazinyl)-3-phenyl-3H-chromeno[2,3-d]pyrimidin- 4(10H)-oneisoxazoles (14), 5-chloro-1-(4-oxo-3-phenyl-4,10-dihydro-3H-chromeno[2,3-d]pyrimidin-2-yl)-3-phenyl-2, 3-dihydro-1H-pyrazole-4-carbonitrile (17), pyrimidines (28a, b), pyridines (29a - 29e, 30, 33a, 33b), benzo[b][1, 4]oxazepin-2- amines (32a, b), 3-chloro-4-(2-imino-2H-chromen-3-yl)-1-phenyl-4-(phenylamino) azetidin-2-one (34a- 34e) and 2-(2- imino-2H-chromen-3-yl)-3-phenyl-2-(phenyl amino)thiazolidin-4-onee (35a -35e). The structures of these compounds were established by elemental analysis, IR, MS and NMR spectral analysis. Keywords: 2-Imino-2H-chromen-3-ylChromeno[2,3-d]pyrimidin-4-one; -Lactam; Thiazolidin-4-ones 1. Introduction Natural and synthetic coumarin derivatives represent, now- adays, an important group of organic compounds that are used as antibiotics [1,2] fungicides [3] anti-inflammatory [4], anticoagulant [5] and antitumor agents [6,7]. Regard- ing their high fluorescence ability, they are widely used as optical whitening agents, brighteners, laser dyes and also as fluorescent probes [8] in biology and medicine [9]. Also, The 4 H-chromene derivatives ethyl 4-((ethoxy- carbonyl) (cyano) methyl)-2-amino-6-bromo-4H-chromene- 3-carboxylate (HA 14-1) has demonstrated promising an- tifungal activities [10], antiviral agent [11], antiprolifera- tion agent [12]. Due to the unique biological and pharma- cological activity, chromene derivatives have attracted considerable attention thus; different processes for the synthesis of chromenes have been reported during the past few years. The importance of the chromone nucleus is evi- denced by the continued appearance of new and improved methods for their synthesis, despite the several existing methods for the synthesis of chromene derivatives [13-20], there still is demand for general synthetic strategies which can efficiently provide variously substituted chromene systems. 2. Experimental 2.1. Instruments All melting points are measured using Galenkanp melting point apparatus and are uncorrected. Elemental analyses were carried out at the Microanalytical Center of Cairo University. IR (KBr pellets υ = cm–1) spectra were de- termined in 1650 FT-IR instrument (Cairo University), 1H-NMR spectra (δ = ppm) were accomplished using 300 MHz NMR Spectrometer and mass spectroscopy were re- corded on GCMS-QP-1000 EX spectrometer (Cairo Uni- versity). 2.2. Material and Reagents Hydrazine hydrate, phenyhydrazine, benzaldehyde and its substituted derivatives, aniline and its substituted de- rivatives, thioglycolic acid, acetylacetone, phosoryl chlo- ride, ethyl acetoacetate and chloroacetyl chloride were purchased from Alderich Chemical Co. Triethylamine, thiourea, urea, o-phenylenediamine, o- aminothiophenol and 2-cyanomethy benzimidazole were purchased from British Drug Houses (BDH). Acetophenone, malononitrile, 2-chloro acetamide, pip- eridine, ethoxymethylene-malononitrile, sodium azide and *Corresponding author. Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF 45 5-amino-(1H)-1,2,4-triazole were purchased from Merck Co., Germany. 2.3. Solvents Dimethyformamide, benzene, pyridine, ethanol and ace- tone were purchased from El-Nasr Pharmaceutical and Chemical Co. (ADWIC), Egypt. 2.4. Organic Preparations Preparation of 2-ethoxy-2, 3-dihydro-3-phenylchro- meno[2,3-d]pyrimidin -4 -o ne ( 2) . A mixture of 1 (2.64 g, 0.01 mol) and triethoxymethane (1.48 mL, 0.05 mol) in 20 mL of dimethylformamide was refluxed 8 h. Then the reaction mixture was poured into 150 mL of crushed ice then the resultant solid was collected by filtration to pro- vide 2 (1.5 g, 60%) as a pale yellow solid; m.p. 285˚C - 287˚C. 1HNMR (CDCl3): δ 1.2 (t, 3H, CH3), 3.9 (q, 2H, CH2) and 6.7 - 7.9 (m, 11H, Ar-H). MS: m/z 320 ([M]+, 65%). Calcd for C19H16N2O3 (320.34). Calcd: C 71.24, H 5.03, N 8.74 %. Found: C 70.14, H 5.01, N 7.88%. Preparation of 2-hydrazinyl-2, 3-dihydro-3-phenyl chromeno[2,3-d]pyrim-idin-4-one (3). A mixture of 2 (3.20 g, 0.01 mol) and hydrazine hydrate (0.5 mL, 0.01 mol) in 30 mL ethanol containing 0.1 mL piperidine was refluxed for 8 hr. The reaction mixture was concentrated under reduced pressure and the residue washed with aci- dified cold water and then triturated with methanol. The formed pale yellow product was filtered, washed well with methanol. Yield 66%, m.p. 250˚C - 252˚C. IR: 1670 (C =O), 3282 (NH) and 3432 (NH2). 1H NMR (DMSO): 4.82 (s, br, 2H, NH2, D2O, exchangeable), 5.1 (s, 1H, CH- methine), 6.7 - 7.9 (m, 11H, Ar-H) and 8.85 (s, br, 1H, NH, D2O, exchangeable). MS: m/z 309([M+3] + , 45%). Calcd for C17H14N4O2 (306.32): C 66.66, H 4.61, N 18.29%. Found: C 66.01, H 3.20, N 17.66 %. Preparation of 3-amino-3-(3-amino-5-oxo-1-(4-oxo- 3-phenyl-3,4-dihydro-2H-chromeno[2,3-d]pyrimidin-2- yl)-1H-pyrazol-4(5H)-ylidene)propanenitrile (5), and its derivatives, (7 - 10, 12, 13a, b). General procedu re: To a solution of hydrazide 3 (3.06 g, 0.01 mol) and ethyl 3-amino-2,4-dicyanobut-2-enoate (1.79 mL, 0.01 mol) in 30 mL ethanol containing 0.1 mL piperidine was refluxed for 6 h. then allowed to cool. The formed solid was filtered off, washed with methanol to af- ford the pyrazoile derivative 5. Analogously, diethyl malo- nate (1.60 mL, 0.01mol), ethyl 2-cyanoacetate (1.13 mL), ethyl 3-oxobutanoate (1.30 mL), pentane-2,4-dione (1.00 mL), 2-(ethoxymethylene) malononitrile (1.22 gm) and 2-cyano-N-phenylacetamide (1.60 gm) were reacted with compound 3 to yield (7 - 10, 12, 13a, b), respectively. 3-A m i n o - 3-( 3- amin o-5 - oxo- 1-( 4-oxo-3-pheny l- 3 ,4 - dihdro-2H-chromeno-[2,3-d]pyrimidin-2-yl)-1H-pyra- zol-4(5H)-ylidene)propanenitrile (5). Yellow crystals (MeOH), yield 70%, m.p. 250˚C - 252˚C. IR: 1699 (CO), 2219 (CN), 3422 (NH2). 1H NMR (DMSO): 2.9 (s, 2H, CH2), 3.5 (s, 2H, NH2), 3.7 (s, 2H, NH2), 6.7 - 7.9 (m, 10H, Ar-H). MS: m/z 439 ([M+], 60%). Calcd for C23H17N7O3 (439.43). C 62.87, H 3.90, N 22.31%. Found: C 61.01, H 3.20, N 21.89%. 1-(3,4-dihydro-4-oxo-3-phenyl-2H-chromeno[2,3-d]- pyrimidin-2-yl)pyrazo-lidine-3,5-dione, (7). Pale yel- low crystals (MeOH), yield 66%. m.p. 280˚C -282˚C. IR: υ (cm–1) 1685 - 1705 (C=O), 3212 (NH), 1HNMR (CDCl3): δ 3.2 (s, 2H, CH2), 5.8 (s, 1H, CH-methine ), 8.1 (s, 1H, NH of pyrazole ring) and 6.1 - 7.8 (m, 10H, Ar-H). MS: m/z 374 ([M]+, 55%). Calcd for C20H14N4O4 (374.35). Calcd: C 64.17, H 3.77, N 14.97%. Found: C 63.33, H 2.91, N 14.15%. Compound 5. Yield 70%, m.p. 257˚C - 259˚C. IR: 1699 (CO), 2219 (CN), 3422 (NH2). 1H NMR (DMSO): 2.9 (s, 2H, CH2), 3.5 (s, 2H, NH2), 3.7 (s, 2H, NH2), 6.7 - 7.9 (m, 10H, Ar-H). MS: m/z 439 ([M+], 60%). Calcd for C23H17N7O3 (439.43). C 62.87, H 3.90, N 22.31 %. Found: C 61.01, H 3.20, N 21.89%. Compound 8. Pale green crystals (Ethanol), yield 50%, m.p. 250˚C - 252˚C, IR: υ (cm–1) 1685-1705(C=O), 3432 (NH2), MS: m/z 373 ([M]+,65%). Calcd for C20H15N5O3 (373.36). Calcd: C 64.34, H 4.05, N 18.76%. Found: C 63.33, H 3.91, N 17.15%. Compound 9. Brown crystals (Ethanol), yield 55%, m.p. 190˚C - 192˚C. IR: υ (cm–1) 1685-1705 (C=O), 1HNMR (CDCl3): δ 1.1 (s, 3H, CH3), 2.5 (s, 2H, CH2), 5.8 (s, 1H, CH-methine), 6.7 - 7.9 (m, 10H, Ar-H). MS: m/z 370 ([M-2]+, 60%). Calcd for C23H16N4O3 (372.38). Calcd: C 67.73, H 4.33, N 15.05%. Found: C 67.17, H 3.31, N 14.15%. Compound 10. Pale green crystals (Ethanol), yield 59 %, m.p. 270˚C-2˚C. IR: υ (cm–1) 1685 - 1705(C=O), MS: m/z 369 ([M-1]+, 66%). Calcd for C22H18N4O2 (370.4). Calcd: C 71.34, H 4.90, N 15.13%. Found: C 70.17, H 3.31, N 14.15%. Preparation of 1-phenyl-tetrazolo[4’,5’:2,3] pyimi- do[4,5-b]chromen-12-one, (11). To a stirred cold solution of 3 (0.306 g, 0.001 mol) in 30 mL of glacial acetic acid, a cold solution of sodium nitrite (0.7 g, 0.01 mol) in 10 mL of H2O was added drop wise stirring at 5˚C. The mixture was stirred for further four hours at room temperature. The solid that precipitated was collected by filtration, washed with water and air dried to afford 55% yield of the tetrazolo derivative 11. Yield 55%, m.p. 250˚C - 252˚C. IR: 1699 (CO). MS: m/z 317 ([M+], 60%). Ca lcd for C17H11N5O2 (317.3). C 64.35, H 3.49, N 22.07%. Found: C 63.01, H 2.20, N 21.89%. Compound 12. Yellow crystals (Ethanol), yield 55%, m.p. 259˚C - 261˚C. IR: υ (cm–1) 1685 - 1705(C=O), 2219 (CN), 3212 - 3423 (NH2), MS: m/z 382 ([M + 1]+, 50%). Calcd for C21H14N6O2 (382.37). Calcd: C 65.96, H 3.69, N Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF 46 21.98%. Found: C 64.17, H 2.31, N 20.15%. Compound 13a. Yellow crystals (Ethanol), yield 62%, m.p. 150˚C - 152˚C. IR: υ (cm–1) 1685 - 1705(C=O), 3212 - 3432 (NH2), 1HNMR (CDCl3): δ 4.1 (s, 2H, NH2), 4.3 (s, 1H, NHPh), 6.2 (s, 1H, = CH-), 6.7 - 7.9 (m, 10H, Ar-H). MS: m/z 448([M]+,40%). Calcd for C26H20N6O2 (448.48). Calcd: C 69.63, H 4.49, N 18.74%. Found: C 68.17, H 3.31, N 17.15%. Compound 13b. Pale yellow crystals (MeOH), yield 50%, m.p. 180˚C - 182˚C. IR: υ (cm–1) 1685 - 1705(C=O), 3212 - 3432 (NH2), MS: m/z 456 ([M + 1]+, 35%). Calcd for C23H17N7O2S (455.49). Calcd: C 60.65, H 3.76, N 71.53, S 7.04%. Found: C 59.17, H 2.66, N 70.26, S 6.54%. Preparation of 2-(2-benzylidenehydrazinyl)-3-phenyl- 3H-chromeno[2,3-d]pyri-midin-4(10H)-one, (14). A mix- ture of the 2-hydrazinyl-3-phenyl-2H-chromeno [2,3-d]pyri- midin-4(3H)-one 3 (3.06 g, 0.01 mol) and benzaldehyde (1.06, 0.01 mol) in 20 mL ethanol containing 0.1 mL of piperidine was refluxed for 8 hr. The reaction mixture was concentrated, cooled then poured into ice/H2O mixture the solid product thus so formed was filtered, washed for several times with water to afford 14 (1.66 g, 61% ) as a brown solid; m.p. 225˚C - 227˚C. IR: υ (cm-1) 1705 (C=O), 3322 (NH). 1HNMR (CDCl3): δ 6.7 - 7.9 (m, 14H, Ar-H), 8.11 (s, 1H, CH=) and 10.11 (s, 1H, NH). MS: m/z 397 ([M]+, 65%). Calcd for C24H18N4O2 (394.43). Calcd: C 73.08, H 4.60, N 14.20 %. Found: C 72.14, H 4.01, N 13.88%. Preparation of 5-oxo-1-(4-oxo-3-phenyl-4, 10-dihy- dro-3H-chromeno[2,3-d]pyrimidin-2-yl)-3-phenylpyra zolidine-4-carbonitrile, (16). A solution of benzylidene- hydrazinyl 14 (3.94 g, 0.01 mol) and ethyl 2-cyanoacetate (1.13 mL) in 30 mL ethanol containing 0.1 mL piperidine was refluxed for 6 h. then allowed to cool. The formed solid was filtered off, washed with methanol to afford the pyrazole derivative 16 (2.45 g, 61%) as a brown solid; m.p. 295˚C - 297˚C. IR: υ (cm–1) 1670 (C=O), 2217 (CN), 3432 (NH). 1HNMR (CDCl3): δ 3.98 (s, 1H, NH), 4.12 (s, 1H, C-3 pyrazole), 4.21 (s, 1H, C-4 pyrazole) and 6.7 - 7.9 (m, 14H, Ar-H). MS: m/z 462 ([M + 1]+, 55%). Calcd for C27H19N5O3 (461.47). Calcd: C 70.27, H 4.15, N 15.18 %. Found: C 69.12, H 4.01, N 14.11%. Preparation of 5-chloro-1-(4-oxo-3-phenyl-4,10-dihy- dro-3H-chromeno[2,3-d]pyrimidin-2-yl)-3-phenyl-2, 3- dihydro-1H-pyrazole-4-carbonitrile, (17). A suspen- sion of 1.0 g of 16 in 10 ml of phosphoryl chloride was heated at 90˚C for 5 hr and, after cooling, crushed ice was added. The solution was then made basic with concen- trated ammonium hydroxide (pH = 8) and the solid that precipitated was collected by filtration, washed with water and purified by crystallization to give 17 as a Yellow solid; m.p. 189˚C - 191˚C. IR: υ (cm–1) 1700 (C=O), 2217 (CN), 3425 (NH). 1HNMR (CDCl3): δ 3.98 (s, 1H, NH), 4.61 (s, 1H, C-3 pyrazole) and 6.7 - 7.9 (m, 14H, Ar-H). MS: m/z 478 ([M-1]+, 16%). Calcd for C27H18N5O2Cl (479.92). Calcd: C 67.57, H 3.78, N 14.59%. Found: C 66.12, H 3.21, N 14.51%. Preparation of 2-(4-amino-3-phenyl-2, 3-dihydro- 1H-pyrazolo[4,3-e][1, 2, 4]-triazolo[1,5-a] pyrimidin- 1-yl)-3-phenyl-3H-chromeno[2,3-d]pyrimidin-4(10H)- one, (18). Amixtuer of 17 (4.79 g, 0.01 mol) and 5- amino-1,2,4-1H-triazole (0.84 g) in 30 mL ethanol con- taining 0.1 mL piperidine was refluxed for 3 hr. then allowed to cool. The formed brownish solid was filtered off, washed with methanol to afford the pyrazoile deriva- tive 18 (2.4 g, 50%) as a brownish solid; m.p. 156˚C - 158˚C. IR: υ (cm–1) 1670 (C=O), 3432 (NH). 1HNMR (DMSO): δ 3.98 (s, br, 1H, NH), 5.21 (s, 1H, C-3 pyra- zole), 6.13 (s, br, 2H, NH2) and 6.7 - 7.9 (m, 14H, Ar-H) and 8.71 (s, 1H, methine proton of triazole ring). MS: m/z 527 ([M]+, 25%). Calcd for C29H21N9O2 (527.54). Calcd: C 66.03, H 4.01, N 23.90 %. Found: C 65.31, H 3.22, N 22.84%. Preparation of 4-amino-7-(methylthio)-1-(4-oxo-3- phenyl-4,10-dihy dr o- 3H-chromeno[2,3-d]py r imidin-2- yl)-3-phenyl-2,3-dihydro-1H-dipyrazolo [1,5-a:4’,3’-e] pyrimidine-6-carbonitrile, (19). Amixtuer of 17 (4.79 g, 0.01 mol) and 5-amino-3-(methylthio)-1H-pyrazole-4- car- bonitrile (1.54 g, 0.01 mol ) in 30 mL ethanol containing 0.1 mL piperidine was refluxed for 5 h. then allowed to cool. The formed pale yellow solid was filtered off, washed with methanol to afford the pyrazoile derivative 19 (2.4 g, 50%) as a pale yellow solid (MeOH); m.p. 240˚C - 242˚C. IR: υ (cm–1) 1670 (C=O), 2221 (CN), 3432 (NH). 1HNMR (DMSO): δ 3.54 (s, 3H, SCH3), 3.98 (s, br, 1H, NH), 5.21 (s, 1H, C-3 pyrazole), 6.13 (s, br, 2H, NH2) and 6.7 - 7.9 (m, 14H, Ar-H) and 8.71 (s, 1H, methine proton of triazole ring). MS: m/z 597 ([M]+, 35 %). Calcd for C32H23N9O2S (597.65). Calcd: C 64.31, H 3.88, N 21.09 %. Found: C 63.56, H 2.52, N 20.78%. Preparation 5-methoxy-1-(4-oxo-3-phenyl-4, 10-di- hydro-3H-chromeno[2,3-d]py-rimidin-2-yl)-3-phenyl- 2,3-dihydro-1H-pyrazole-4-carbonitrile, (20). A solu- tion of 10 mmol of freshly prepared sodium methoxide and 1.0 mmol of the chloro derivatives 17 in 10 ml of anhydrous methanol was refluxed for 4 hr then the reac- tion mixture was evaporated to dryness in vacuo. The crude residue was treated with water and neutralized with 10% hydrochloric acid, and the solid precipitate, collected by filtration, was purified by crystallization to obtain 20 as yellow crystals (DMSO-HCl); yield 30%. m.p. 288˚C - 290˚C. IR: υ (cm–1) 1699 (C=O), 2219 (CN), 3422 (NH). 1HNMR (DMSO): δ 3.82 (s, 3H, OCH3), 3.98 (s, br, 1H, NH), 4.61 (s, 1H, C-3 pyrazole) and 6.7 - 7.9 (m, 14H, Ar-H). MS: m/z 475 ([M]+, 62%). Calcd for C28H23N9O2S (597.65). Calcd: C 64.31, H 3.88, N 21.09 %. Found: C 63.56, H 2.52, N 20.78%. Prepar atio n 5-azido-1-(4-oxo-3-phenyl-4, 10-dihy- Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF 47 dro-3H-chromeno[2,3-d]pyri-midin-2-yl)-3-phenyl-2,3 -dihydro-1H-pyrazole-4-carbonitrile, (21). A solution of the chloro compound 17 (1.437 g, 3 mmol) in acetone (5 mL) was stirred and ice-cooled. The solution of NaN3 (0.13 g, 2 mmol) in water (1 mL) was added drop wise in the solution and this mixture was stirred for 1 hr at room temperature. After evaporation of acetone, the crude product was separated by filtration and recrystallized from dichlormethane. We have obtained (0.9 g, 62%) of 21 as Yellow solid with mp 150˚C - 152˚C [21, 22]. IR: υ (cm–1) 1699 (C=O), 2217 (CN), 3422 (NH). 1HNMR (DMSO): δ 3.98 (s, br, 1H, NH), 4.61 (s, 1H, C-3 pyrazole) and 6.7 - 7.9 (m, 14H, Ar-H). MS: m/z 486 ([M]+, 32%). Calcd for C27H18N8O2 (486.48). Calcd: C 66.66, H 3.73, N 23.03%. Found: C 65.76, H 2.56, N 22.73%. Preparation of 2-(4-amino- 3-phenyl-2,3-dihydro- pyrazolo [3 ,4 -c]pyrazol-1(6H)-yl)-3-phenyl-3H-chro- meno[2,3-d]pyrimidin-4(10H)-one, (23). A mixture of 17 (3.20 g, 0.01 mol) and hydrazine hydrate (0.5 mL, 0.01 mol) in 20 mL DMF containing 0.1 mL piperidine was refluxed for 8 hr. The reaction mixture was concentrated under reduced pressure and the residue washed with aci- dified cold water and then triturated with methanol. The formed yellow product was filtered, washed well with methanol. Yield 58%, m.p. 150˚C - 152˚C. IR: 1670 (C=O), 3432 (NH) and 3455 (NH2). 1H NMR (DMSO): 3.98 (s, br, 1H, NH), 5.21 (s, 1H, CH- methine), 6.7 - 7.9 (m, 11H, Ar-H) and 9.21 (s, 1H, NH). MS: m/z 475([M] +, 64%). Calcd for C27H21N7O2 (475.50): C 68.20, H 4.45, N 20.62%. Found: C 67.03, H 3.32, N 19.23%. Preparation of 3-(2-imino-2H- chromen-3-yl)-1-phe- nyl-3-(phenylamino) prop-2-en-1-one, (24a) and its derivatives (24b-e). General procedure: A mixture of the 2-imino-N- phenyl-2H-chromene-3-carboxamide 1 (2.64 g, 0.01 mol) and acetophenone (1.2 ml, 0.01 mol), p-hydroxyacetophe- none (1.36 g, 0.01 mol), p-nitroacetophenone (1.65 g, 0.01 mol), o-nitroacetophenone (1.65 g, 0.01 mol) and p-chloro- acetophenone (1.54 g, 0.01 mol) respectively, in 20 mL DMF containing 0.1ml of piperidine was refluxed for 8 hr. The reaction mixture was concentrated, cooled then poured into ice/H2O mixture the solid product thus so formed was filtered, washed for several times with water to afford 24a - 24e derivatives. Compound 24a. Yellow crystals (MeOH), yield 60%. m.p. 182˚C - 184˚C. IR: υ (cm–1) 1665 - 1705 (C=O), 3322 (NH), 1HNMR (DMSO): δ 4.1 (s, 1H, NH-amine), 6.5 (s, 1H, CH-ethylene), 7.1-7.6 (m, 15H, Ar-H), 11.5 (s, 1H, =NH). MS: m/z 366 ([M]+,40%). Calcd for C24H18N2O2 (366.41). Calcd: C 78.67, H 4.95, N 7.65%. Found: C 77.17, H 4.01, N 8.15%. Compound 24b. Pale green crystals (MeOH), yield 55%, m.p. 220˚C - 222˚C. IR: υ (cm–1) 1665 - 1705(C=O), 3445 (OH), 1HNMR (CDCl3): δ 4.1 (s, 1H, NH-amine), 6.5 (s, 1H, CH-ethylene), 7.1 - 7.6 (m, 14H, Ar-H), 11.5 (s, 1H, =NH) and 9.9 (s, 1H, phenolic OH). MS: m/z 383 ([M + 1]+, 60%). Calcd for C24H18N2O3 (382.41). Calcd: C 75.38, H 4.74, N 7.33%. Found: C 74.17, H 3.91, N 6.15%. Compound 24c. Brown crystals (Ethanol), yield 65%, m.p. 190˚C - 192˚C. IR: υ (cm–1) 1665 - 1705(C=O), 1HNMR (DMSO): δ 4.1(s, 1H, NH-amine), 6.5(s, 1H, CH-ethylene), 7.1 - 7.6 (m, 14H, Ar-H), 11.5 (s, 1H, =NH). MS: m/z 411([M]+, 62%). Calcd for C24H17N3O4 (411.41). Calcd: C 70.07, H 4.16, N 10.21%. Found: C 69.17, H 3.31, N 9.15%. Compound 2 4d. Brown crystals (Ethanol), yield 59 %, m.p. 170˚C -172˚C. IR: υ (cm–1) 1665 - 1705(C=O). MS: m/z 410 ([M-1]+, 52%). Calcd for C24H17N3O4 (411.41). Calcd: C70.07, H 4.16, N 10.21%. Found: C 69.17, H 3.31, N 9.15%. Compound 24e. Yellow crystals (Ethanol), yield 55%, m.p. 255˚C - 257˚C. IR: υ (cm–1) 1665 - 1705(C=O), 1HNMR (DMSO): δ 4.1 (s, 1H, NH-amine), 6.5 (s, 1H, CH-ethylene), 7.1 - 7.6 (m, 14H, Ar-H), 11.5 (s, 1H, =NH). MS: m/z 400 ([M]+,60%). Calcd for C24H17N2O2Cl (400.86). Calcd: C 71.91, H 4.27, N 6.99, Cl8.84%. Found : C 70.17, H 3.31, N 5.15, Cl7.18%. Preparation of 2-imino-N,N'-diphenyl-2H-chrom- ene-3-carboxamidines, (25a) and its derivatives (25b- 25e). General procedure: A mixture of equimolar amount of compound 1 (2.64 g, 0.01 mol) and aniline (0.93 ml, 0.01 mol), p-hydroxyaniline (1.09 g, 0.01 mol), p-nitroaniline (1.38 g, 0.01 mol), o-nitroaniline ( 1.38 g, 0.01 mol and p-chloroaniline (1.27 g, 0.01 mol), respectively in 30 mL ethanol containing 0.1 mL of piperidine was refluxed for 5 hr. The reaction mixture was concentrated, poured into ice/H2O mixture, and the solid product thus formed, fil- tered, washed for several times with water and crystallized from methanol. Compound 25a. Pale brown crystals (Methanol), yield 58%, m.p. 205˚C - 207˚C. IR: υ (cm–1) 3380(NH), 1HNMR (CDCl3): δ 4.1 (s, 1H, NH-amine), 7.1 - 7.6(m, 15H, Ar- H), 11.5(s, 1H, =NH). MS: m/z 339 ([M]+, 65%). Calcd for C22H17N3O(339.39). C 77.86, H 5.05, N 12.38%. Found: C 76.66, H 4.52, N 11.12%. Compound 25b. Yellow crystals (Methanol), yield 60%, m.p. 210˚C - 212˚C. IR: υ (cm–1) 3380(NH), 3445 (OH), 1HNMR (DMSO): δ 4.1(s, 1H, NH-amine), 7.1- 7.6(m, 14H, Ar-H), 11.5 (s, 1H, = NH) and 9.9 (s, 1H, phenolic OH). MS: m/z 357 ([M+2]+, 50%). Calc d for C22H17N3O2 (355.39). C 74.35, H 4.82, N 11.82%. Found : C 73.45, H 3.52, N 10.12%. Compound 25c. Pale yellow crystals (Methanol), yield 64%, m.p. 285˚C - 287˚C. 1HNMR (DMSO): δ 4.1 (s, 1H, NH-amine), 7.1 - 7.6 (m, 14H, Ar-H), 11.5 (s, 1H, = NH), MS: m/z 385 ([M + 1]+, 55%). Calcd for C22H16N4O3Cl (384.39). C 68.74, H 4.20, N 14.58%. Found: C 67.72, H Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF 48 3.52, N 13.12%. Compound 25d. Yellow crystals (Methanol), yield 60%, m.p. 215˚C - 117˚C. MS: m/z 384([M]+, 55%). Calcd for C22H16N4O3Cl (384.39). C 68.74, H 4.20, N 14.58%. Found: C 67.72, H 3.52, N 13.12%. Compound 25e. Pale brown crystals (methanol), yield 55%, m.p. 230˚C - 232˚C. 1HNMR (DMSO): δ 4.1 (s, 1H, NH-amine), 7.1 - 7.6 (m, 14H, Ar-H), 11.5 (s, 1H, =NH), MS: m/z 372([M − 1]+, 70%). Cal cd for C22H16N3OCl (373.83). C 70.68, H 4.31, N 11.24, Cl 9.48%. Found : C 69.45, H 3.52, N 11.12, Cl 8.48%. Preparation of 4,5-dihydro-5-(2-imino-2H-chromen- 3-yl)-N,3-diphenyl-1H-pyrazol-5 -a mine, (26a) a- nd it s derivatives (26b an d 27). General procedure: A mixture of 24a (3.66 g, 0.01 mol) and hydrazine hydrate (0.05 ml, 0.01 mol), phenyl hydrazine (1.08 ml, 0.01 mol) and hydroxylamine hy- drochloride (0.69 gm, 0.01 mol) containing 0.1 ml pipe- ridine was refluxed for 8 hr. The reaction mixture was concentrated under reduced pressure and the residue washed with acidified cold water and then triturated with methanol, and the solid product thus formed, filtered, washed for several times with water and crystallized from methanol. Compound 26a. Pale brown crystals (methanol), yield 66%, m.p. 155˚C - 157˚C. IR: 3117 - 3293 (NH). 1H NMR (DMSO): 2.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 7.1 (s, H, NH hydrazide), 7.2 - 7.8 (m, 15H, Ar-H), 11.5 (s, 1H, =NH). MS: m/z 383([M+3]+, 55%). Calcd for C24H20N4O (380.44): C 75.77, H 5.30, N 14.73%. Found: C 74.01, H 4.20, N 13.66%. Compound 26b. Brown crystals (Methanol), yield 50%, m.p. 295˚C - 297˚C. IR: 3117 - 3293 (NH). 1H NMR (DMSO): 1.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 7.1 (s, H, NH hydrazide), 7.2 - 7.8 (m, 20H, Ar-H) and 11.5 (s, 1H, =NH). MS: m/z 456 ([M+], 75%). Calcd for C30H24N4O (456.54): C 78.92, H 5.30, N 12.27%. Found: C 77.01, H 4.20, N 11.66%. Compound 27. Pale brown crystals (Methanol), yield 55%, m.p. 230˚C - 232˚C. IR: 3117 - 3293 (NH). 1H NMR (DMSO): 1.9 (s, 2H, CH2), 4.1(s, 1H, NH-amine), 7.2 - 7.8 (m, 15H, Ar-H) and 11.5 (s, 1H, = NH). MS: m/z 383 ([M+2] +, 30%). Calcd for C24H19N3O2 (381.43): C 75.57, H 5.02, N 11.02%. Found: C 74.01, H 4.20, N 10.66%. Preparation of 4,5-dihydro-4-(2-imino- 2H-chromen- 3-yl)-6-phenyl-4-(phenyl-amino) pyrimi-din-2(1-H)-one, (28a), and its derivative (28b). General procedure: A mixture of equimolar amounts of 24a (3.66 g, 0.01 mol) and urea (0.60 g, 0.01 mol) or thiourea (0.76 g, 0.01 mol) and 0.1 mL piperidine was refluxed for 8 h. in 30 mL of ethanol. The solvent was evaporated under vacuum, and the residue was poured to 30 ml acidified cold water and then triturated with metha- nol. The product were filtered and crystallized from etha- nol. Compound 28a. Brown crystals (Ethanol); yield 67%, m.p. 196˚C - 198˚C. IR: 1685 - 1705 (C=O), 3063 - 3288 (NH). 1H NMR (DMSO): 2.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 7.2 - 7.8 (m, 15H, Ar-H), 8.1 (s, H, NH-amide) and 11.5 (s, 1H, = NH), MS: m/z 408 ([M]+, 45%). Calcd for C25H20N4O2 (408.45): C 73.51, H 4.94, N 13.72%. Found: C 72.01, H 3.20, N 12.66%. Compound 28b. Brown crystals (Methanol), yield 59%, m.p. 210˚C - 112˚C. IR: 1230 (C=S), 3063 - 3288 (NH), MS: m/z 422 ([M-2]+, 35%). Calcd for C25H20N4OS (424.52): C 70.73, H 4.75, N 13.20%. Found: C 69.01, H 3.20, N 12.66%. Preparation of 1, 2, 5, 6-tetrahydro-6-(2-imino-2H- chromen-3-yl)-2-oxo-4-phenyl-6-(phenylamino) pyrine- carbonitrile, (29a) and its derivatives (29b - 29e) and (30). General procedure: A mixture of 24a (3.66 g, 0.01 mol) and cyanoacetamide (0.84 g, 0.01 mol) in 30 ml of ethanol in the presence of 0.1 ml of piperidine was re- fluxed for 8 h. The reaction mixture was concentrated under vacuum and the residue washed with acidified cold water and then triturated with methanol. The solid product formed was filtered and crystallized from ethanol to afford 29a in 65% yield. In analogously, the Chalcone 24a was reacted with cyanothioacetamide (1.0 g, 0.01 mol), 2- cyano-N-p-tolylacetamide (1.74 g, 0.01 mol), 2-cyanoace- tohydrazide (0.99 g, 0.01 mol), 2-cyano-N-phenylace- tamide (1.6 g, 0.01 mol) and 2-chloroacetamide (0.93 g, 0.01 mol) to yield the pyridine derivatives 29b -29e and 30 respectively. Compound 29a. Yellow crystals (Methanol), yield 56%, m.p. 184˚C - 186˚C. IR: 1689 - 1705 (C=O), 2220 (CN), 3188 - 3244(NH). 1H NMR (DMSO): 2.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 7.2 - 7.8 (m, 15H, Ar-H), 8.1 (s, H, NH-amide) and 11.5 (s, 1H, =NH), MS: m/z 432 ([M]+, 65%). Calcd for C27H20N4O2 (432.47). C 74.98, H 4.66, N 12.95%. Found: C 73.01, H 3.20, N 11.66%. Compound 29b. Pale yellow crystals (Methanol), yield 50%, m.p. 240˚C - 242˚C. IR: 1251 (C=S), 2218 (CN), 3188 - 3244 (NH). MS: m/z 448 ([M]+, 45%). Calcd for C27H20N4OS (448.54). C 72.30, H 4.49, N 12.49, S 7.15%. Found: C 71.11, H 3.21, N 11.12, S 6.55%. Compound 29c. Pale red crystals (Methanol), yield 60%, m.p. 235˚C - 238˚C, IR: 1689 – 1705 (C=O), 2210 (CN), 3188 - 3244 (NH), MS: m/z 522 ([M] +, 40%). Calcd for C34H26N4O2 (522.6): C 78.14, H 5.01, N 10.72%. Found: C 77.01, H 4.20, N 9.66%. Compound 29d. Pale red crystals (Methanol), yield 75%, m.p. 217˚C - 219˚C. IR: 1689 – 1705 (C=O), 2210 (CN), 3188 - 3344 (NH and NH2). MS: m/z 447 ([M] +, 35%). Calcd for C27H21N5O2 (447.49): C 71.47, H 4.73, N 15.65%. Found: C 70.01, H 3.20, N 14.66%. Compound 29e. Yellow crystals (Methanol), yield 35%, Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF 49 m.p. 280˚C - 282˚C. IR: 1689 - 1705 (C=O), 2210 (CN), 3188 - 3344 (NH and NH2). 1H NMR (DMSO): 1.9 (s, 2H, CH2), 2.1 (s, 2H, NH2), 4.1 (s, 1H, -NHPh), 7.2 - 7.8 (m, 20H, Ar-H) and 11.5 (s, 1H, =NH), MS: m/z 510 [M + 2]+, 50 %). Calcd for C33H24N4O2 (508.57). C 77.93, H 4.76, N 11.02%. Found: C 76.41, H 3.10, N 10.06%. Compound 30. Brown crystals (Methanol), yield 50%, m.p. 220˚C - 222˚C. IR: 1699 - 1705 (C=O), 3188 - 3244 (NH). 1H NMR (DMSO): 1.9 (s, 2H, CH2), 4.1 (s, 1H, NH- amine), 7.2 - 7.8 (m, 15H, Ar-H), 8.1 (s, H, NH) and 11.5 (s, 1H, =NH), MS: m/z 441 ([M]+, 55%). Calcd for C26H20N3O2Cl (441.91). C 70.67, H 4.56, N 9.51, Cl 8.02%. Found: C 69.11, H 3.20, N 8.26, Cl 7.61%. Preparation of 2-(2-imino-2H-chromen-3-yl)-4-phe- nyl-2-(phenylamino)pyrido-[1,2-a]benzim-idazo-lo-5- carbonitriles, (31). A mixture of 24a (3.66 g, 0.01 mol), 2-cyanomethylbenzimidazole (1.57 g, 0.01 mol) and 0.1 mL of piperidine in 30 mL of ethanol was refluxed for 8 h. The solvent was evaporated under vacuum and the residue was washed by acidified cold water and then triturated with methanol. The solid product was filtered and crys- tallized from ethanol, as brown crystals, yield 65%, m.p. 278˚C - 280˚C. IR: 2220 (CN), 3188 - 3244 (NH). 1H NMR (CDCl3): 2.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 6.9 - 7.6 (m, 19H, Ar-H) and 11.5 (s, 1H, =NH), MS: m/z 505 ([M] +, 35%). Ca lcd for C33H23N5O (505.57): C 78.40, H 4.59, N 13.85%. Found: C 77.01, H 3.20, N 12.66%. Preparation of 2,3-dihydro-2-(2-imino-2H-chrom-e- n-3-yl)-N,4-diphenyl-benzo[b][1,4]oxazepin-2-amine, (32a) and its derivatives, (32b). Genera l procedure : A mixture of an equimolar amounts of 24a (3.66 g, 0.01 mol) and (1.09 g, 0.01 mol) o- aminophenol and (1.25 g, 0.01 mol) o-aminothiophenol respectively, in 30 mL ethanol containing 0.1 mL of piperi- dine was refluxed for 8 hr. The reaction mixture was con- centrated, cooled then poured into ice/H2O mixture. The solid product thus so formed was filtered, washed for sev- eral times with water and crystallized from ethanol. Compound 32a. Brown crystals (Ethanol), yield 60%, m.p. 282˚C - 284˚C, IR: 3289(NH), 1H NMR (DMSO): 2.9(s, 2H, CH2), 4.1(s, 1H, NH-amine), 6.7 - 7.3 (m, 19H, Ar-H) and 11.5(s, 1H, = NH), MS: m/z 457 ([M] +, 55%). Calcd for C30H23N3O2 (457.52): C 78.75, H 5.07, N 9.18%. Found: C 77.01, H 4.20, N 8.66%. Compound 32b. Yellow crystals (Ethanol), yield 45 %, m.p. 270˚C-272˚C. IR: 3188-3244(NH), 1H NMR (CDCl3): 1.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 6.9 - 7.6 (m, 19H, Ar-H) and 11.5 (s, 1H, =NH), MS: m/z 476 ([M+3]+, 45%). Calcd for C30H23N3OS (473.59). C 76.08, H 4.90, N 8.87, S 6.77%. Found: C 77.01, H 3.20, N 12.66, S 5.12%. Preparation of 2-(dicyanomethylene)-1,2,5,6-tetra- hydro-6-(2-imino-2H-chromen-3-yl)-4-phenyl-6-(phe- nylamino)pyridine-3-carbonitrile, (33a) and its de- rivatives (33b). General procedure: A mixture of 24a (0.366 g, 0.001 mol) and 2-aminoprop-1-ene-1,1,3-tricarbonitrile (0.132 g, 0.001 mol) in 30 mL of ethanol in the presence of 0.1 mL of piperidine was refluxed for 8 h. The reaction mix- ture was concentrated under vacuum and the residue washed with acidified cold water and then triturated with methanol. The solid product formed was filtered and crystallized from methanol to afford 33a in 60% yield. In analogously, the Chalcone 34a was reacted with ethyl 3-amino-2, 4-dicyanobut-2-enoate (1.79 g, 0.01 mol) to yield the pyridine derivative 33b. Compound 33a. Yellow crystals (Methanol), yield 60%, m.p. 184˚C - 186˚C. IR: 2219 (CN), 3289 (NH). 1H NMR (DMSO): 2.9 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 6.7 - 7.3 (m, 15H, Ar-H), 8.1 (s, H, NH) and 11.5 (s, 1H, =NH), MS: m/z 482 ([M+2]+, 35%). Calcd for C30H20N6O (480.52). C 74.99, H 4.20, N 17.49%. Found: C 73.01, H 3.20, N 16.66%. Compound 33b. Pale yellow crystals (Methanol), yield 45%, m.p. 240˚C - 242˚C. IR: 2219 (CN), 3289 (NH). 1H NMR (DMSO): δ 1.2 (t, 3H, CH3), 2.2 (s, 2H, CH2), 3.7 (q, 2H, CH2), 4.1 (s, 1H, NH-amine), 6.7 - 7.9 (m, 15H, Ar-H) and 11.5 (s, 1H, =NH), MS: m/z 527 ([M]+, 35%). Calcd for C32H25N5O3 (527.57). Calcd: C 72.85, H 4.78, N 13.27%. Found: C 71.14, H 3.01, N 12.88%. Preparation of 3-chloro-4- (2-imino-2H-chromen-3- yl)-1-phenyl-4-(phenyl-amino) azetidin-2-one, (34a) and its derivatives, (34b - 34e). Compound 34a. Brown crystals (Methanol) yield 60%, m.p. 280˚C - 282˚C. IR: 1705 (C=O), 3289 (NH). 1H NMR (DMSO): 4.1 (s, 1H, NH-amine), 5.2 (s, 1H, CH, C-3 of β-lactam ring), 7.1 - 7.6 (m, 15H, Ar-H) and 11.5 (s, 1H, =NH), MS: m/z 415 ([M]+, 40%), Calc d for C24H18N3O2Cl (415.87): C 69.31, H 4.36, N 10.10, Cl 8.5279%. Found: C 68.19, H 3.21, N 9.18, Cl 7.12%. Compound 34b. Pale yellow crystals (Methanol), yield 63%, m.p. 275˚C - 277˚C. IR: 1705(C=O), 3289 (NH), 3388(OH). 1H NMR (DMSO): 4.1 (s, 1H, NH-amine), 5.2 (s, 1H, CH, C-3 of β-lactam ring), 7.1 - 7.6 (m, 14H, Ar-H), 11.5 (s, 1H, =NH) and 10.1 (s, 1H, phenolic OH). MS: m/z 431([M]+, 55%), Calcd for C24H18N3O3Cl (431.87): C 66.75, H 4.20, N 9.73, Cl 8.21%. Found: C 65.09, H 3.52, N 9.08, Cl 7.12%. Compound 34c. Pale brown crystals (Methanol), yield 70%, m.p. 210˚C - 212˚C. IR: 1705 (C=O), 3289 (NH). 1H NMR (CDCl3): 4.1 (s, 1H, NH-amine), 5.2 (s, 1H, CH, C-3 of β-lactam ring), 7.1 - 7.6 (m, 14H, Ar-H) and 11.5 (s, 1H, =NH). MS: m/z 460([M]+, 40%), Calcd for C24H17N4O4Cl (460.87): C 62.55, H 3.72, N 12.16, Cl 7.69%. Found: C 61.07, H 2.22, N 11.18, Cl 6.11%. Compound 34d. Brown crystals (Ethanol), yield 62%, m.p. 175˚C - 177˚C. IR: 1705 (C=O), 3289 (NH). MS: m/z 461 ([M + 1]+, 55%), Calcd for C24H17N4O4Cl (460.87): C 62.55, H 3.72, N 12.16, Cl 7.69%. Found: C 61.17, H 2.25, Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF 50 N 11.48, Cl 6.21%. Compound 34e. Yellow crystals (Ethanol), yield 55%, m.p. 250˚C - 252˚C. IR: 1705 (C=O), 3289 (NH). 1H NMR (CDCl3): 4.1 (s, 1H, NH-amine), 5.2 (s, 1H, CH, C-3 of β-lactam ring), 7.1 - 7.6(m, 14H, Ar-H) and 11.5 (s, 1H, =NH). MS: m/z 450([M]+, 65%), Calcd for C24H17N3O2Cl2 (450.32): C 64.01, H 3.81, N 9.33, Cl 7.11%. Found: C 63.07, H 2.82, N 8.18, Cl 6.41%. Preparation of 2-(2-imino-2H- chromen-3-yl)-3-phe- nyl-2-(phenylamino) thiazolidin-4-one, (35a) and its derivatives (35b - 35 e ). General procedure: An equimolar mixture of 25a (0.99 g, 0.003 mol) and thioglycolic acid (0.276 mL, 0.003 mol) in dry benzene (20 mL) was refluxed for 10 h. The reac- tion mixture was evaporated to dryness under reduced pressure. The thiazolidinone was separated off, washed with ether and crystallized from ethanol. Analogously, 25b - 25e reacted with thioglycolic acid to yield 35b - 35e. Compound 35a. Yellow crystals (ethanol), yield 60%, m.p. 185˚C - 187˚C. IR: 1230 (CS), 1695 (C=O), 3289 (NH). 1H NMR (DMSO): 3.8 (s, 2H, CH2), 4.1(s, 1H, NH- amine), 6.8 - 7.8 (m, 15H, Ar-H) and 11.5 (s, 1H, =NH). MS: m/z 411 ([M − 2]+, 35%). Calcd for C24H19N3O2S (413.49): C 69.71, H 4.63, N 10.16, S 7.91%. Found: C 68.46, H 3.17, N 9.46, S 6.55%. Compound 35b. Yellow crystals (Methanol), yield 70%, m.p. 215˚C - 117˚C. IR: 1230 (CS), 1695 (C=O), 3289 (NH). 1H NMR (DMSO): 3.8 (s, 2H, CH2), 4.1 (s, 1H, NH- amine), 6.8 - 7.8 (m, 15H, Ar-H), 11.5 (s, 1H, =NH) and 10.1 (s, 1H, phenolic OH). MS: m/z 429([M]+, 44%). Calcd for C24H19N3O3S (429.49): C 67.12, H 4.46, N 9.78, S 7.47%. Found: C 66.46, H 3.17, N 8.46, S 6.25%. Compound 35c. Pale brown crystals (Ethanol), yield 76%, m.p. 198˚C - 200˚C. IR: 1230 (CS), 1695 (C=O), 3289 (NH). 1H NMR (DMSO): 3.8 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 6.8 - 7.8 (m, 14H, Ar-H) and 11.5 (s, 1H, =NH). MS: m/z 457 ([M-1]+, 55%). Calcd for C24H18N4O4S (458.49): C 62.87, H 3.96, N 12.22, S 6.99%. Found: C 61.66, H 3.17, N 11.16, S 5.55%. Compound 35d. Yellow crystals (Ethanol), yield 65%, m.p. 290˚C - 292˚C. IR: 1230 (CS), 1695 (C=O), 3289 (NH). MS: m/z 458 ([M]+, 55%). Calcd for C24H18N4O4S (458.49): C 62.87, H 3.96, N 12.22, S 6.99%. Found: C 61.12, H 3.74, N 11.18, S 6.25%. Compound 35e. Brown crystals (Methanol), yield 62%, m.p. 235˚C - 237˚C. IR: 1230 (CS), 1695 (C=O), 3289 (NH). 1H NMR (DMSO): 3.8 (s, 2H, CH2), 4.1 (s, 1H, NH-amine), 6.8 - 7.8 (m, 14H, Ar-H) and 11.5 (s, 1H, =NH). MS: m/z 447 ([M]+, 65%). Calcd for C24H18N3O2SCl (447.94): C 64.35, H 4.05, N 9.38, S 7.91%. Fo und: C 63.63, H 3.57, N 9.16, S 6.55%. 3. Results and Discussion The synthetic procedures adopted to obtain the target compounds are depicted in Schemes 1 - 10. The starting compound, 2-imino-N-phenyl-2H-chromene-3-carboxami- de 1, was prepared according to the previously reported procedure [18]. Thus, refluxing of compound 1 with triethyl orthoformate in dimethylformamide affording 2-ethoxy-2,3- dihydro-3-phenylchromeno[2,3-d]pyrimidin-4-one 2, (Sche- me 1). The structure of the later product was based on IR, 1H NMR, and mass spectra. The IR spectrum of 2 showed the lack of any absorption of the NH functions, and the 1H NMR spectrum (CDCl3) displayed a triplet at δ 1.2 and a quartet at 3.9 ppm due to the ethoxy protons and a multi- plet at 6.7 - 7.9 ppm due to aromatic protons, respectively. The MS of 2 showed the [M]+ ion at m/z 320 (65%). Fur- thermore, compound 2 was allowed to react with hydrazine hydrate in ethanolic solution using piperidine as a catalyst yielding 2-hydrazinyl-2, 3-dihydro-3-phenyl-chromeno[2,3- d] pyrimidin-4-one, 3. The structure of 3 was confirmed based on elemental and spectroscopic analysis. The IR spectrum of 3 showed the presence of absorption bands at υ 1670, 3282 and 3432 cm–1 due to CO, NH and NH2 groups, respectively. The 1H NMR spectrum (DMSO) displayed three signals at δ 4.82, 5.1 and 8.85 due to NH2, pyrimidine proton and NH respectively, and a multiplet at 6.7 - 7.9 ppm, for aromatic protons. While, the MS of 3 showed m/z at 309 ([M + 3]+, 45%). Compound 3 was utilized as a key intermediate for the synthesis of some new pyrazole derivatives based on mild and efficient reaction of compound 3 with some of labo- ratory available compounds. Thus, Compound 3 reacted with ethyl 3-amino-2,4-dicyanobut-2-enoate in boiling DMF containing a catalytic amount of piperidine. Two isomeric products seemed possible for this reaction 5 or 6, (Scheme 2). Structure 6 ruled out based on analytical and spectroscopic data (IR, 1H NMR and MS). Thus, the IR spectra of the product 5 showed the presence of absorption bands at υ 1699, 2219 and 3422 cm–1 due to C=O, CN and NH2 groups, respectively. Accordingly, the 1H NMR spectrum (DMSO) of the product 5 showed three singlet signals at δ 2.9, 3.5 and 3.7 due to the methylene and two NH2 groups respectively, and a multiplet at 6.7 - 7.9 ppm, due to aromatic protons. The MS of 5 displayed [M]+ at m/z 439 (60%). It is note worth that, trial to cyclise 5 in boiling pyridine was unsuccessful, (Scheme 2). Also, compound 3 reacted with diethyl malonate in boil- ing DMF containing a catalytic amount of piperidine af- forded 1-(3,4-dihydro-4-oxo-3-phenyl-2H-chromeno[2, 3-d]pyrimidin-2-yl)pyrazolidine-3,5-dione 7, (Scheme 3). The structure of 7 was confirmed based on elemental and O NHPh NH O 1 CH(OC2H5)3 O N N OPh OEt 2 H2NNH2.H2O Ethanol/Pip. O N N OPh NHNH2 DMF,Piperidine Schemes 1. Synthetic route to variously substituted 2, 3- dihydro-3 -phe ny lchrome no[2,3 - d] pyrimidin-4-one. Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF Copyright © 2012 SciRes. OJSTA 51 spectroscopic analysis. The IR spectra 7 showed the pres- ence of absorption band centered between υ 1685 - 1705, 3212 cm-1 due to CO and NH functions respectively. Accordingly, the 1H NMR spectrum (CDCl3) of 7 showed three singlet signals at δ 3.2, 5.8 and 8.1 ppm due to the methylene, methine and NH pyrazole ring protons re- spectively, and a multiplet at δ 6.1 - 7.8 for aromatic pro- tons. The MS of 7 displayed m/z at 374 ([M]+, 55%). Similarly, compound 3 was allowed to react with ethyl 2-cyanoacetate, ethyl 3-oxobutanoate and pentane-2,4-dione to afford the corresponding pyrazole derivatives 8 - 10 respectively, (Schemes 3). Likewise, compound 3 was reacted with 2-(ethoxyme- thylene) malononitrile and 2-cyano-N-phenylacetamide gave the corresponding pyrazole derivatives 12, 13a, b respectively, (Scheme 4). While the reaction of com- pound 3 with nitrous acid afforded the tetrazolo[1,5- a]pyrimidine derivatives 11, (Scheme 4). The structure of these compounds was in agreement with analytical and spectroscopic data. The IR spectrum of 11 showed the presence of absorption bands at υ 1699 cm–1 due to CO group. While, the MS of compound 11 showed m/z at 317([M]+, 60%), ( Scheme 4). On the other hand, the hydrazine 3 reacted with aro- matic aldehydes afforded 2-(2-benzylidenehydrazinyl)-3- phenyl-3H-chromeno[2,3-d]pyrimidin-4-one 14, which was cyclized to 5-oxo-1-(4-oxo-3-phenyl-4,10-dihydro- 3H-chromeno[2,3-d]pyrimi-din-2-yl)-3-phenylpyrazolid- ine-4-carbonitrile 16 by treatment with ethyl cyanoace- tate. It seemed that the addition of active methylene hy- drogen of ethyl cyanoacetate to the imino carbon of 14 gave the intermediate 15, which subsequently cyclized via elimination of ethanol molecule yielding 16. The MS of 16 showed m/z at 462 (M + 1)+, 55%. Its IR spectrum revealed absorption bands at υ 3432 cm-1 (NH), 2217cm-1 (CN) and 1670 cm-1 (CO). The 1H NMR spectrum showed a multiplet signals at δ 6.71 - 7.92 for aromatic protons and three singlet signals at δ 3.98, 4.12 and 4.21 due to -NH, the two methine protons of pyrazole ring, respectively, (Scheme 5). The compound 16 was transformed in to the chloro derivatives 17, by treatment with phosphoryl chlo- ride (Scheme 5). The MS of 17 showed at m/z at 478 ([M − 1]+, 16%). The IR spectrum of 17 showed the presence of absorption band at ν 1700 cm-1 due to C=O, 2217 cm-1 due to CN and 3425 cm-1 due to NH function. The 1HNMR of compound 17 showed singlet signals at δ ppm 3.98 and 4.61 due to NH and methane proton of pyrazole ring. Compound 17 was utilized as a useful starting material for the synthesis of a variety heterocycle-isolated coumarin derivatives based on mild and efficient reaction of com- pound 17 with some of laboratory available compounds. Thus, compound 17 reacted with 5-amine-1H-1,2,4-tri- azole in boiling DMF containing a catalytic amount of piperidine afforded 2-(4-amino-3-phenyl-2,3-dihydro-1H- pyrazolo[4,3- e][1 ,2,4 ] tr iazolo[1,5-a]pyrimidin-1-yl)-3-phe- nyl-3H-chromeno[2,3-d]pyrimidin-4(10H)-one 18, (Sche- me 6). Similarly, compound 17 reacted with 5-amino-3- (methylthio)-1H-pyrazole-4-carbonitrile yielded 4-amino- 7-( meth yl th io )-1-(4-oxo-3-p hen yl -4 ,10-dihydro-3H-chro- meno[2,3-d]pyrimidin-2-yl)-3-phenyl-2, 3-dihydro-1H-di- pyrazolo[1,5-a:4’,3’-e]pyrimidine-6-carbonitrile 19. The O N N OPh 6 NNH2 O H2NCN DMF, Piperidine Reflux CN CO2C2H5 H2N NC O N N N Ph O N N NH2 H2N O 3 O N N OPh NN O H2N CN NH2 5 CN Pyridene/Reflux -EtOH 4 Scheme 2. Synthetic rpute to prepare isolated pyrazole derivatives and reaction conditions. Y- CH2CO2Et O N N OPh N Y NH 2 O ON N O Ph 7;X =O, Z = NH 8;X=NH 2,Z=N 9;X=CH 3,Z=N NZ O X DMF, Piperidine Reflux O N N OPh NNCH3 H3C MeMe OO 10 DMF, Piperidine Reflu x 3 Y=-CO 2Et, CN, -COCH3, Scheme 3. Synthetic rpute to isolated oxazole 7 and pyrazole derivatives 8 - 10, and reaction conditions. 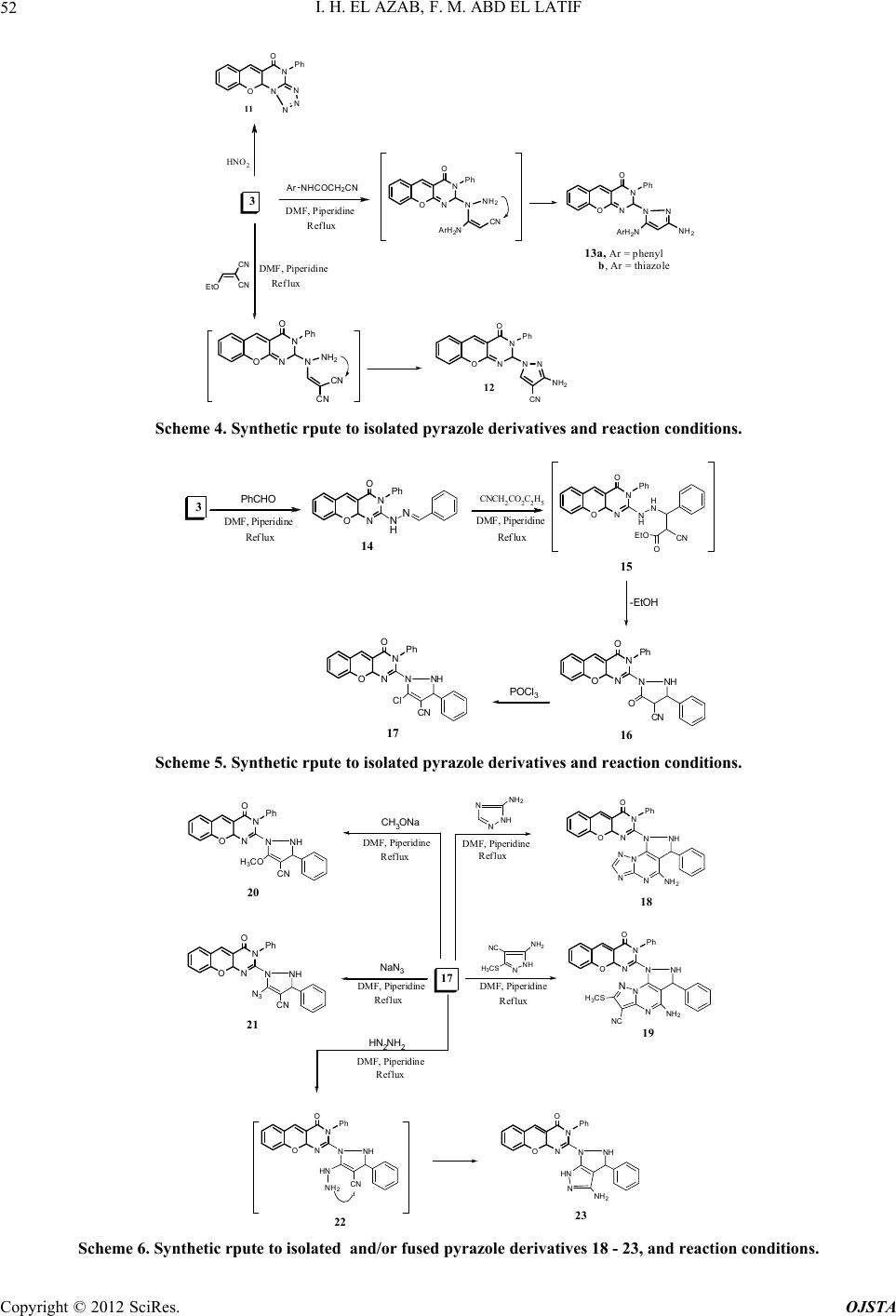 I. H. EL AZAB, F. M. ABD EL LATIF 52 3 HN O2 O N OPh NN N N EtO CN CN O N N O Ph NN NH2 CN 11 12 NHCOCH2CN O N N O Ph NN NH2 ArH2N Ar DMF, Piperidine DMF, Piperidine Reflux Refl ux 13a,Ar= phenyl b, Ar = thiazole O N N O Ph N CN NH2 ArH2N O N N OPh N CN NH 2 CN Scheme 4. Synthetic rpute to isolated pyrazole derivatives and r e ac tion conditions. 3O N OPh NN H NO N OPh NN H H N O N OPh NNNH O CN PhCHO DMF, Piperidine Ref lux CNCH2CO2C2H5 DMF, Piperidine Ref lux POCl 3 O N OPh NNNH Cl CN 14 15 16 17 CN EtO O -EtOH Scheme 5. Synthetic rpute to isolated pyrazole derivatives and r e ac tion conditions. 17 CH3ONa DMF, Piperidine Reflux O N OPh NNNH H3CO CN 20 O N OPh NNNH N3CN 21 NaN3NNH NH2 NC H3CS O N OPh NNNH 19 N N N NH 2 NC H3CS O N OPh NNNH 18 N N N NNH 2 N NNH NH 2 DMF,PiperidineDMF,Piperidine DMF,Piperidine Reflux HN2NH2 Reflux Reflux Reflux DMF, Piperidine O N OPh NNNH HN CN 22 NH2 O N OPh NNNH 23 HN NNH 2 Scheme 6. Synthetic rpute to isolated and/or fused pyrazole derivatives 18 - 23, and reaction conditions. Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF Copyright © 2012 SciRes. OJSTA 53 structure of these compounds was confirmed based on elemental and spectroscopic analysis. (See experimental section). Also, the chloro derivative 17 was converted to the 5-methoxy-1-(4-oxo-3-phenyl-4,10-dihydro-3H-chro- meno[2,3-d]pyrimidin-2-yl)-3-phenyl-2,3-dihy-dro-1H- pyrazole-4-carbonitrile 20 by refluxing with MeONa, (Scheme 6). Structure 20 confirmed based on analytical and spectroscopic data (IR, 1H NMR and MS). Thus, the IR spectra of the product 20 showed the presence of ab- sorption bands at υ 1699, 2219 and 3422 cm–1 due to C=O, CN and NH groups, respectively. Accordingly, the 1H NMR spectrum (DMSO) of the product 20 showed three singlet signals at δ 3.82, 3.98 and 4.61 due to the -OCH3, -NH, and methine protons respectively, and a multiplet at 6.7 - 7.9 ppm, due to aromatic protons. The MS of 20 displayed [M]+ at m/z 475 (62%). We prepared 5-azido-1- (4-oxo-3-phenyl-4,10-dihydro -3H-c hromeno[2,3-d]pyrimi- din-2-yl)-3-phenyl-2, 3-dihydro-1H-pyrazole-4-carboni- trile 21 from the chloro compound 17 by reaction with NaN3 in acetone [21, 22] .The structure of 21 was in agree- ment with analytical and spectroscopic data. The IR spectrum of 21 showed the presence of absorption bands at υ 1699 cm–1 due to CO, 2217 cm–1 due to CN and 3422 cm–1 due to NH groups. While, the MS of compound 21 showed m/z at 486([M]+, 32%). In further reactions, chloro compound 17 on treatment with hydrazine hydrate in DMF containing a catalytic amount of piperidine at reflux temperature afforded 2-(4-amino-3-phenyl-2,3-di- hydro-pyrazolo[3,4 -c]pyrazol-1(6H)-yl)-3-phenyl-3H-chro- meno[2,3-d] pyrimidin-4(10H)-one 23, The formation of 23 may be proceeded via an initial elimination of HCl molecule to give the intermediate 22, which cyclized by nucleophilic addition of the amino function into the cyano group yielded 23, (Scheme 6). The IR spectrum of 23 showed the presence of absorption bands at υ 1670 cm–1 due to (CO) and 3432 - 3455 cm–1 due to (NH, NH2) with the absence of any characteristic absorption of (CN) group. The 1H NMR spectrum of 23 showed four singlet at δ 3.98, 5.21, 6.53 and 9.21 ppm due to the -NH amine, methine of pyrazole ring, amino and imino group respectively, and a multiplet at δ 6.97 - 7.81 for aromatic protons. The MS of 23 displayed m/z at 475 (M+, 15%). On the other hand, the carboxamide 1 easily condensed with acetophenone derivatives in DMF containing a cata- lytic amount of piperidine at reflux temperature to afford chromenochalcones 24a - 24e via elimination of water (Scheme 7). The IR spectrum of 24a showed absorption bands at υ 1665 - 1705 and 3322 due to CO and NH groups respectively, the Ms of 24a showed m/z at 366 ([M]+, 40%). While, the 1HNMR spectrum of 24a (DMSO) showed three singlet signals at δ 4.1, 6.5 and 11.5 ppm due to the NH- amine, methylene and NH-imino groups re- spectively, and a multiplet at δ 7.1 - 7.6 for aromatic pro- tons. Likewise, compound 1 reacts with some aromatic amines to afford the corresponding Schiff's base 25a - 25e, (Sche- me 7). The IR spectrum of 25a showed the presence of absorption bands at υ 3380, due to NH function, with the absence of any characteristic absorption of a C=O group. The 1H NMR spectrum (CDCl3) displayed two signals at δ 4.1, 3.7 and 11.5 due to NH-amine, and NH-imino groups respectively, and a multiplet at 7.1 - 7.6 ppm, for aromatic protons. While, the MS of 25a showed m/z at 339([M]+, 65%). The reactivity of exocyclic C = C conjugated with the carbonyl group in 24a - 24e was investigated by reaction with hydrazines, hydroxylamine, urea, thiourea and some laboratory available active methylene compounds. The nature of the products obtained characterized by elemental and spectroscopic data, indicates that the reaction pro- ceeded via condensation followed by a nucleophilic attack through α, β-unsaturated ketonic group. Pyrazoles/isoxazole derivatives 26a - 26b and 27 were synthesized by treating 24a with equimolar ratios of hydrazine hydrate (or phenyl- hydrzine or hydroxylamine respectively) in ethanol con- taining a catalytic amount of piperidine, (Scheme 8). The structure of these compounds was established based on analytical and spectroscopic data. The IR spectra 26a showed the presence of absorption band centered between υ 3117 - 3293 cm–1 due to NH function, with the absence of any characteristic absorption of a C=O group. Accord- ingly, the 1H NMR spectrum (DMSO) of 26a showed four singlet signals at δ 2.9, 4.1, 7.1 and 11.5 ppm due to the proton at C-4 of pyrazole, NH- amine, NH- hydrazide and =NH imino respectively, and a multiplet at δ 7.2 - 7.8 for aromatic protons. The MS of 26a displayed m/z at 383 ([M+3]+, 55%). The activation exerted by the carbonyl group on the exocyclic double bond in 24a renders them available for the cyclocondensation addition of various amino compounds such as urea and thiourea. Thus, when the chalcone 24a was reacted with an equimolar quantity of urea or thiourea respectively, (Scheme 7) an initial condensation of one amino group with the carbonyl func- tion occurred releasing water, followed by a nucleophilic addition of the second amino group to the double bond forming 4,5-dihydro-4-(2-imino-2H-chromen-3-yl)-6-phe- nyl-4-(phenylamino)pyrimidin-2(1H)-ones, 28a and/or thione 28b. The structures of the synthesized compounds were confirmed by analytical and spectroscopic data (IR, 1H NMR and MS). The IR spectra 28a showed the pres- ence of absorption bands centered between υ 1685 - 1705, 3063 - 3288 cm–1 due to C=O and NH functions, respec- tively. The 1H NMR spectrum (DMSO) of 28a showed four singlet signals at δ 2.9, 4.1, 8.1 and 11.5 ppm due to the proton at C-4 of pyrimidine, NH-amine, NH-amide and NH-imino protons , respectively and a multiplet at δ 7.2 - 7.8 for aromatic protons. The MS of 28a showed a peak at m/z 408 ([M]+, 45%). New pyridine derivatives  I. H. EL AZAB, F. M. ABD EL LATIF 54 have been prepared via condensation of 24a with the active methylene groups of cyanoacetamide, cyanothioace- tamide, 2-cyano-N-p-tolylacetamide, 2-cyanoacetohy-dra- zide and 2-cyano-N-phenylacetamide respectively, followed by a nucleophilic addition of the amino/imino group to the double bond, afforded 1,2,5,6-tetrahydro-6-(2-imino-2H- chromen-3-yl)-2-oxo(thioxo)-4-phenyl-6-(phenylamino) pyridine-3-carbonitriles 29a - 29e , respectively (Scheme 8). The IR spectrum of 29a showed the presence of ab- sorption bands at υ 1689 - 1705, 2220 and 3188 - 3244 cm–1 due to C=O, CN, and NH functions, respectively, and the 1H NMR spectrum (DMSO) displayed three singlet signals at δ 2.9, 4.1 and 8.1 due to the proton at C-5 of pyridine, NH-amine and NH amide, respectively, and a multiplet at 7.2 - 7.6 ppm, respectively. The MS of 29a showed a peak at m/z 432 ([M]+, 65%). Similarly, compound 24a reacted with 2-chloroaceta- mide to afford 3-chloro-5, 6-dihydro-6-(2-imino-2H-chro- men-3-yl)-4-phenyl-6-(phenylamino)pyridin-2(1H)-one 30, (Scheme 9). Also, 2-(cyanomethyl) benzimidazole, reacted with the chalcone 24a, in ethanol using piperidine as a catalyst gave the 2-(2-imino- 2H-chromen-3-yl)-4- phenyl-2-(phenylamino) pyrido[1,2-a]ben-zimidazolo-5- carbonitriles, derivative probably via initial condensation of the activated methylene group with the carbonyl func- tion releasing water, followed by a nucleophilic addition of the NH group at the double bond of compound 24a to afford 31, (Scheme 9). The structure of 31 was assigned based on IR, 1H NMR, and mass spectra. The IR spectrum showed the presence of absorption bands at υ 2220 cm–1 and 3188 - 3244 cm–1 due to CN and NH functions re- spectively, while that due to C=O was completely disap- peared, and the 1H NMR spectrum (CDCl3) displayed three singlet at δ 2.9, 4.1 and 11.5 due to the C-3 proton, NH-amine and NH-imino protons respectively, and a multiplet at δ 6.9 - 7.6 ppm, for aromatic protons. The MS showed the [M]+ ion at m/z 505 (35%). Similarly, the chalcone 24a was reacted with amino phenols in ethanolic solution containing a catalytic amount of piperidine to give 2,3-dihydro-2-(2-imino-2H-chromen-3-yl)-N,4-diphe- nyl-benzo[b][1,4]azepine derivatives , 32a, b (Scheme 9). The IR spectrum of 32a showed the presence of absorp- tion bands at υ 3289 cm–1 due to NH function, the 1H NMR spectrum (DMSO) of 32a displayed three singlet signals at δ 2.9, 4.1 and 11.5 due to the C-3 protons, NH- amine and NH-imino protons respectively, and a multiplet at 6.7 - 7.3 ppm for aromatic protons. While, the MS of 32a showed a peak at m/z 457 ([M]+, 55%). Also, com- und 24a was reacted with 2-aminoprop-1-ene-1, 1,3-tri- carbonitrile and/or ethyl 3-amino-2,4-dicyanobut-2-enoate to afford 2-(dicyano- methylene)-1,2,5,6-tetrahydro-6-(2- imino-2H-chromen-3-yl)-4-phenyl-6-(phenylamino)pyri- dine-3-carbonitrile, 33a and 2-(1-cyano-2-oxobutylidene)- 1,2,5,6-tetrahydro-6-(2-imino-2H-chromen-3-yl)-4-phenyl- 6-(phenylamino) pyridine-3-carbonitrile 33b, respectively. The structure of these products was established based on the IR spectrum of 33a which showed the presence of ab- sorption bands at υ 2219 and 3289 cm–1 due to C=O and NH functions, respectively. The 1H NMR spectrum (DMSO) of 33a displayed two singlet signals at δ 2.9 and 4.1 due to the C-3 protons and NH-amine and a multiplet at 6.7 - 7.3 ppm for aromatic protons. While, the MS of 33a showed a peak at m/z 482 ([M + 2]+, 35%). On the other hand, the new synthesized Schiff’s bases 2-imino-N, N’-diphenyl-2H-chromene-3-carboxamidines, 25a - 25e, were used to prepare new compounds. Thus, cy- cloaddition of chloroacetyl chloride to 25a - 25e pro- ceeded smoothly in ethanolic solution containing triethy- lamine as a catalyst to give the β-lactam, derivatives 34a - 34e, (Sc heme 10 ). The IR spectra of 34a - 34e showed the 24a -e a,Ar= Ph b,Ar=p-(OH)C6H4 c,Ar=p-(NO2)C6H4 d,Ar=o-(NO 2)C 6H4 e, Ar=p-(Cl)C6H4 ONH 1 ArNH2ONH NHPh NAr 25a-e DMF/Reflex DM F/Ref lex Scheme 7 O NHPh Ar Ar COCH3 Scheme 7. Synthetic rpute to chalcones 24a - e and Schiff’s bases 25a - e. Copyright © 2012 SciRes. OJSTA 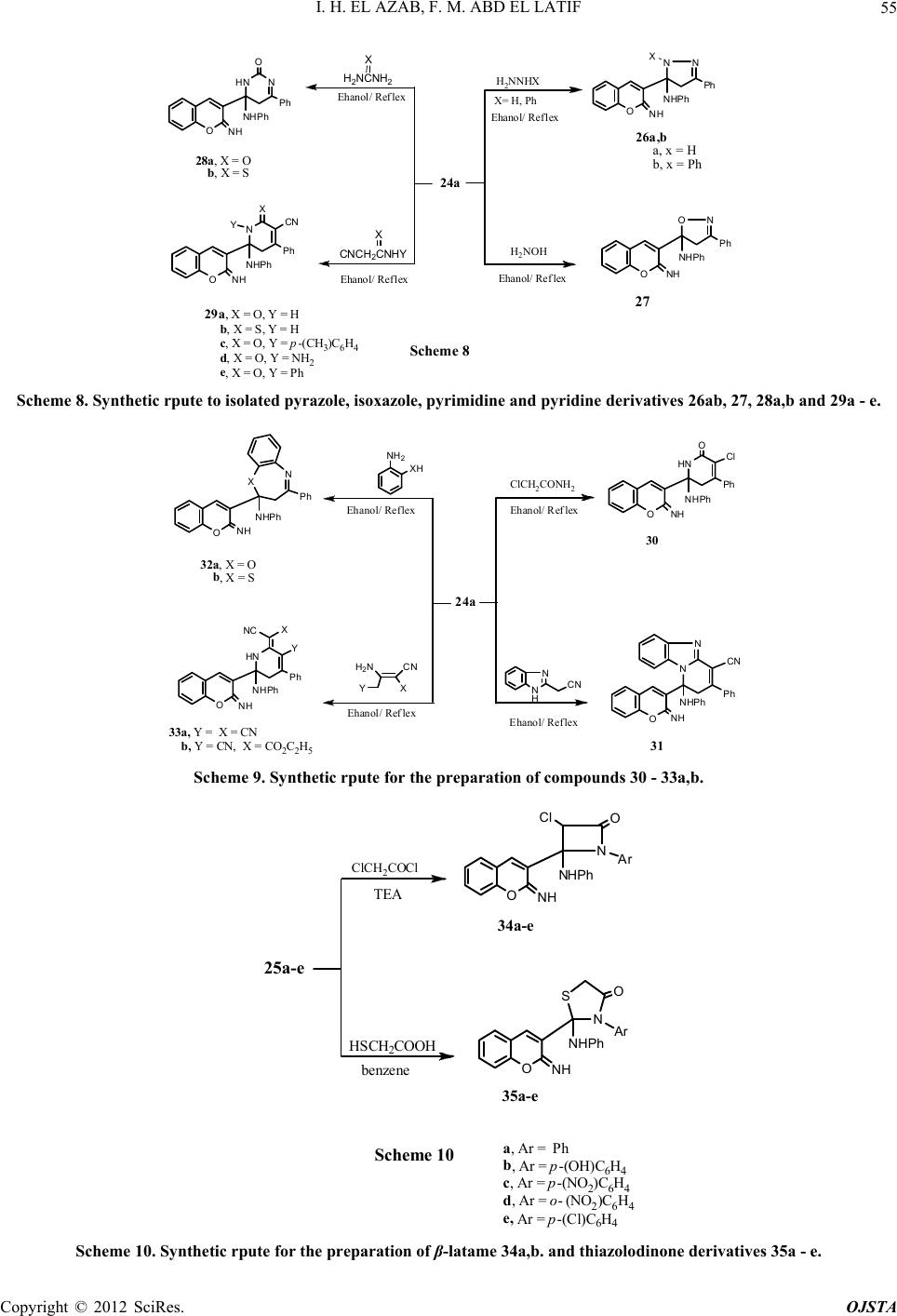 I. H. EL AZAB, F. M. ABD EL LATIF 55 24a H2NNHX X= H,Ph NN NHPh Ph NO NH Ph Ph H2NOH HN N NHPh Ph O 28a,X=O b,X=S ONH ONH ONH 26a ,b a, x = H b, x = Ph 27 N NHPh Ph X ONH CN 29a,X=O,Y=H b,X=S,Y=H c,X=O,Y=p-( CH 3)C6H4 d, X =O,Y =NH 2 e,X=O,Y=Ph Ehanol/Reflex X CNCH2CNHY Y Ehanol/Ref lex Ehanol/Reflex Ehanol/ Reflex Scheme 8 H2NCNH2 XX Scheme 8. Synthetic rpute to isolated pyrazole, isoxazole, pyrimidine and pyridine derivatives 26ab, 27, 28a,b and 29a - e. CN X H2N Y 33a, Y= X=CN b, Y=CN,X=CO 2C2H5 Ehanol/Ref lex HN NHPh Ph ONH Y X NC ClCH2CONH2 Ehanol/Ref lex 30 HN NHPh Ph O ONH Cl N H NCN Ehanol/Reflex N N CN Ph NHPh ONH NH 2XH N X Ph NHPh ONH 32a,X=O b,X=S Ehanol/ Reflex 31 24a Scheme 9. Synthetic rpute for the pr e par ation of compounds 30 - 33a,b. 34a-e ONH 25a-e ONH 35a-e ClCH2COCl HSCH2COOH benzene TEA NAr Cl NHPh O N S Ar NHPh O Scheme 10a,Ar= Ph b,Ar=p-(OH)C6H4 c,Ar=p-(NO2)C6H4 d,Ar=o-(NO 2)C6H4 e, Ar = p-(Cl)C6H4 Scheme 10. Synthetic rpute for the preparation of β-latame 34a,b. and thiazolodinone derivatives 35a - e. Copyright © 2012 SciRes. OJSTA  I. H. EL AZAB, F. M. ABD EL LATIF Copyright © 2012 SciRes. OJSTA 56 presence of absorption bands due to C=O and NH func- tions respectively. The 1H NMR spectrum (DMSO) of 34c showed three singlet signals at δ 4.1, 5.2 and 11.5 ppm for NH-amine, the proton at C3 of the β-lactam unit and NH-imino protons respectively and a multiplet at δ 7.1 - 7.6 for aromatic protons. The MS of 34c showed the [M]+ ion at m/z 460 (40%). Also, the Schiff’s bases 25a - 25e were reacted with equimolar ratio of thioglycolic acid in boiling benzene using water separator system, the thiol group of thioglycolic acid could be added to the imino carbon atoms of Schiff’s bases 25a - 25e followed by smooth cyclization to afford 2-(2-imino-2H-chromen-3- yl)-3-phenyl-2-(phenylamino) thiazolidin-4-ones, 35a - 35e (Scheme 10). The IR spectra of 35 showed bands due to CS, CO and NH groups, respectively. While, the 1HNMR spectrum of 35a (DMSO) showed three singlet signals at δ 3.8, 4.1 and 5.8 for the methylene of thiazolidinone, NH- amine and NH-imino protons respectively and a multiplet at δ 6.8 - 7.8 for aromatic protons. In the MS of 35a showed m/z at 411 ([M-2]+, 35%). 4. Conclusion Despite the several existing methods for the synthesis of chromene derivatives, there still is demand for general strategies, which can efficiently provide variously sub- stituted chromene systems. Thus, this work opened a new avenue for the synthesis of a variety of 2-imino-N-ph- enyl-2H-chromene-3-carboxamide, derivatives. REFERENCES [1] A. M. El-Agrody M. S. Abd El-Latif, N. A. El-Hady, A. H. Fakery and A. H. Bedair, “Heteroaromatization with 4-Hy- droxycoumarin Part II: Synthesis of Some New Pyrano[2, 3-d]pyrimidines,[1,2,4]triazolo[1,5-c]pyrimidines and Py- rimido[1,6-b]-[1,2,4]triazine Derivatives,” Molecules, Vol. 6, No. 6, 2001, pp. 519-527. doi:10.3390/60600519 [2] K. Mazaahir, S. Shilpi, M. R. K. Khalilur and S. T. Sha- ranjit, “Aqua Mediated Synthesis of Substituted 2-Amino- 4H-chromenes and in Vitro Study as Antibacterial Agents,” Bioorganic & Medicinal Chemistry Letters, Vol. 15, No. 19, 2005, pp. 4295-4298. doi:10.1016/j.bmcl.2005.06.041 [3] A. Luke, P. Soizic, S. Brigitte, M. Sylvie, K. Michel, T. C. Stewart, T. François and L. J. Yves, European Journal of Medicinal Chemistry, 2009, pp. 1-9. [4] B. Andrea, V. Nicole, T. Jakob, H. Sonja, B. Stefan and E. M. Christa, “Synthesis and Pharmacological Evaluation of Coumarin Derivatives as Cannabinoid Receptor Antago- nists and Inverse Agonists,” Bioorganic & Medicinal Che- mistry, Vol. 17, No. 7, 2009, pp. 2842-2851. doi:10.1016/j.bmc.2009.02.027 [5] P. Vassiliki, K. K. Ioannis, M. Panagiotis, P. Nicole and A. Ioanna, “Synthesis and Free Radical Scavenging Activity of Some New Spiropyranocoumarins,” Bioorganic & Me- dicinal Chemistry Letters, Vol. 18, No. 21, 2008, pp. 5781- 5784. doi:10.1016/j.bmcl.2008.09.065 [6] N. Tadigoppula, K. Tanvir, N. Shweta, G. Neena and G. Suman, Bioorganic & Medicinal Chemistry, Vol. 13, No. 13, 2005, pp. 6543-6550. [7] K. William, K. Shailaja, S. C. Jiang, H. Zhang, J. H. Zhao, S. J. Jia, L. F. Xu, C. Crogan-Grundya, R. Denis, N. Bar- riault, L. Vaillancourt, S. Charron, J. Dodd, G. Attardo, D. Labrecque, S. Lamothe, H. Gourdeau, B. Tseng, J. Drewea and S. X. Cai, “Discovery of 4-Aryl-4H-chromenes as a New Series of Apoptosis Inducers Using a Cell- and Cas- pase-Based High-Throughput Screening Assay. 2. Struc- ture—Activity Relationships of the 7- and 5-, 6-, 8-Posi- tions,” Bioorganic & Medicinal Chemistry Letters, Vol. 15, No. 21, 2005, pp. 4745-4751. doi:10.1016/j.bmcl.2005.07.066 [8] G. Danielle, V. Samuel, L. M. Vijay, C. Frédéric, V. Guillaume, M. Florence, J. Philippe and G. René, “The Synthesis of New, Selected Analogues of the Pro-Apop- totic and Anticancer Molecule HA 14-1,” Tetrahedron Letters, Vol. 49, No. 20, 2008, pp. 3276-3278. doi:10.1016/j.tetlet.2008.03.070 [9] T. El-Sayed Ali, S. A.-A. Abdel-Aziz, H. M. El-Shaaer, F. I. Hanafy and A. Z. El-Fauomy, Turkish Journal of Chem- istry, Vol. 32, 2008, pp. 365-374. [10] M. K. Mostafa, H. F. A. Ashraf, A. E. Fathy and M. E. Ahmed, IL Farmaco, Vol. 57, 2002, pp. 715-722. [11] A. G. Martinez and L. J. Marco, “Friedländer Reaction on 2-Amino-3-cyano-4H-pyrans: Synthesis of Derivatives of 4H-Pyran [2,3-b] Quinoline, New Tacrine Analogues,” Bioorganic & Medicinal Chemistry Letters, Vol. 7, No. 24, 1997, pp. 3165-3170. doi:10.1016/S0960-894X(97)10165-2 [12] C. P. Dell and C. W. Smith, European Patent No. EP537949, 1993. [13] R. L. Dorta, A. Martin, E. Suárez and C. Betancor, “En- dipitous Acid-Catalyzed Rearrangement of 13-Methoxy- 1,6,8-trioxadispiro[4.1.5.3]pentadecane to 3-Chroman-5- ylpropan-1-ol,” The Journal of Organic Chemistry, Vol. 62, No. 7, 1997, pp. 2273-2274. doi:10.1021/jo962065j [14] Y. S. Vyacheslav, S. M. Vladimir and A. I. Roman, “Re- actions of 3-(Polyfluoroacyl)chromones with Hydroxyl- amine. The First Synthesis of 3-Cyano-2-(polyfluoroal- kyl)chromones,” Tetrahedron Letters, Vol. 47, No. 48, 2006, pp. 8543-8546. doi:10.1016/j.tetlet.2006.09.136 [15] D. Brian, Z. Liren, T. Junko, P. Joseph, H. Sarah, C. Brett, E. H. Christopher, W. Jenny, N. Christi, M. Ajay, S. David, W. Warren and S. G. Val, Bioorganic & Medicinal Che- mistry Letters, Vol. 16, 2006, pp. 4237-4242. [16] Y.-W. Guo, Y.-L. Shi, H.-B. Li and M. Shi, “Reactions of Salicyl N-Tosylimines or Salicylaldehydes with Diethyl Acetylenedicarboxylate for the Synthesis of Highly Func- tionalized Chromenes,” Tetrahedron, Vol. 62, No. 25, 2006, pp. 5875-5882. doi:10.1016/j.tet.2006.04.011 [17] Y. Seiji, M. Mikiko, M. Yohei, M. Masahiro and H. Yo- shiro, Tetrahedron Letters, Vol. 45, 2004, pp. 6971-6973. [18] E. H. E. Galal and H. H. E. Ahmed, “Activated Nitriles in Heterocyclic Synthesis. Novel Synthesis of 5-Imino-5H- [1]benzopyrano[3,4-c]pyridine-4(3H)-thiones and Their Oxo Analogues,” Bulletin of the Chemical Society of Ja- pan, Vol. 63, No. 4, 1990, pp. 1230-1232.  I. H. EL AZAB, F. M. ABD EL LATIF 57 doi:10.1246/bcsj.63.1230 [19] M. M. Rafat, Z. S. Hoda and H. E. Mohamed, Gazzetta Chimica Italiana, 1992, 122. [20] M. K. Sergiy, E. B. Igor, M. S. Konstantyn, P. C. Valentyn and V. B. Yaroslav, “A New Pathway to 3-Hetaryl-2-oxo- 2H-chromenes: On the Proposed Mechanisms for the Re- action of 3-Carbamoyl-2-iminochromenes with Dinu- cleophiles,” Molecules, Vol. 5, No. 10, 2000, pp. 1146- 1165. doi:10.3390/51001146 [21] S. Andrea, V. Daniel, L. André and F. Ľubomír, ARKIVOC, Vol. 6, 2001, pp. 122-128. [22] T. Steinfuhrer, A. Hantschmann, M. Pietsch and M. Weis- senfels, Liebigs Ann. Chem., Vol. 23, 1992. Copyright © 2012 SciRes. OJSTA
|