Journal of Cancer Therapy
Vol. 3 No. 6 (2012) , Article ID: 25482 , 11 pages DOI:10.4236/jct.2012.36149
Breast Cancer Therapies Present and Future
![]()
1Experimental Therapeutics, Department of Pathology and Laboratory Medicine, BC Cancer Agency, Vancouver, Canada; 2Department of Biology, Langara College, Vancouver, Canada; 3Neuro-Oncology Branch, National Cancer Institute, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, USA.
Email: *edwardsli@mail.nih.gov
Received September 1st, 2012; revised October 2nd, 2012; accepted October 13th, 2012
Keywords: Breast Cancer; Brain Metastasis; Drug Combinations
ABSTRACT
Significant advances in breast cancer treatment have been made where it is now possible to treat localized disease to a curable state. However, for approximately 30% of women with primary disease, metastatic breast cancer (MBC) or recurrent disease, treatment has remained challenging. Major obstacles in the effective treatment of breast cancer in these populations include: 1) the molecular heterogeneity of the disease; 2) treatment of MBC and more specifically brain metastasis; and 3) defining combination therapies that address the evolution of resistance with disease relapse. The acknowledgement of these difficulties has led to an effort to further understand the roadblocks to therapy with the anticipation that more appropriate treatments will result. Here we describe the current state of breast cancer treatment, and the potential for improved therapy.
1. Introduction
Breast cancer is the leading cancer diagnosis in women in both North America and Europe [1]. Although advances in early detection and treatment have led to a decrease in the overall mortality rate [2], our initial victory, alarmingly, has revealed major roadblocks to the treatment of breast cancer. This illustrates that our current understanding of breast cancer as a disease is not yet complete. One such roadblock is the revelation that breast cancer is a collection of diseases and not a single entity, second, surprisingly, we are also observing an increase in the incidence of brain metastasis, of which, breast cancer accounts for 15% - 25%, second only to lung cancer in metastasis to the brain [3]. Finally, it is clear that using a single drug or drug regimen to treat all patients is not possible. Rather, researchers and clinicians are beginning to see the value in personalizing breast cancer treatment. Already, molecular markers such as the Estrogen receptor (ER) and Her2/neu have been successfully targeted in patients with positive disease, using drugs such as tamoxifen and trastuzumab respectively. However, standard treatments fail in patients with metastatic breast cancer (MBC) and disease relapse. We are seeing an aging population of patients in whom early detection/diagnosis and better treatment modalities have increased disease free survival time. However, for many of these patients with relapse, it is not uncommon for the disease to evolve a resistance to existing drugs. Treating breast cancer patients without regard to the above roadblocks can ultimately influence clinical outcome [4].
Here we assess the current and potential future prospects for breast cancer therapy, and consider the strengths and weaknesses of therapy in the context of the biological pitfalls that breast cancer harbors. These include breast cancer heterogeneity, brain metastasis, and treatment resistance. We discuss how effective the current breast cancer treatments are and what the potential new treatments may be. In conclusion, we demonstrate how a more informed rationale in determining drug combination therapy will be beneficial for the development of future treatments.
2. Existing Drug Regimens
Clinically, the choice of therapies for a particular patient is determined by the patient’s age, health, stage of disease and prior treatment. Additionally, tumor size, nodal involvement, hormone receptor (HR) status, and Her2/ neu expression play very important roles in determining the use of chemotherapy, endocrine therapy and/or targeted therapies. Table 1 summarizes the clinically available regimens for the treatment of breast cancer patients, which include first, second and third generation nonspecific cytotoxic agents, endocrine therapies, and specific biological agents. Patients presenting with HR-
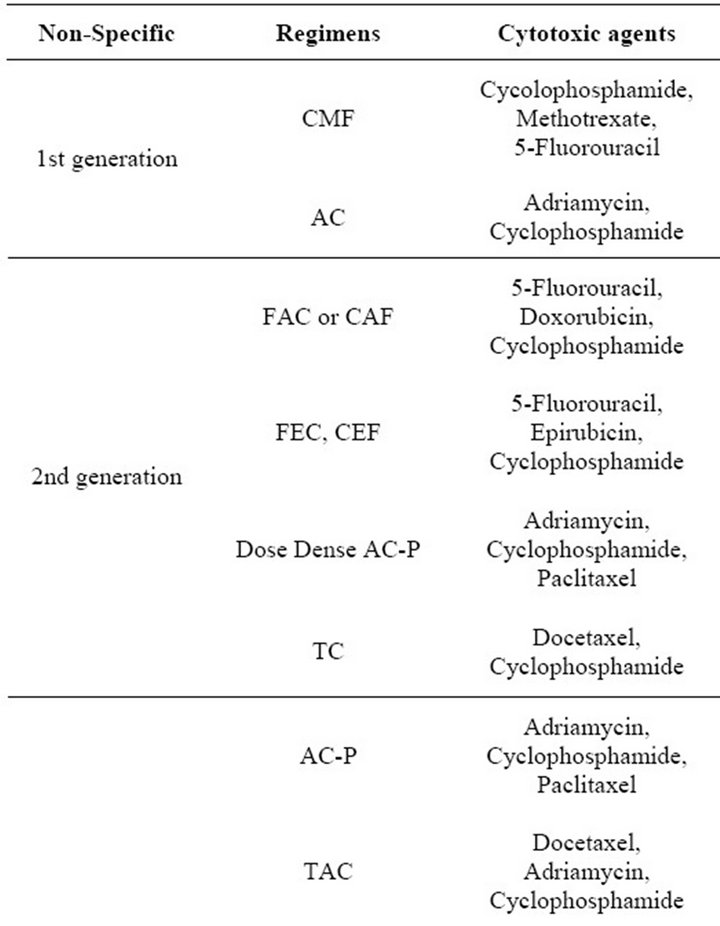
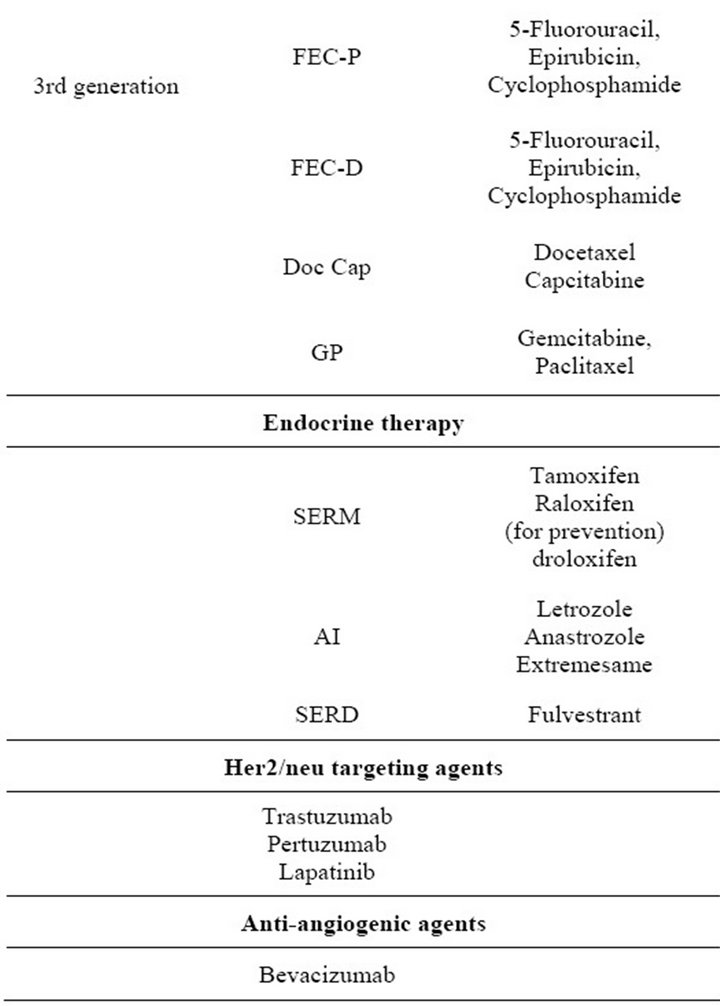
Table 1. Drug therapy available for patients with breast cancer.
negative disease, progression on endocrine therapy or metastatic disease are typically treated with nonspecific anthracyclineand/or taxane-based chemotherapeutic regimens [5,6]. Neo-adjuvant chemotherapy includes anthracyclines (doxorubicin or epirubicin) followed by surgery and radiation. Adjuvant chemotherapy is more variable, but still anthracycline or taxane based [5,6]. ER positivity is a predictive marker for endocrine therapy. Options for endocrine therapy in breast cancer patients include Selective Estrogen Response Modifiers (SERMs), aromatase inhibitors (AIs), or Selective Estrogen Receptor Down-regulators (SERDs) [5-7]. Selection of endocrine therapy depends on menopausal status and concern about possible side-effects.
In addition to the non-targeted cytotoxic agents described above, breast cancers are also treated with targeted agents. The approved targeted treatments available for breast cancer are in the form of antibodies or small molecule inhibitors that antagonize Her2/neu or vascular endothelial growth factor (VEGF). It is estimated that 25% of breast cancer patients have tumors which overexpress the receptor tyrosine kinase Her2/neu [8,9]. Pharmacologic agents or therapeutic antibodies targeting Her2/neu include trastuzumab (targeting Her2/neu positive tumours) [10-12], and lapatinib (targeting both Epidermal growth factor receptor (EGFR) and Her2/neu signalling) [11,13-15]. For patients with Her2/neu overexpression, trastuzumab is a first-line treatment that is typically used in combination with non-anthracyclinebased chemotherapies such as the taxanes: paclitaxel and more commonly docetaxel, or vinorelbine, capecitabine, platinum compounds or gemcitabine [16,17]. In 2007 lapatinib was approved for use in patients with advanced breast cancer pretreated with trastuzumab or anthracyclines [18].
VEGF is associated with poor prognosis and is clearly associated with angiogenesis [19,20]. Therefore, agents have been designed to suppress angiogenesis (e.g. bevacizumab which targets VEGF [11,21-23] or sunitinib, which is a small molecule tyrosine kinase inhibitor that targets the VEGF receptors 1, 2 and 3 [21,22,24-26]) and have been considered for use as part of an adjuvant treatment regimen [27-31]. Currently only bevacizumab is approved for use in breast cancer patients.
Dual EGFR and Her2/neu inhibitors are also under development. These include, afatinib [32,33] and neartinib [34,35], which are second generation tyrosine kinase inhibitors (TKIs) specific for Her2/neu and EGFR.
Aflibercept, which is designed to bind VEGF-A and B, has recently completed a phase I clinical trial of 54 patients and has been recommended for further study in breast cancer [36]. Finally, multi-targeted TKIs such as sunitinib (VEGFR, PDGF, KIT) [37,38], sorefinib (Raf, Flt3, c-kit, RET, VEGFR-2, -3 and PDGF) [39,40], axitinib (VEGFR-1, -2 -3, PDGFR, cKit) [41], vandetanib (VEGFR, EGFR, RET) [42], pazopanib (VEGFR-1, -2, -3, PDGFRa/b, c-kit) [43] are approved for use in some cancers and are currently being evaluated in the context of breast cancer clinical trials.
3. Novel Targeted Therapy and Personalized Medicine
In the age of gene expression analysis, it is becoming increasingly clear that breast cancer is a heterogeneous disease. Breast cancer is now recognized not as a single entity, but as at least five molecularly distinct neoplastic disorders; 1) normal like; 2) triple-negative or basal-like, (TNBC); 3) Her2/neu-positive; 4) luminal-A; and 5) luminal-B breast cancer [44-46]. Presently, this molecular distinction is aiding doctors and their patients in making treatment decisions. However, as gene expression profiling is becoming more prolific, even this five subtype system is rapidly becoming limited. New, more personalized therapy is being developed on the basis of molecular subtypes, mechanisms of carcinogenesis and epigenetic changes seen in breast cancer. Selected examples of novel targets currently being evaluated using small molecule inhibitors or siRNA are discussed below.
3.1. Phosphatase and Tensin Homolog Deleted on Chromosome 10 (PTEN)/ Phosphatidylinositol 3-Kinase (PI3K)/AKT Pathway Inhibitors
PTEN is a dual protein/lipid phosphatase and its main substrate phosphatidyl-inositol 3,4,5 triphosphate (PIP3) is the product of PI3K activity. PI3K is downstream of Insulin like growth factor (IGF-1) and Her2/neu, and leads to the activation of AKT and mTOR, among other signaling pathways in a cell. PI3K/AKT is involved in growth, survival, motility, angiogenesis, epithelial to mesenchymal transition (EMT), metabolism and drug resistance (anti-ER resistance and anti-Her2 resistance). Numerous studies show that the PI3K/AKT pathway signaling is at the heart of many phenotypic changes in breast cancer as well as other cancers [47,48]. Greater than 70% of breast cancers are observed to have mutations, amplifications, deletions, methylations, and post-translational modifications in this pathway [48]. These changes can be the result of PTEN loss or mutations that activate this pathway. Targeting the PI3K/AKT pathway can take many forms. There are several TKIs that are already FDA approved for use clinically or in clinical trials that can act as inhibitors of the PI3K pathway at the receptor level (trastuzumab, lapatanib). Isoform specific (CAL- 101) and pan-PI3K (SF1126, BKM120, GDC-0941, XL- 147) inhibitors are being investigated and several are in phase I and II clinical trials. Mammalian target of rapamycin (mTOR) is a serine-threonine kinase member of the cellular PI3K pathway, which is involved in transcriptional/translational control, cell cycle progression and survival. mTOR is activated by Her2/neu, ER and IGF signaling. Inhibition of mTOR has received a lot of attention and several small molecule inhibitors and antibodies have been developed. These include rapamycin analogs, or rapalogs, such as everolimus (RAD001) and antibodies against mTOR such as temsirolimus and ridaforolimus. Everolimus has been studied in combination with letrozole in women with operable ER-positive breast cancer as a neo-adjuvant therapy. Everolimus was able to increase the efficacy of letrozole in this patient population where the response rate for the combination was 11% higher than in single agent letrozole therapy [49]. Pan-PI3K inhibitors with dual PI3K/mTOR inhibitory activity such as XL-765, SF-1126, BEZ-235, GDC-0941, and GSK1059615 are being pursued as a possible targeted treatment in vitro and in vivo. Specific AKT inhibitors are also available and these include AT-13148 and A-443654, MK2206, GSK690693. Our laboratory has extensively worked with ILK inhibitors. The role of ILK in the P13K pathway is somewhat controversial, but it is widely accepted that ILK is upstream of AKT in the PI3K pathway and acts to phosphorylate AKT on serine 473. Although clinical studies are not underway, the ILK inhibitor QLT0267 has been evaluated in preclinical models of breast cancer with promising results [50,51].
3.2. Integrin Linked Kinase (ILK)
ILK functions as a scaffold protein associated with integrins [52-56], as well as a serine/threonine protein kinase localized to focal adhesions (FAs) [57-60]. In both cases the endogenous role of ILK is in signalling pathways involved in cell-matrix interactions. In a normal cell, ILK is required to maintain tissue homeostasis. ILK has been associated with multiple signal transduction cascades involving downstream molecules such as AKT, GSK-3, HIF-1 alpha, Wnt, E-cadherin, TWIST and VEGF, pathways [55]. These pathways are known regulators of key cancer processes such as EMT, angiogenesis and apoptosis. Over-expression or increased activity of ILK in epithelial cells is related to epithelial-mesenchymal transition (EMT) [61-64] and deregulated growth [55,65-67], while targeted inhibition of ILK induces apoptosis and cell cycle arrest [55,56,68,69]. ILK is an attractive therapeutic target and methods to silence ILK expression (e.g. use of siRNA targeting ILK mRNA) or to inhibit its kinase activity should prove valuable as a tool in the treatment of a variety of human cancers, including breast cancer [55,56,66,67,70-73]. Our own lab was able to show that silencing of ILK using siRNA in breast tumor cells leads to attenuation of PI3K signalling as indicated by a decrease in AKT phosphorylation [50]. More recently we were able to suppress Her2/neu expression in breast cancer cell lines through the inhibition of ILK using siRNA [51]. This was later confirmed by another lab [74].
3.3. Epigenetic Modulation as a Future Treatment of Breast Cancer
In the same way that genome-wide studies for somatic mutations are changing the landscape of marker identification and leading to high-throughput analyses of candidate genes, large-scale epigenomic methods have been developed for the characterization of global epigenetic alterations in cancer. Methods such as Quantitative multiplex-methylation specific PCR or genome-wide profiling for methylations or histone modification are commonly used for epigenomic studies [75]. Epigenetic modifications that alter gene expression include DNA methylations, histone modification (acetylations, methylations, phosphorylations, ubiquitination, nucleosomal remodelling), polycomb/trithorax protein complexing, and small non-coding or anti-sense RNA (microRNA) expression and activity. Many compounds have been discovered to target proteins that control DNA methylation, histone acetylation, and histone methylation. Some of them are already being used clinically with encouraging effects. Some research has focused on the role of epigenetic gene silencing, important in the pathogenesis of breast cancer, in which acetylation or deacetylation of DNA modifies the expression of tumor-suppressing genes. Current research in epigenetics as it relates to breast cancer is briefly described.
3.4. Histone Modifications: Histone Deacetylase Inhibitors
Histone Deacetylase (HDAC) removes acetyl groups from histones which results in chromatin coiling that effectively inhibits transcription. HDAC based gene regulation is seen in genes that are important to cell survival, proliferation, differentiation and apoptosis. In addition to its job in chromatin condensation HDACs also play a role in recruiting transcription factors to promoter sites, and HDAC has been found complexed with the methyl CpG binding domain (MBD) family of proteins at sites of DNA methylation. In cancer, HDACs can be abnormally expressed leading to hypoacetylation and can modulate chromatin configurations leading to the silencing of genes involved in tumor suppression. In breast cancer, abnormal histone modification in combination with DNA hypermethylation is frequently associated with epigenetic silencing of tumor suppressor genes and genomic instability, therefore HDAC inhibitors (HDACi) could represent a new treatment option for breast cancer. HDACi, including both class-specific inhibitors (entinostat and romidepsin) and pan-HDACi (vorinostat, belinostat and panobinostat), have been developed recently. Vorinostat and romidepsin are two examples of HDACi that are clinically approved for use in T-cell lymphoma and are now being considered for breast cancer. A phase I trial confirmed an acceptable toxicity profile and recommended phase II trial escalating doses of the HDACi vorinostat with weekly doxorubicin [76]. In 2009 Munster et al., illustrated that HDACi are associated with a transcriptional down-regulation in ER positive cells while in ER-negative cells HDACi re-establish expression making them sensitive to anti-estrogen therapies [76]. In 2011 Muster went on to show that in patients with ER positive MBC on endocrine therapy, these patients may benefit with the combination of vorinostat and tamoxifen which can reverse hormone therapy resistance [77]. Ramaswamy et al., showed that a combination of vorinostat with paclitaxel and bevacizumab used in patients with MBC induced a partial or complete response in more than 50% of patients [78]. Other HDACi including entinostat and panobinostat are currently in phase I or II clinical trials.
4. Brain Metastasis and Breast Cancer
Central nervous system (CNS) tumors due to metastasis now account for the majority of tumors in the CNS [79], outnumbering primary brain tumors 10:1 [80]. Although the incidence of brain metastasis from breast cancer (BMBC) is estimated to be 10% to 16% [81,82] this has now been shown to be a gross underestimation of the true incidence which is closer to 30% [83] when autopsy patients are included in BMBC. Evidence suggests that common features of patients who experience brain metastasis from breast cancer include tumors that are triple negative (lacking receptors for progesterone, estrogen and Her2/neu) or the molecular subtype to triple negative breast cancer, basal-like breast cancer or Her2/neu positive breast cancers. Her2/neu patients appear to be at the greatest risk with Her2/neu positive patients accounting for 25% of the population [84]. Further risk factors associated with the development of BMBC include but are not limited to, high p53 levels, number of metastatic disease locations, patient age (median 40), node positive and tumor relapse starting in bone or the liver [85-87]. Within 2 to 3 years of a patient diagnosis of a primary tumor, the appearance of CNS metastasis becomes apparent [88]. Interestingly, breast cancer patients develop brain metastasis after the development of metastasis to the liver, lung and bone [89]. Estimates suggest that almost half the patients with BMBC present with headache [90]. Further clinical manifestations range from vomiting, nausea, decreased cognitive function to ataxia and seizures [91]. Given the restrictive nature of the blood-brain barrier (BBB)-(the filter that separates the circulating blood and restricts entry of various molecules while allowing the specific entry of certain metabolites into the brain) it is still unclear how breast cancer cells gain entry to the brain. On the other hand, several factors are known to be involved in the promotion of breast cancer invasion and migration, these include platelet derived growth factor (PDGF), interleukin (IL)-6, tumor necrosis factor (TNF)-α, IL-3 and insulin like growth factor (IGF-1) [92]. BMBC is a late stage event in breast cancer and traditionally, the survival rate is 20% after 1 year [93].
5. Treatment of Brain Metastasis
The use of chemotherapy in the treatment of BMBC has been largely ineffective, reducing chemotherapeutic treatment to a palliative role [94]. The ineffectiveness of chemotherapy is thought to be due to two reasons: 1) the presence of the BBB hinders access to the tumor; 2) BMBC occurs late in the development of breast cancer when chemoresistance has evolved. Therefore, the standard of treatment for BMBC is radiotherapy, surgery or radiosurgical treatments. Chemotherapy is used as a last attempt at care when radiotherapy and surgical options fail. Although chemotherapy is not a first line treatment in BMBC, improved patient survival has been demonstrated when chemotherapy was used as an adjuvant to radiotherapy [95]. The BBB has also determined the choice of chemotherapeutic agents that can be used in the treatment of BMBC. Drugs such as paclitaxel, vincristine, docetaxel and doxorubicin are drugs that when used in normal doses are largely ineffective, due to a poor showing of drug concentration in the brain [96]. The entry of chemotherapeutic agents into the brain is based upon criteria such as molecular weight, drug solubility and the drugs ability to bind to plasma proteins [97]. Given this information, drugs such as Tremodar®, topotecan and capecitabine have been shown to cross the BBB at clinically relevant concentrations [98,99].
5.1. Tremodar®
Tremodar® is an alkylating agent used in the treatment of primary brain tumors i.e. glioblastoma (GBM). Tremodar® (temozolomide) is well tolerated by patients even with longer dosing schedules. In addition, temozolomide has been shown to improve patient survival in the treatment of GBMs in conjunction with radiotherapy [100]. However, temozolomide for the treatment of BMBC has been shown to have no effect when used as a single agent, and this was shown in a phase II clinical trial [101]. Although it should be noted that the number of patients in these studies was small, other studies also confirmed no improvement in survival with temozolomide treatment against BMBC [102]. It has since been observed that temozolomide is an alkylating agent and breast cancers highly expresses O(6)-methyl-guanine-DNA-methyl-transferase (MGMT) which functions to repair DNA damage resulting from alkylating agents. Thus for at least breast cancer, there is likely a high degree of intrinsic resistance to temozolomide [103]. Temozolomide appears to give the best results when used in some type of combination therapy. For example, temozolomide, in combination with whole brain radiotherapy (WBRT) resulted in 79% with a positive response and an increase in patient survival [104,105]. Another combination using temozolomide which is proving to be effective in BMBC is temozolomide and capecitabine. With an overall response rate of 18%, this combination was well tolerated and cognitive function did not decrease in patients that had stable disease or responded to treatment. It should be noted that all patients were treated with WBRT prior to temozolomide and capecitabine combination therapy [106]. It would be of interest to do a head to head comparison of temozolomide after WBRT or temozolomide and capecitabine after WBRT.
5.2. Capecitabine
Capecitabine is an anti-metabolite that is truly a last line of defense drug. It has been used in combination (See temozolomide) and has also been used as a single agent. Interestingly, one study indicated that in recurrent or refractory BMBC, capecitabine as a single agent after WBRT resulted in an overall response rate of 45% with a median progression free survival of 7.3 months [107]. Capecitabine has also been shown to have some effect even after other chemotherapeutic regimens had failed [108]. Although there is evidence of the effects of capecitabine, it has not been shown in humans to cross the BBB, however this has been shown in mice. This is an important caveat particularly when capecitabine is used in combination therapy, as data is needed to determine if the effects of combination therapy are truly the result of both drugs that cross the BBB or perhaps an interaction that enhances or that may hinder the ability of one drug to cross the BBB.
5.3. Topotecan
Topotecan is a derivative of camptothecin that inhibits the activity of topoisomerase I. Although topotecan does appear to cross the BBB very well, concrete studies of the use of topotecan for BMBC still need to be performed. The majority of studies with topotecan have been in a combination regimen, however, these studies have had very low numbers of patients that only have CNS involvement due to metastasis. The ineffectiveness of chemotherapeutic agents with respect to BMBC is largely due to the BBB, followed closely by chemoresistance due to late stage disease progression. Chemotherapeutic agents that due cross the BBB have not been sufficiently studied in clinical trials or with sufficient patient numbers to determine the true effectiveness of these agents. This also holds true for more targeted therapeutic agents such. Drug studies and clinical trials that focus on CNS involvement due to metastasis and a concern for BMBC as the focus would greatly enhance our understanding, particularly for exploring options such as targeting the micro-environment and angiogenesis in BMBC.
6. Defining Drug Combination Therapies
Therapeutic approaches in the treatment of breast cancer have always included combinations whether combinations of treatment modalities (surgery, radiation, and systemic control using hormonal treatment or chemotherapy or both) or the combination of systemic therapies which may include combinations of non-specific chemotherapeutic agents, chemotherapeutic agents and targeted biological agents or non-specific biological agents in combination. The use of multiple drugs, in theory, helps to avoid the development of resistant subpopulations and makes the eradication of a range of different malignant cell populations feasible especially in the context of such a heterogeneous disease such as breast cancer. Combination therapies may also help to resolve the issue with targeted therapies that show redundancies. Clinicians and scientists have understood these benefits based on empirical data demonstrating improved treatment outcomes when using multiple drugs. This principle is being applied to targeted agents and only now are researchers beginning to realize the benefits of combining classical chemotherapeutic agents with novel drugs targeting specific molecular pathways. As an example, the combination of Perjeta, trastuzumab and docetaxel chemotherapy was recently approved for use in patients and was able to significantly extend progression-free survival in people with previously untreated Her2/neu-positive MBC compared to trastuzumab plus docetaxel chemotherapy (6.1 month improvement in median progression free survival, 18.5 vs 12.4 months) [109,110].
Analysis of drug interactions is a complicated procedure with multiple assessment strategies available [110, 111]. Widely used strategies to determine whether various drug combinations result in synergistic, antagonist or additive effects include the statistical method, the arithmetic sum, the fractional product concept, the isobologram method, the median effect principle (MEP), and response surface modeling. The MEP method describes the relationship between a measured response within a population of cells (fraction affected (fa) versus the fraction unaffected (fu)) and the fraction of the dose (D) required to achieve a specified effect level and is represented by the formula:
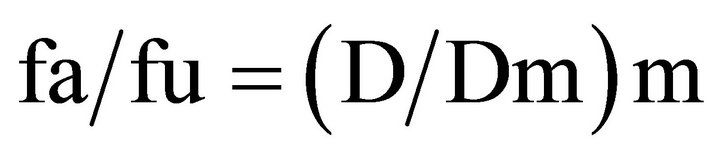
Equation (1) shown above indicates where Dm is the dose required to achieve a 50% effect level and m is a coefficient indicating the sigmoidicity of the dose-effect curve. The right side of the equation 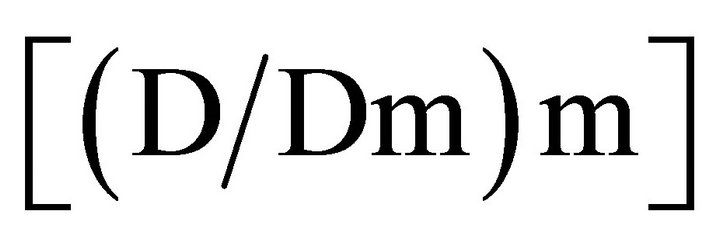 represents the dose, and the left side of the equation [fa/fu] represents the effect of the interaction. This analysis can be used to determine the combination index (CI) which provides a measure of whether the drug-drug interactions are additive (CI = 1), antagonistic (CI > 1) or synergistic (CI < 1). The CI can be calculated at any effect level and the effect used can be derived on the basis of any measured endpoint (e.g. cell viability, cell number, apoptosis induction, intracellular enzyme inhibition, inhibition of VEGF secretion, etc). To determine CI values, a commercially available program CalcuSyn can be used to calculate CI values for a broad range of effect levels and, on the basis of this analysis, fa verses CI plots can be generated. The latter plots are important to consider and recognize that drug-drug interactions are dependent on the effect level. Some drug-drug interactions are highly synergistic but synergy is only observed at low effect levels, while at high effect levels the interaction proves to be antagonistic. It is also now well established that drug-drug interactions are also influenced by drug-drug ratio, drug sequencing, experimental endpoint assessment and experimental conditions. These variables make it very challenging to determine whether a specific drug combination is providing therapeutic effects which are better than would be expected based on the action of the individual drugs (i.e. synergistic).
represents the dose, and the left side of the equation [fa/fu] represents the effect of the interaction. This analysis can be used to determine the combination index (CI) which provides a measure of whether the drug-drug interactions are additive (CI = 1), antagonistic (CI > 1) or synergistic (CI < 1). The CI can be calculated at any effect level and the effect used can be derived on the basis of any measured endpoint (e.g. cell viability, cell number, apoptosis induction, intracellular enzyme inhibition, inhibition of VEGF secretion, etc). To determine CI values, a commercially available program CalcuSyn can be used to calculate CI values for a broad range of effect levels and, on the basis of this analysis, fa verses CI plots can be generated. The latter plots are important to consider and recognize that drug-drug interactions are dependent on the effect level. Some drug-drug interactions are highly synergistic but synergy is only observed at low effect levels, while at high effect levels the interaction proves to be antagonistic. It is also now well established that drug-drug interactions are also influenced by drug-drug ratio, drug sequencing, experimental endpoint assessment and experimental conditions. These variables make it very challenging to determine whether a specific drug combination is providing therapeutic effects which are better than would be expected based on the action of the individual drugs (i.e. synergistic).
The use of CI values also highlights an important practical aspect of selecting drug combinations that combine to provide synergistic effects: synergistic drug combinations can produce effects comparable to those achieved with the single agents, but at substantially lower doses. This interpretation suggests that one value of selecting a synergistic drug combination arises as a result of potential reductions in toxicity. This suggests the potential of also treating patients longer due to a well tolerated drug combination and a greater positive response rate.
7. Conclusion
As our understanding of breast cancer has advanced in the realm of early detection and diagnosis, we have seen better patient outcomes with respect to localized disease. However, better treatments are required, certainly in the context of MBC and BMBC. The many drugs that are currently being used or being studied for the treatment of breast cancer are slowly yielding some benefit. It is becoming increasingly clear that drug combination therapy holds the most promise for patients suggesting our efforts should be concentrated here. Rationally designed combinations of targeted and non-targeted therapies are the cornerstone of novel treatments for breast cancer. For this reason a comprehensive understanding of combination assessment strategies is critical to the continued success in the treatment of this disease.
REFERENCES
- American Cancer Society, “Breast Cancer Facts & Figures 2011-2012,” American Cancer Society, Atlanta, 2012.
- J. Ferlay, et al., “Estimates of the Cancer Incidence and Mortality in Europe in 2006,” Annals of Oncology, Vol. 18, 2007, pp. 581-592. doi:10.1093/annonc/mdl498
- American Cancer Society, “Cancer Facts & Figures 2012,” American Cancer Society, Atlanta, 2012.
- B. Groner and N. E. Hynes, “Unfavorable Drug interactions in Targeted Breast Cancer Therapy,” Cancer Cell, Vol. 18, No. 5, 2010, pp. 401-402. doi:10.1016/j.ccr.2010.10.027
- C. Andreetta, A. M. Minisi, M. Misicoria and F. Puglisi, “First-Line Chemotherapy with or without Biologic Agents for Metastatic Breast Cancer,” Critical Reviews in Oncology/Hematology, Vol. 76, No. 2, 2010, pp. 99-111. doi:10.1016/j.critrevonc.2010.01.007
- V. Guarneri and P. F. Conte, “The Curability of Breast Cancer and the Treatment of Advanced Disease,” European Journal of Nuclear Medicine and Molecular Imaging, Vol. 31, No. 1, 2004, pp. S149-S161. doi:10.1007/s00259-004-1538-5
- C. K. Baumann and M. Castiglione-Gertsch, “Clinical Use of Selective Estrogen Receptor Modulators and Down Regulators with the Main Focus on Breast Cancer,” Minerva Ginecologica, Vol. 61, No. 6, 2009, pp. 517-539.
- W. Dean-Colomb and F. J. Esteva, “Her2-Positive Breast Cancer: Herceptin and Beyond,” European Journal of Cancer, Vol. 44, No. 18, 2008, pp. 2806-2812. doi:10.1016/j.ejca.2008.09.013
- D. J. Slamon, “Proto-Oncogenes and Human Cancers,” New England Journal of Medicine, Vol. 317, No. 15, 1987, pp. 955-957. doi:10.1056/NEJM198710083171509
- M. Kurosumi, “Recent Trends of HER-2 Testing and Trastuzumab Therapy for Breast Cancer,” Breast Cancer, Vol. 16, No. 4, 2009, pp. 284-287. doi:10.1007/s12282-009-0159-z
- N. Normanno, et al., “Target-Based Therapies in Breast Cancer: Current Status and Future Perspectives,” Endocrine-Related Cancer, Vol. 16, No. 3, 2009, pp. 675-702. doi:10.1677/ERC-08-0208
- N. L. Spector and K. L. Blackwell, “Understanding the Mechanisms behind Trastuzumab Therapy for Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer,” Journal of Clinical Oncology, Vol. 27, No. 34, 2009, pp. 5838-5847. doi:10.1200/JCO.2009.22.1507
- J. E. Frampton, “Lapatinib: A Review of Its Use in the Treatment of HER2-Overexpressing, Trastuzumab-Refractory, Advanced or Metastatic Breast Cancer,” Drugs, Vol. 69, No. 15, 2009, pp. 2125-2148.
- J. S. Frenel, et al., “Lapatinib in Metastatic Breast Cancer,” Womens Health, Vol. 5, No. 6, 2009, pp. 603-612. doi:10.2217/whe.09.54
- V. Roy and E. A. Perez, “Beyond Trastuzumab: Small Molecule Tyrosine Kinase Inhibitors in HER-2-Positive Breast Cancer,” Oncologist, Vol. 14, No. 11, 2009, pp. 1061-1069. doi:10.1634/theoncologist.2009-0142
- J. A. Ligibel and E. P. Winer, “Trastuzumab/Chemotherapy Combinations in Metastatic Breast Cancer,” Seminars in Oncology, Vol. 29, No. 3, 2002, pp. 38-43. doi:10.1016/S0093-7754(02)70125-5
- A. Lin and H. S. Rugo, “The Role of Trastuzumab in Early Stage Breast Cancer: Current Data and Treatment Recommendations,” Current Treatment Options in Oncology, Vol. 8, No. 1, 2007, pp. 47-60. doi:10.1007/s11864-007-0008-2
- Q. Ryan, et al., “FDA Drug Approval Summary: Lapatinib in Combination with Capecitabine for Previously Treated Metastatic Breast Cancer That Overexpresses HER-2,” Oncologist, Vol. 13, No. 10, 2008, pp. 1114- 1119.
- P. Manders, et al., “The Prognostic Value of Vascular Endothelial Growth Factor in 574 Node-Negative Breast Cancer Patients Who Did Not Receive Adjuvant Systemic Therapy,” British Journal of Cancer, Vol. 87, No. 7, 2002, pp. 772-778. doi:10.1038/sj.bjc.6600555
- G. W. Sledge Jr., “Vascular Endothelial Growth Factor in Breast Cancer: Biologic and Therapeutic Aspects,” Seminars in Oncology, Vol. 29, No. 3, 2002, pp. 104-110. doi:10.1016/S0093-7754(02)70133-4
- D. F. Hayes, K. Miller and G. Sledge, “Angiogenesis as Targeted Breast Cancer Therapy,” Breast, Vol. 16, Suppl. 2, 2007, pp. S17-S19.
- P. K. Shahi, A. S. Lovelle and G. P. Manga, “Tumoral Angiogenesis and Breast Cancer,” Clinical & Translational Oncology, Vol. 11, No. 3, 2009, pp. 138-142. doi:10.1007/S12094-009-0329-7
- T. A. Traina, “Bevacizumab in the Treatment of Metastatic Breast Cancer,” Oncology, Vol. 23, No. 4, 2009, pp. 327-332.
- L. Q. Chow and S. G. Eckhardt, “Sunitinib: From Rational Design to Clinical Efficacy,” Journal of Clinical Oncology, Vol. 25, No. 7, 2007, pp. 884-896. doi:10.1200/JCO.2006.06.3602
- S. P. Ivy, J. Y. Wick and B. M. Kaufman, “An Overview of Small-Molecule Inhibitors of VEGFR Signaling,” Nature Reviews Clinical Oncology, Vol. 6, No. 10, 2009, pp. 569-579. doi:10.1038/nrclinonc.2009.130
- D. Pytel, T. Sliwinski, T. Poplawski, D. Ferriola and I. Majsterek, “Tyrosine Kinase Blockers: New Hope for Successful Cancer Therapy,” Anti-Cancer Agents in Medicinal Chemistry, Vol. 9, No. 1, 2009, pp. 66-76.
- O. Dizdar and K. Altundag, “Emerging Drugs in Metastatic Breast Cancer,” Expert Opinion on Emerging Drugs, Vol. 14, No. 1, 2009, pp. 85-98. doi:10.1517/14728210802625671
- G. M. Higa, “Breast Cancer: Beyond the Cutting Edge,” Expert Opinion on Pharmacotherapy, Vol. 10, No. 15, 2009, pp. 2479-2498. doi:10.1517/14656560903167957
- G. M. Higa and J. Abraham, “Biological Mechanisms of Bevacizumab-Associated Adverse Events,” Expert Review of Anticancer Therapy, Vol. 9, No. 7, 2009, pp. 999- 1007. doi:10.1586/era.09.68
- P. G. Morris and C. A. Hudis, “Personalizing Therapy for Metastatic Breast Cancer,” Expert Review of Anticancer Therapy, Vol. 9, No. 9, 2009, pp. 1223-1226. doi:10.1586/era.09.89
- P. G. Morris, H. L. McArthur and C. A. Hudis, “Therapeutic Options for Metastatic Breast Cancer,” Expert Opinion on Pharmacotherapy, Vol. 10, No. 6, 2009, pp. 967-981. doi:10.1517/14656560902834961
- N. U. Lin, et al., “A Phase II Study of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker, in Patients with HER2-Positive Metastatic Breast Cancer Progressing after Trastuzumab,” Breast Cancer Research and Treatment, Vol. 133, No. 3, 2012, pp. 1057-1065. doi:10.1007/s10549-012-2003-y
- M. Schuler, et al., “A Phase II Trial to Assess Efficacy and Safety Of Afatinib in Extensively Pretreated Patients with HER2-Negative Metastatic Breast Cancer,” Breast Cancer Research and Treatment, Vol. 134, No. 3, 2012, pp. 1149-1159. doi:10.1007/s10549-012-2126-1
- Y. Ito, et al., “Safety, Efficacy and Pharmacokinetics of Neratinib (HKI-272) in Japanese Patients with Advanced Solid Tumors: A Phase 1 Dose-Escalation Study,” Japanese Journal of Clinical Oncology, Vol. 42, No. 4, 2012, pp. 278-286. doi:10.1093/jjco/hys012
- K. K. Wong, et al., “A Phase I Study with Neratinib (HKI-272), an Irreversible Pan ErbB Receptor Tyrosine Kinase Inhibitor, in Patients with Solid Tumors,” Clinical Cancer Research, Vol. 15, No. 7, 2009, pp. 2552-2558. doi:10.1158/1078-0432.CCR-08-1978
- N. Isambert, et al., “Phase I Dose-Escalation Study of Intravenous Aflibercept in Combination with Docetaxel in Patients with Advanced Solid Tumors,” Clinical Cancer Research, Vol. 18, No. 6, 2012, pp. 1743-1750. doi:10.1158/1078-0432.CCR-11-1918
- C. H. Barrios, et al., “Phase III Randomized Trial of Sunitinib versus Capecitabine in Patients with Previously Treated HER2-Negative Advanced Breast Cancer,” Breast Cancer Research and Treatment, Vol. 121, No. 1, 2010, pp. 121-131. doi:10.1007/s10549-010-0788-0
- J. Bergh, et al., “First-Line Treatment of Advanced Breast Cancer with Sunitinib in Combination with Docetaxel Versus Docetaxel Alone: Results of a Prospective, Randomized Phase III Study,” Journal of Clinical Oncology, Vol. 30, No. 9, 2012, pp. 921-929. doi:10.1200/JCO.2011.35.7376
- J. Baselga, et al., “Sorafenib in Combination with Capecitabine: An Oral Regimen for Patients with HER2- Negative Locally Advanced or Metastatic Breast Cancer,” Journal of Clinical Oncology, Vol. 30, No. 13, 2012, pp. 1484-1491. doi:10.1200/JCO.2011.36.7771
- C. Isaacs, et al., “Phase I/II Study of Sorafenib with Anastrozole in Patients with Hormone Receptor Positive Aromatase Inhibitor Resistant Metastatic Breast Cancer,” Breast Cancer Research and Treatment, Vol. 125, No. 1, 2011, pp. 137-143. doi:10.1007/s10549-010-1226-z.
- H. S. Rugo, et al., “Randomized, Placebo-Controlled, Double-Blind, Phase II Study of Axitinib plus Docetaxel Versus Docetaxel plus Placebo in Patients with Metastatic Breast Cancer,” Journal of Clinical Oncology, Vol. 29, No. 18, 2011, pp. 2459-465. doi:10.1200/JCO.2010.31.2975
- K. Boer, et al., “Vandetanib with Docetaxel as SecondLine Treatment for Advanced Breast Cancer: A DoubleBlind, Placebo-Controlled, Randomized Phase II Study,” Investigational New Drugs, Vol. 30, No. 2, 2012, pp. 681-687. doi:10.1007/s10637-010-9538-8.
- S. K. Taylor, et al., “A Phase II Study of Pazopanib in Patients with Recurrent or Metastatic Invasive Breast Carcinoma: A Trial of the Princess Margaret Hospital Phase II Consortium,” Oncologist, Vol. 15, No. 8, 2010, pp. 810-818. doi:10.1634/theoncologist.2010-0081
- N. R. Bertos and M. Park, “Breast Cancer—One Term, Many Entities?” Journal of Clinical Investigation, Vol. 121, No. 10, 2011, pp. 3789-3796. doi:10.1172/JCI57100
- M. Hergueta-Redondo, J. Palacios, A. Cano and G. Moreno-Bueno, “New Molecular Taxonomy in Breast Cancer,” Clinical & Translational Oncology, Vol. 10, No. 12, 2008, pp. 777-785. doi:10.1007/s12094-008-0290-x
- J. Peppercorn, C. M. Perou and L. A. Carey, “Molecular Subtypes in Breast Cancer Evaluation and Management: Divide and Conquer,” Cancer Investigation, Vol. 26, No. 1, 2008, pp. 1-10. doi:10.1080/07357900701784238
- K. Polyak, “Breast Cancer: Origins and Evolution,” Journal of Clinical Investigation, Vol. 117, No. 11, 2007, pp. 3155-3163. doi:10.1172/JCI33295
- T. W. Miller, B. N. Rexer, J. T. Garrett and C. L. Arteaga, “Mutations in the Phosphatidylinositol 3-Kinase Pathway: Role in Tumor Progression and Therapeutic Implications in Breast Cancer,” Breast Cancer Research, Vol. 13, No. 6, 2011, pp. 224-236. doi:10.1186/bcr3039
- J. Baselga, et al., “Phase II Randomized Study of Neoadjuvant Everolimus plus Letrozole Compared with Placebo plus Letrozole in Patients with Estrogen Receptor-Positive Breast Cancer,” Journal of Clinical Oncology, Vol. 27, No. 16, 2009, pp. 2630-2637. doi:10.1200/JCO.2008.18.8391
- J. Kalra, et al., “QLT0267, a Small Molecule Inhibitor Targeting Integrin-Linked Kinase (ILK), and Docetaxel Can Combine to Produce Synergistic Interactions Linked to Enhanced Cytotoxicity, Reductions in P-AKT Levels, Altered F-Actin Architecture and Improved Treatment Outcomes in an Orthotopic Breast Cancer Model,” Breast Cancer Research, Vol. 11, No. 3, 2009, pp. R25-R40. doi:10.1186/bcr2252
- J. Kalra, et al., “ILK Inhibition Engenders Suppression of Her2/neu Expression through a Pathway Involving TWIST and YB-1,” Oncogene, Vol. 29, No. 48, 2010, pp. 6343-6356. doi:10.1038/onc.2010.366
- S. Dedhar, B. Williams and G. Hannigan, “IntegrinLinked Kinase (ILK): A Regulator of Integrin and Growth-Factor Signaling,” Trends in Cell Biology, Vol. 9, No. 8, 1999, pp. 319-323. doi:10.1016/S0962-8924(99)01612-8
- K. R. Legate and R. Fassler, “Mechanisms That Regulate Adaptor Binding to Beta-Integrin Cytoplasmic Tails,” Journal of Cell Science, Vol. 122, 2009, pp. 187-198. doi:10.1242/jcs.041624
- K. R. Legate, E. Montanez and R. Fussler, “ILK, PINCH and Parvin: The tIPP of Integrin Signaling,” Nature Reviews Molecular Cell Biology, Vol. 7, No. 1, 2006, pp. 20-31. doi:10.1038/nrm1789
- P. C. McDonald, A. B. Fielding and S. Dedhar, “Integrin-Linked Kinase—Essential Roles in Physiology and Cancer Biology,” Journal of Cell Science, Vol. 121, 2008, pp. 3121-3132. doi:10.1242/jcs.017996.
- S. Persad and S. Dedhar, “The Role of Integrin-Linked Kinase (ILK) in Cancer Progression,” Cancer and Metastasis Reviews, Vol. 22, No. 4, 2003, pp. 375-384. doi:10.1023/A:1023777013659
- F. Li, Y. Zhang and C. Wu, “Integrin-Linked Kinase Is Localized to Cell-Matrix Focal Adhesions but Not CellCell Adhesion Sites and the Focal Adhesion Localization of Integrin-Linked Kinase Is Regulated by the PINCHBinding ANK Repeats,” Journal of Cell Science, Vol. 112, 1999, pp. 4589-4599.
- S. N. Nikolopoulos and C. E. Turner, “Integrin-Linked Kinase (ILK) Binding to Paxillin LD1 Motif Regulates ILK Localization to Focal Adhesions,” Journal of Biological Chemistry, Vol. 276, No. 26, 2001, pp. 23499- 23505. doi:10.1074/jbc.M102163200
- Y. Tu, Y. Huang, Y. Zhang, Y. Hua and C. Wu, “A New Focal Adhesion Protein That Interacts with IntegrinLinked Kinase and Regulates Cell Adhesion and Spreading,” Journal of Cell Biology, Vol. 153, No. 3, 2001, pp. 585-598. doi:10.1083/jcb.153.3.585
- [61] Y. Yang, et al., “Structural Basis of Focal Adhesion Localization of LIM-Only Adaptor PINCH by IntegrinLinked Kinase,” Journal of Biological Chemistry, Vol. 284, No. 9, 2009, pp. 5836-5844. doi:10.1074/jbc.M805319200
- [62] E. R. Abboud, et al., “Integrin-Linked Kinase: A Hypoxia-Induced Anti-Apoptotic Factor Exploited by Cancer Cells,” International Journal of Oncology, Vol. 30, No. 1, 2007, pp. 113-122.
- [63] N. Ahmed, et al., “Integrin-Linked Kinase Expression Increases with Ovarian Tumour Grade and Is Sustained by Peritoneal Tumour Fluid,” Journal of Pathology, Vol. 201, No. 2, 2003, pp. 229-237. doi:10.1002/path.1441
- [64] A. Oloumi, T. McPhee and S. Dedhar, “Regulation of E-Cadherin Expression and Beta-Catenin/Tcf Transcriptional Activity by the Integrin-Linked Kinase,” Biochimica et Biophysica Acta, Vol. 1691, No. 1, 2004, pp. 1- 15.
- [65] A. Oloumi, S. Syam and S. Dedhar, “Modulation of Wnt3a-Mediated Nuclear Beta-Catenin Accumulation and Activation by Integrin-Linked Kinase in Mammalian Cells,” Oncogene, Vol. 25, No. 59, 2006, pp. 7747-7757. doi:10.1038/sj.onc.1209752
- [66] G. Radeva, et al., “Overexpression of the Integrin-Linked Kinase Promotes Anchorage-Independent Cell Cycle Progression,” Journal of Biological Chemistry, Vol. 272, No. 21, 1997, pp. 13937-13944. doi:10.1074/jbc.272.21.13937.
- [67] N. Yoganathan, et al., “Integrin-Linked Kinase, a Promising Cancer Therapeutic Target: Biochemical and Biological Properties,” Pharmacology & Therapeutics, Vol. 93, No. 2-3, 2002, pp. 233-242. doi:10.1016/S0163-7258(02)00192-4.
- [68] T. N. Yoganathan, et al., “Integrin-Linked Kinase (ILK): A ‘Hot’ Therapeutic Target,” Biochemical Pharmacology, Vol. 60, No. 8, 2000, pp. 1115-1119. doi:10.1016/S0006-2952(00)00444-5.
- [69] M. S. Duxbury, et al., “RNA Interference Demonstrates a Novel Role for Integrin-Linked Kinase as a Determinant of Pancreatic Adenocarcinoma Cell Gemcitabine Chemoresistance,” Clinical Cancer Research, Vol. 11, No. 9, 2005, pp. 3433-3438. doi:10.1158/1078-0432.CCR-04-1510
- [70] S. Persad, et al., “Inhibition of Integrin-Linked Kinase (ILK) Suppresses Activation of Protein Kinase B/Akt and Induces Cell Cycle Arrest and Apoptosis of PTEN-Mutant Prostate Cancer Cells,” Proceedings of the National Academy of Sciences, Vol. 97, No. 7, 2000, pp. 3207-3212.
- [71] L. A. Edwards, et al., “Integrin-Linked Kinase (ILK) in Combination Molecular Targeting,” Cancer Treatment and Research, Vol. 119, 2004, pp. 59-75. doi:10.1007/1-4020-7847-1_4
- [72] A. B. Fielding and S. Dedhar, “The Mitotic Functions of Integrin-Linked Kinase,” Cancer and Metastasis Reviews, Vol. 28, No. 1-2, 2009, pp. 99-111. doi:10.1007/s10555-008-9177-0
- [73] G. Hannigan, A. A. Troussard and S. Dedhar, “IntegrinLinked Kinase: A Cancer Therapeutic Target Unique among Its ILK,” Nature Reviews Cancer, Vol. 5, No. 1, 2005, pp. 51-63. doi:10.1038/nrc1524
- [74] S. Hehlgans, M. Haase and N. Cordes, “Signalling via Integrins: Implications for Cell Survival and Anticancer Strategies,” Biochimica et Biophysica Acta, Vol. 1775, No. 1, 2007, pp. 163-180.
- [75] S. M. Pontier, et al., “Integrin-Linked Kinase Has a Critical Role in ErbB2 Mammary Tumor Progression: Implications for Human Breast Cancer,” Oncogene, Vol. 29, No. 23, 2010, pp. 3374-3385. doi:10.1038/onc.2010.86
- [76] P. K. Lo and S. Sukumar, “Epigenomics and Breast Cancer,” Pharmacogenomics, Vol. 9, No. 12, 2008, pp. 1879- 1902. doi:10.2217/14622416.9.12.1879
- [77] P. N. Munster, et al., “Phase I Trial of Vorinostat and Doxorubicin in Solid Tumours: Histone Deacetylase 2 Expression as a Predictive Marker,” British Journal of Cancer, Vol. 101, No. 7, 2009, pp. 1044-1050. doi:10.1038/sj.bjc.6605293
- [78] P. N. Munster, et al., “A Phase II Study of the Histone Deacetylase Inhibitor Vorinostat Combined with Tamoxifen for the Treatment of Patients with Hormone Therapy-Resistant Breast Cancer,” British Journal of Cancer, Vol. 104, No. 12, 2011, pp. 1828-1835. doi:10.1038/bjc.2011.156
- [79] B. Ramaswamy, et al., “Phase I-II Study of Vorinostat plus Paclitaxel and Bevacizumab in Metastatic Breast Cancer: Evidence for Vorinostat-Induced Tubulin Acetylation and Hsp90 Inhibition in Vivo,” Breast Cancer Research and Treatment, Vol. 132, No. 3, 2012, pp. 1063- 1072. doi:10.1007/s10549-011-1928-x
- [80] R. Weil, D. Palmieri, J. Bronder, A. Stark and P. Steeg, “Breast Cancer Metastasis to the Central Nervous System,” American Journal of Pathology, Vol. 167, No. 4, 2005, pp. 913-920. doi:10.1016/S0002-9440(10)61180-7
- [81] Y. Tsukada, A. Fouad, J. W. Pickren and W. W. Lane, “Central Nervous System Metastasis from Breast Carcinoma. Autopsy Study,” Cancer, Vol. 52, No. 12, 1983, pp. 2349-2354.
- [82] V. Patanaphan, O. M. Salazar and R. Risco, “Breast Cancer: Metastastic Patterns and Their Prognosis,” Southern Medical Journal, Vol. 81, No. 9, 1988, pp. 1109-1112. doi:10.1097/00007611-198809000-00011
- [83] S. Y. Cho and H. Y. Choi, “Causes of Death and Metastatic Patterns in Patients with Mammary Cancer,” American Journal of Clinical Pathology, Vol. 73, No. 2, 1980, pp. 232-234.
- [84] H. Kennecke, et al., “Metastatic Behavior of Breast Cancer Subtypes,” Journal of Clinical Oncology, Vol. 28, 2010, pp. 3271-3277. doi:10.1200/JCO.2009.25.9820
- [85] C. Sezgin, E. Gokmen, M. Esassolak, N. Ozdemir and E. Goker, “Risk Factors for Central Nervous System Metastasis in Patients with Metastatic Breast Cancer,” Medical Oncology, Vol. 24, No. 2, 2007, pp. 155-161. doi:10.1007/BF02698034
- [86] K. Miller, T. Weathers, L. Hanley, et al., “Occult Central Nervous System Involvement in Patients with Metastatic Breast Cancer: Prevalence, Predictive Factors and Impact on Overall Survival,” Annals of Oncology, Vol. 14, 2003, pp. 1072-1077. doi:10.1093/annonc/mdg300
- [87] L. A. Carey, et al., “Central Nervous System Metastases in Women after Multimodality Therapy for High Risk Breast Cancer,” Breast Cancer Research and Treatment, Vol. 88, No. 3, 2004, pp. 273-280. doi:10.1007/s10549-004-0999-3
- [88] B. C. Pestalozzi, et al., “Identifying Breast Cancer Patients at Risk for Central Nervous System (CNS) Metastases in Trials of the International Breast Cancer Study Group (IBCSG),” Annals of Oncology,, Vol. 17, No. 6, 2006, pp. 935-944. doi:10.1093/annonc/mdl064
- [89] A. DiStefano, Y. Y. Yap, G. N. Hortobagyi and G. R. Blumenschein, “The Natural History of Breast-Cancer Patients with Brain Metastases,” Cancer, Vol. 44, No. 5, 1979, pp. 1913-1918. doi:10.1002/1097-0142(197911)44:5<1913::AID-CNCR2820440554>3.0.CO;2-D
- [90] P. A. Forsyth and J. B. Posner, “Headaches in Patients with Brain Tumors: A Study of 111 Patients,” Neurology, Vol. 43, No. 9, 1993, pp. 1678-1683. doi:10.1212/WNL.43.9.1678
- [91] C. M. Issa, R. Semrau, R. Kath and K. Hoffken, “Isolated Brain Metastases as the Sole Manifestation of a Late Relapse in Breast Cancer,” Journal of Cancer Research and Clinical Oncology, Vol. 128, No. 1, 2002, pp. 61-63. doi:10.1007/s004320100286
- [92] N. U. Lin, J. R. Bellon and E. P. Winer, “CNS Metastasis in Breast Cancer,” Journal of Clinical Oncology, Vol. 22, No. 17, pp. 3608-3617. doi:10.1200/JCO.2004.01.175
- [93] R. J. Weil, D. C. Palmieri, J. L. Bronder, A. M. Stark and P. S. Steeg, “Breast Cancer Metastasis to the Central Nervous System,” American Journal of Pathology, Vol. 167, No. 4, 2005, pp. 913-920. doi:10.1016/S0002-9440(10)61180-7.
- [94] J. Engel, et al., “Determinants and Prognoses of Locoregional and Distant Progression in Breast Cancer,” International Journal of Radiation Oncology*Biology*Physics, Vol. 55, 2003, pp. 1186-1195. doi:10.1016/S0360-3016(02)04476-0
- [95] R. Soffietti, R. Ruda and E. Trevisan, “Brain Metastases: Current Management and New Developments,” Current Opinion in Oncology, Vol. 20, No. 6, 2008, pp. 676-684. doi:10.1097/CCO.0b013e32831186fe
- [96] S. S. Lee, et al., “Brain Metastases in Breast Cancer: Prognostic Factors and Management,” Breast Cancer Research and Treatment, Vol. 111, No. 3, 2008, pp. 523- 530. doi:10.1007/s10549-007-9806-2
- [97] D. Palmieri, et al., “Brain Metastases of Breast Cancer,” Breast Disease, Vol. 26, No. 1, 2006, pp. 139-147.
- [98] L. L. Muldoon, et al., “Chemotherapy Delivery Issues in Central Nervous System Malignancy: A Reality Check,” Journal of Clinical Oncology, Vol. 25, No. 16, 2007, pp. 2295-2305. doi:10.1200/JCO.2006.09.9861.
- [99] N. U. Lin, J. R. Bellon and E. P. Winer, “CNS Metastases in Breast Cancer,” Journal of Clinical Oncology, Vol. 22, No. 17, 2004, pp. 3608-3617. doi:10.1200/JCO.2004.01.175
- [100] M. L. Wang, W. K. Yung, M. E. Royce, D. F. Schomer and R. L. Theriault, “Capecitabine for 5-FluorouracilResistant Brain Metastases from Breast Cancer,” American Journal of Clinical Oncology, Vol. 24, No. 4, 2001, pp. 421-424. doi:10.1097/00000421-200108000-00026
- [101] W. Wick, M. Platten and M. Weller, “New (Alternative) Temozolomide Regimens for the Treatment of Glioma,” Neuro-Oncology, Vol. 11, No. 1, 2009, pp. 69-79. doi:10.1215/15228517-2008-078
- [102] L. E. Abrey, et al., “A Phase II Trial of Temozolomide for Patients with Recurrent or Progressive Brain Metastases,” Journal of Neuro-Oncology, Vol. 53, No. 3, 2001, pp. 259-265. doi:10.1023/A:1012226718323
- [103] C. Christodoulou, et al., “Phase II Study of Temozolomide in Heavily Pretreated Cancer Patients with Brain Metastases,” Annals of Oncology, Vol. 12, No. 2, 2001, pp. 249-254. doi:10.1023/A:1008354323167
- [104] I. Preuss, et al., “O6-Methylguanine-DNA Methyltransferase Activity in Breast and Brain Tumors,” International Journal of Cancer, Vol. 61, No. 3, 1995, pp. 321- 326. doi:10.1002/ijc.2910610308
- [105] R. Addeo, et al., “Phase 2 Trial of Temozolomide Using Protracted Low-Dose and Whole Brain Radiotherapy for Non Small Cell Lung Cancer and Breast-Cancer Patients with Brain Metastases,” Cancer, Vol. 113, No. 9, 2008, pp. 2524-2531. doi:10.1002/cncr.23859
- [106] E. Rivera, “Phase I Study of Capecitabine in Combination with Temozolomide in the Treatment of Patients with Brain Metastases from Breast Carcinoma,” Cancer, Vol. 107, No. 6, 2006, pp. 1348-1354. doi:10.1002/cncr.22127
- [107] M. Kurt, S. Aksoy, M. Hayran and N. Guler, “A Retrospective Review of Breast-Cancer Patients with Central Nervous System Metastasis Treated with Capecitabine,” ASCO Annual Meeting, Abstract No 1098.
- [108] V. Guarneri, A. Frassoldati, S. Giovannelli, F. Borghi and P. Conte, “Primary Systemic Therapy for Operable Breast Cancer: A Review of Clinical Trials and Perspectives,” Cancer Letters, Vol. 248, No. 2, 2007, pp. 175-185. doi:10.1016/j.canlet.2006.07.001
- [109] J. A. O’Shaughnessy, “The Evolving Role of Capectiabine in Breast Cancer,” Clinical Breast Cancer, Suppl. 1, 2003, pp. S20-S25.
- [110] J. Baselga, et al., “Pertuzumab plus Trastuzumab plus Docetaxel for Metastatic Breast Cancer,” New England Journal of Medicine, Vol. 366, No. 2, 2012, pp. 109119. doi:10.1056/NEJMoa1113216
- [111] B. C. Pestalozzi, D. Zahrieh, K. N. Price, et al., “Identifying Breast Cancer Patients at Risk for Central Nervous System (CNS) Metastases in Trials of the International Breast Cancer Study Group (IBCSG),” Annals of Oncology, Vol. 17, No. 6, 2006, pp. 935-944. doi:10.1093/annonc/mdl064
- [112] W. Zoli, L. Ricotti, A. Tesei, F. Barzanti and D. Amadori, “In Vitro Preclinical Models for a Rational Design of Chemotherapy Combinations in Human Tumors,” Critical Reviews in Oncology/Hematology, Vol. 37, No. 1, 2001, pp. 69-82. doi:10.1016/S1040-8428(00)00110-4
NOTES
*Corresponding author.