Journal of Cancer Therapy
Vol.3 No.5(2012), Article ID:23459,8 pages DOI:10.4236/jct.2012.35082
Gene Expression Profile, Androgen Independence and Prostate Cancer
![]()
1Molecular Pathology Division, Chemical Pathology and Immunology Unit, Department of Medical Laboratory Sciences, School of Biomedical Science, Faculty of Health Science, Ebonyi State University Nigeria, Ebonyi, Nigeria; 2Medical Microbiology Unit, Department of Medical Laboratory Science, Faculty of Health Science, Ebonyi State University, Abakaliki, Nigeria; 3Histopathology Unitt, Department of Medical Laboratory Sciences, School of Biomedical Science, Faculty of Health Science, Ebonyi State University, Abakaliki, Nigeria.
Email: *Maswello2002@yahoo.com
Received June 14th, 2012; revised July 20th, 2012; accepted July 31st, 2012
Keywords: Prostate Cancer; Androgen Ablation; Gene Expression; Microarrays
ABSTRACT
It is now generally accepted that the burden of disease due to prostate cancer has tremendously increased globally. Current data indicates that prostate cancer is the most common form of cancer in men in the United State of America, and the second leading cause of death due to cancer in men. Progression to androgen independence and subsequent therapeutic resistance and death is a common fate of patients with prostate cancer. This review highlights the gene expression profile of androgen independent prostate cancer and the possible mechanisms that results in transformation to such treatment resistant state.
1. Introduction
It is now generally accepted that the burden of disease due to prostate cancer has tremendously increased globally. Current data indicates that prostate cancer is the most common form of cancer in men in the United State of America, and the second leading cause of death due to cancer [1], (see Figure 1). Approximately 100 men are being diagnosed of this disease daily in the United Kingdom [2]. In both American and British studies, men of African descendant or who emigrated from Africa to either USA or Britain had increased risk of being diagnosed with this disease [3]. Factors contributing to the development of prostate cancer have been suggested. The most outstanding of them are environmental and genetic inheritance [4].
1.1. Genetic Inheritance and Prostate Cancer
Heterocyclic aromatic amines such as 2 amino-1-methyl- 6-phenylimidazol[4,5-b]pyridine (phip) is one of the chemicals found in the cooked red meat which has been reported to be a carcinogen for prostate cancer in animal models [4,5]. Recently, Koutros et al. [6] demonstrated that phip may act as promoters of malignant transformation by altering mitogenic signaling. Several animal and human experimental studies have demonstrated the carcinogenicity of three phips in particular: PhIP (2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine)MeIQx (2-amino-3,8-dimethylimidazo-[4,5-b]quinoxaline), and DiMeIQx (2-amino-3,4,8-trimethylimidazo-[4,5-f] quinoxaline). In animal models, PhIP increases mutation frequency, for instance male lacl transgenic rats that were fed with a diet containing 200 ppm of 2-amino-1-methy l-6-phenylimidazo[4,5-b]pyridine (PhIP), had mutantion frequency that was >20-fold higher than the controls after 61 days of treatment; the authors clearly showed that PhIP-induced mutations were G:C > T:A transversions and deletions of G:C bp respectively [7]. The strongest evidence for phip intake and he risk of prostate cancer was reported in a nested case-control study that included 1126 prostate cancer cases and 1127 controls who were selected for a genome-wide association study for prostate cancer, phip exposure and SNP in the GSTM3 and GSTP1 genes which directly involved in metabolism of the xenobiotics [5]. That study showed that among individuals carrying the AG or AA genotype for GSTM3, that the risk of prostate cancer for those with high intake was increased compared to those with low intake (OR = 1.7, 95% CI 0.8, 3.6, p-interaction = 0.001 and OR = 2.3, 95% CI 1.2, 4.7, p-interaction = 0.001, respectively) [6]. In addition, individuals carrying the GG genotype, risk of prostate cancer was decreased compared to those with
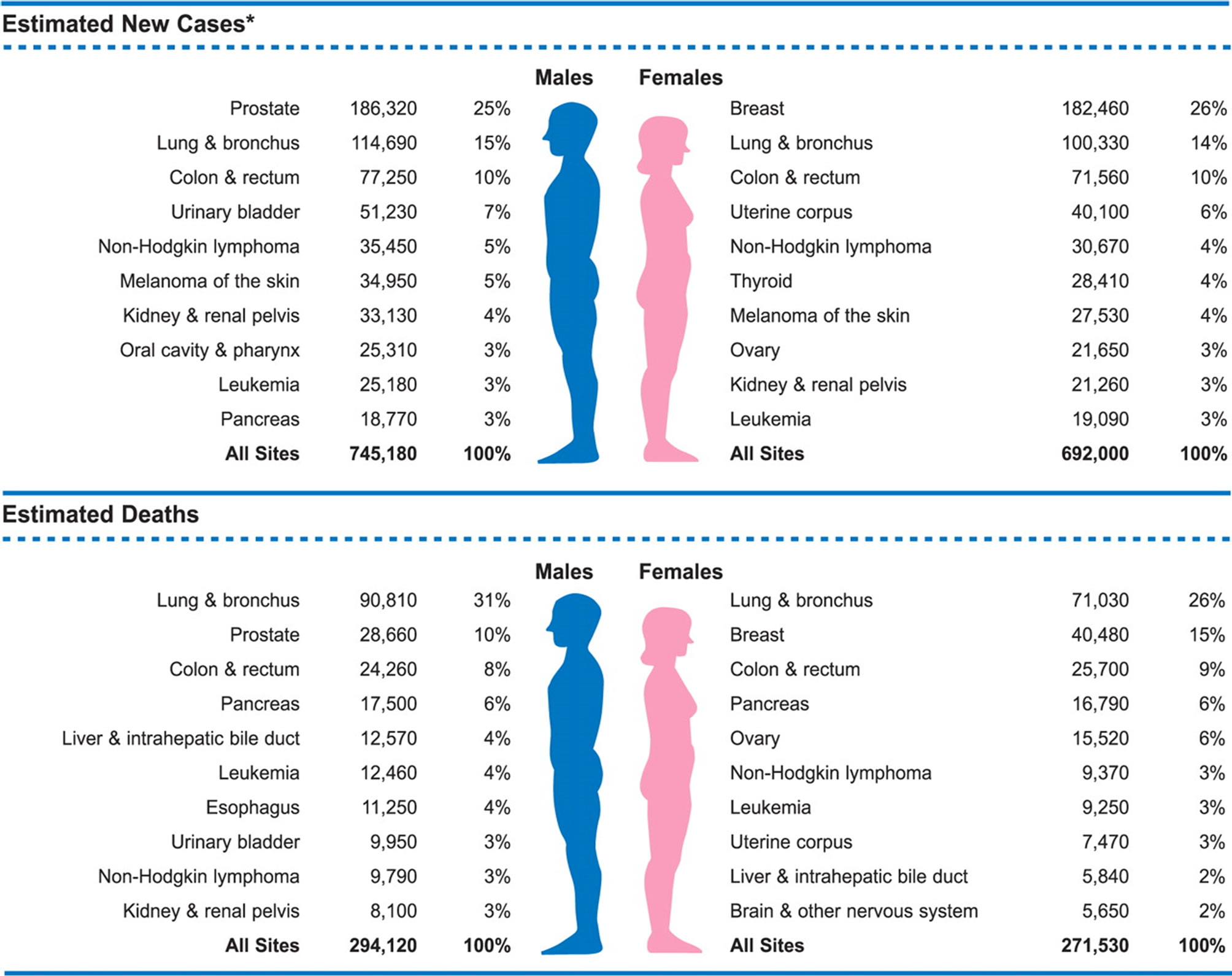
Figure 1. Illustrating the available epidemiologic data for all forms of cancer including prostate cancer. This shows that prostate cancer is the leading cause of cancer diagnosed in men and the second cause of death due to cancer in men [1].
low MeIQx and DiMeIQx intake (OR = 0.6, 95% CI 0.5, 0.8 and OR = 0.7, 95% CI 0.5, 0.8, respectively). Evidence from that study shows that the effect of phip on carcinogenesis may depend on the genotype of the individual in the gens involved in phip metabolism since men carrying the A variant, with high DiMeIQx intake had two-fold greater risk of prostate cancer than those with low intake [6]. Expression of GSTM 3 in prostate tissues acts to detoxify active heterocylic aromatic amines metabolites by conjugation with glutathione, therefore altered expression of the enzyme could lead to differential clearance of activated phip metabolites resulting in an accumulation of DNA damaging species, which could increase the risk for carcinogenesis at this site [6,7].
2. Management Options for Advanced Prostate Tumour
For men with localized prostate cancer, common treatment options include watchful waiting, surgery to remove the prostate gland (radical prostatectomy), external beam radiation therapy EBRT, cryotherapy, androgenablation therapy [8-13]. Advanced localised prostate cancer is usually treated by androgen ablation therapy [14,15]. In this treatment protocol, at least three recognised agents have been used; among them is bicalutamide, which has been shown to be most efficacious and the first line drug for patients with localised prostate cancer.
Androgen Ablation Therapy (AAT)
AAT involves the use of androgen blockades or antagonists such as flutamide, bicalutamide or casodex and nilutamide to competitively and directly block the androgen receptors in the cytosol of the prostate cells. These agents are generally—used either as intermittent therapy—to prevent the flare phenomenon commonly associated with GnRH agonists or monotherapy, in men in whom GnRH agonist could not effectively control their disease [16]. Patients treated with this drug have temporal benefits in symptoms and quality of life over a period of 2 years and subsequently develop androgen independent state which is commonly characterised with treatment resistance and cancer cells spread to bone and lymph node (Figures 2 and 3) and death due to these complications [14-22]. Mechanisms leading to development of androgen independent state and poor treatment outcome in prostate cancer patients are yet to be fully explained.
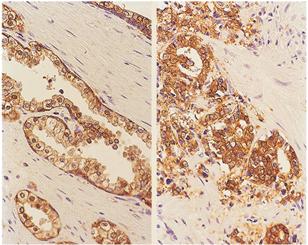
Figure 2. Illustrating the various features of prostate cancer with androgen independent phenotype in comparison with norma prostaic tissue. Immunohistochemical staining for E-cadherin showing secretory glandular structure (left) in the normal prostate and how it is disrupted in invasive prostate cancer which is commonly found in androgen independent prostate cancer states (right) [4].
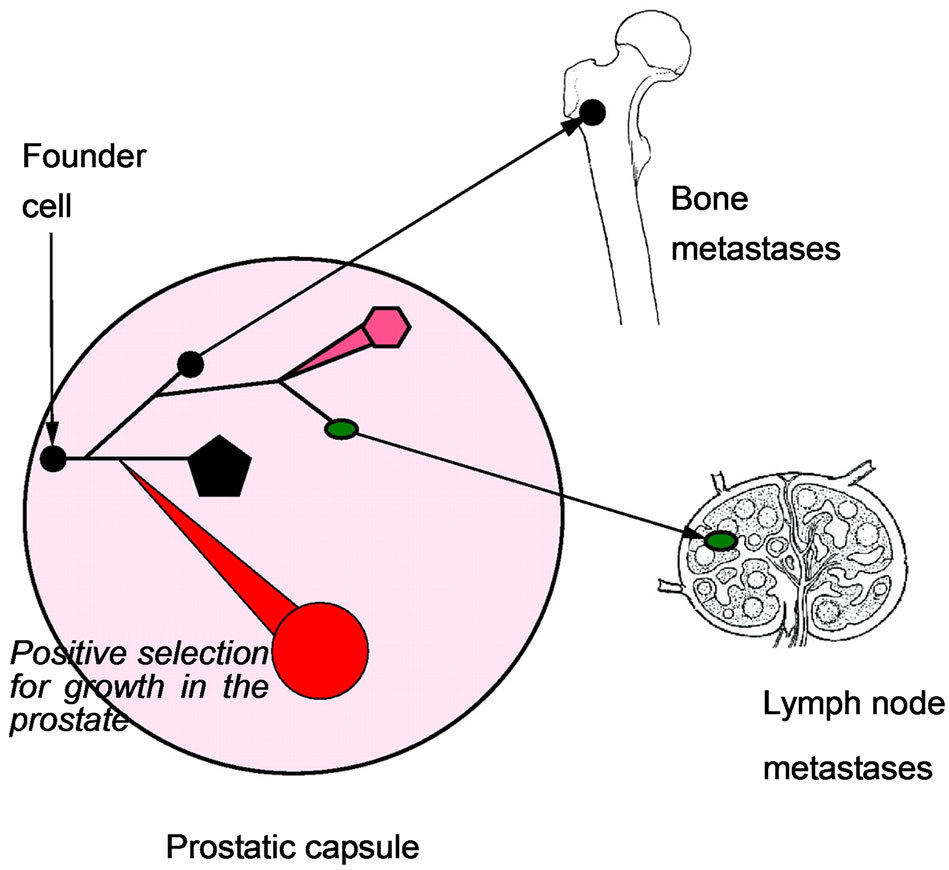
Figure 3. The bones and the lymph nodes are the main initial sites of prostate cancer metastasis [2].
3. Experimental Evidence for Androgen Ablation Therapy and Develo Pment of Drug Resistant Phenotype
Patel et al. [23] studied the characteristic changes that lead to the development of androgen independence in prostate cancer cells earlier dependent on androgens using LNCap cell and sublimes CL1. They transfected the androgen independent sublines CL1 with a green fluorescence protein and injected them into the Mice, and observed extensive local invasion and metastasis to distant organs. In a slightly similar experimental design, Tso et al. [24] observed that the androgen deprived LNCap cells were fast growing, and had ability of potentiating the growth of endothelia cells, and were resistant to both chemotherapy and radiotherapy, with high metastatic potential. The works of Freedland et al. [25] revealed three different growth pattern of androgen refractory cells when transfected and implanted into the Severe Combined Immunodeficiency Disease SCID Mice. And showed that the androgen refractory prostate cancer cells were fast growing with widespread to metastasis. Zhou et al. [26] found that androgen independent prostate cancer cells had decreased apoptosis and acquired higher proliferation rate than the androgen dependent cells. Common to all these studies are increased invasiveness and malignant metastasis, loss of apoptosis and chemotherapeutic resistance. It is not clear what molecular and cellular changes are responsible the transformation to androgen refractory phenotype following androgen ablation therapy.
4. Molecular and Cellular Alterations in Androgen Refractory Prostate Cancer
Evidence has shown that androgen dependent prostate cancer cells required the presence of both epidermal Growth Factor EGF and androgens for optimal cell proliferation, whereas androgen independent or refractory prostate cancer cells exhibited significant growth without these molecules [27]. Freedland et al. [25] observed heterogeneous growth response in prostate cancer cells exposed to androgen ablation, and suggested that there were various alteration in the genes expressions profile of rapidly proliferating prostate cancer cells. Many studies have shown androgen receptor gene over expression following androgen ablation and progression to androgen refractory phenotype, see Figure 4 [23,26,28].
5. Alteration in Androgen Receptor Gene Expression
It has been established that androgen ablation therapy induces initial reduction in the androgen receptor protein before progression to androgen independent state [23] and [26]. Hughe et al. [29] demonstrated that this treat-
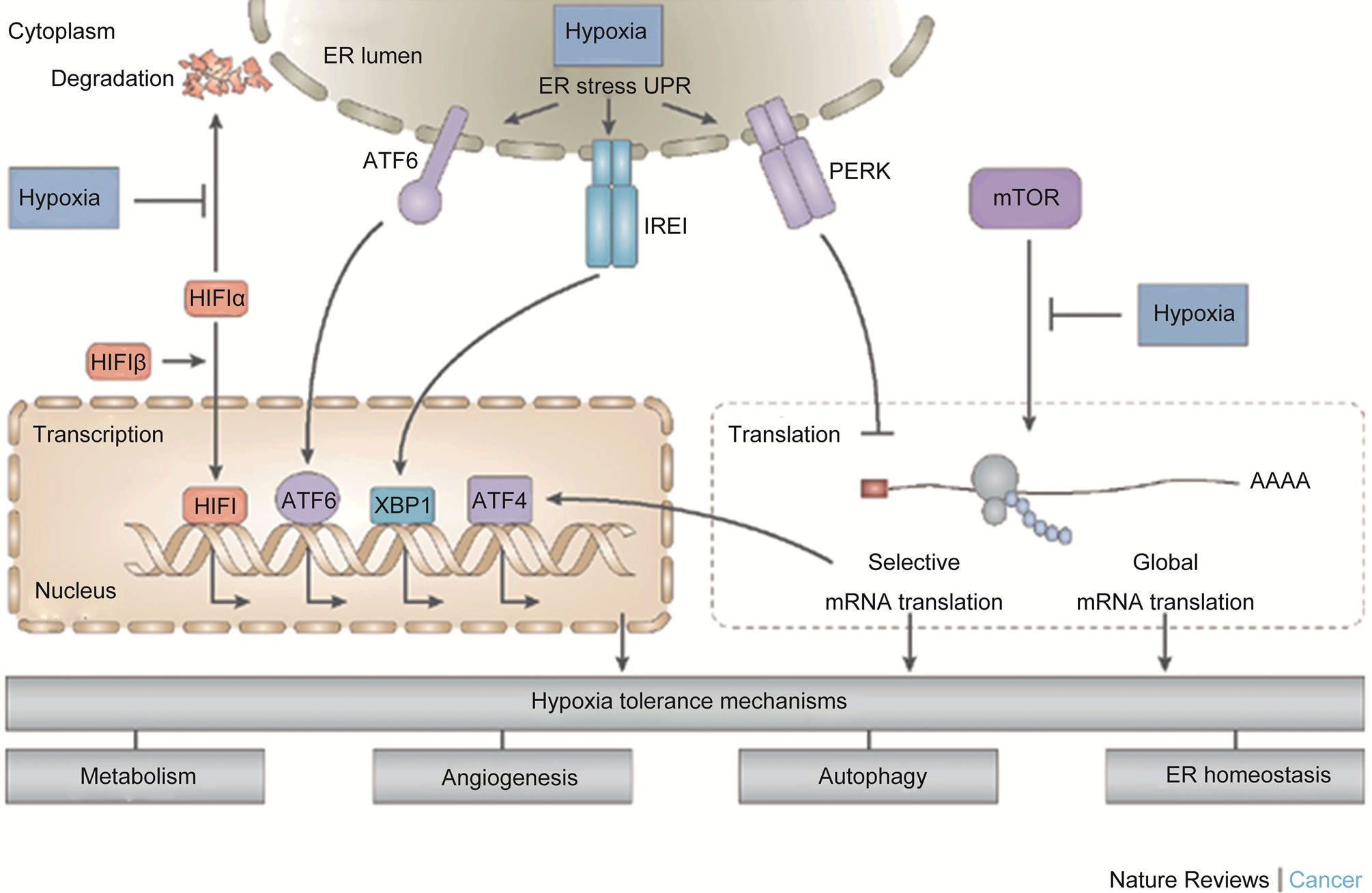
Figure 4. Illustrating the three cellular O2-sensing pathways which are often activated by hypoxia and might play in important rile in transformation to androgen resistance commonly associated with treatment failures after androgene ablation therapy [30]. As it is easily noticed from the figure that the cancer cells may promote pathways that could result in increased angiogenesis. This may in addition contribute to promote further tumour spread to distant organs.
ment intervention might induce some structural changes or mutation in the androgen receptor causing androgen independence. However, in a study involving 44 mutant androgen receptor prostate tumours; 15.9% had loss of function, 6.8% had wild type function, 31.8 % had partial function and 45.5% demonstrated a gain of function [29]. Also, Marceili et al. [31] investigated androgen receptor mutations in 137 specimens of prostate cancer from primary tumour of stage B and D, and observed that only 8% had mutation on in the androgen receptor and that there was no mutation in the 99 patients with stage B prostate cancer. Put together, there may some changes in the androgen receptors of androgen independent prostate cancer cells, but the extent of such changes may not the solely responsible for the development of refractory state. Thus progression to androgen refractory prostate cancer state may not be due to mutations in the androgen recaptors.
6. Evidence from Gene Expression Profile Studies
Data from microarray analyses studies of prostate cancer cells progressing to androgen refractory phenotype indicated that the onset of androgen independence did not require any alteration in the status of androgen receptors, even though too many genes were dysregulated [32]. The gene profiling analysis of androgen refractory prostate cancer cells [33], further threw more lights on the nature of the dysregulations, indicating that the following genes were up regulated; PCDH7, TPTE, TSP4, EPHA3, HGF, MET, EGF, TEM8 while HTAT1P2, CDKN2A, CDKN2B, CDKN1C, PPP2CA, SPRY, etc, were down regulated [33]. Also P53, P21, bax, bcl-2, VEGF, waf1 were all up regulated in the androgen refractory prostate cancer cells in a separate study [26]. Furthermore, the changes responsible for progression to androgen independence in prostate cancer have characterised using real time polymerase chain reaction (RT PCR) [23]. Thus, the androgen independent cells had over expression of phosphatise and tensin homolog gene PTEN, interleukin 6, interleukin 8, transforming growth factor beta, vascular endothelial growth factors VEGF, epidermal growth factor receptors EGFR, bcl-2 and low level of P53 [23], also see Figure 4. In addition, the functions of these genes have been reviewed elsewhere [4,29]. However, a full functional genomics of these delineated perturbed genes may further open a new window into the complex multifactoral acquisition of androgen independence following androgen ablation therapy.
7. The Mechanism of Progression to Treatment Resistant Phenotype
The emergence of cells refractory to androgen ablation and radiation therapy is often the major cause of death in men treated with prostate cancer. It is not yet known the actual initiators of the emergence of these resistant cells. Currently, some hypotheses have evolved to explain the changes that lead to the progression to androgen independence. These include;
1) Increased in the sensitivity of the androgen receptors [5,30,34-37].
2) Decreased and/or loss of specificity of the androgen receptors [4,35].
3) Outlaw pathways [5,33,37].
4) involvement of neuroendocrine cells [37-39].
5) involvement of prostate cancer stem cells [2,36].
6) increasing level of tumour hypoxia [21,40,41].
7.1. Hypersensitivity of the Androgen Receptors
The androgen receptors become more sensitive to exceedingly low concentration of androgens. This could be the effect of over expression of messenger RNAs for androgen receptors in the androgen independent prostatic cells [23,33]. This will result to higher expression of androgen receptors, making it easier and faster for binding of ligand even at lower concentration [34].
A second explanation of this phenomenon is that androgen deprivation may exact a selective ones will subsequently undergo clonal expansion with over expression of androgen receptor genes [25,26,28,33,34].
Additionally, it could be that the androgen independent prostate cells have higher expression of or increased activity of 5 alpha reductase, leading increased conversion of testosterone to DHT with a consequent autocrine action [34]. Hence, differences in the level of androgens between prostatic tissues and the serum has been reported in the androgen refractory cells following androgen ablation therapy; with disproportionally higher levels in the prostatic tissues than that in the serum [5,34].
7.2. Altered Specificity
Here the androgen receptor becomes activated by nonandrogens and even antiandrogens normally present in the serum. Though earlier studies pointed accusing finger on mutations in the androgen receptors as the main cause of promiscuous ligand binding [5,34,35], on the other hand, recent studies held conflicting but more convincing evidences; that the level of mutations in androgen receptors were quite negligible [29,33]. However, microdissection of tumours and Laser Capture techniques studies may resolve the current controversies [5].
7.3. Outlaw Pathways
The process by which steroid hormone receptors become activated by either nonsteroid ligand or ligand independent means was described as outlaw pathways [5]. The following ligands have been shown to activate the androgen receptor of androgen independent prostate cells; insulin like growth factors (IGF), keratinocyte growth factors (KGF), epidermal growth factors (EGF), causing proliferation of the cells [5,34]. Thus making the tumour growth to be androgen independent.
7.4. Bypass Pathways
Mechanisms that can cause proliferation of prostate cells and inhibition of apoptosis in androgen dependent or independent state without the involvement of androgen receptor are known as a bypass pathway [5]. The works of Raffo et al. [35] revealed an elevation of expression of bcl-2 oncoprotein. This protein is normally involved suppression of apoptosis. It is not normally found expressed on the epithelia cells. Hence, bcl-2 may be involved in the protection of androgen independent prostate cancer from apoptosis and the level of expression was strongly correlated with the extent of hormone refractory phenotype [35].
Other players in the bypass pathway are the prostate cancer stem cells and neuroendocrine cells. Evidence has shown that prostate tumour is made up of heterogeneous population of cells with various phenotypes [30]. Many studies have identifies and selected Stem cells from primary and metastic prostate cancer and characterised using expression of CD133 as the target marker [30]. Of the cells, 0.1% of cells in the tumour expressed CD133 phenotype; these cells were shown to be capable of self renewal, proliferation, differentiation and invasion, [2, 39]. That report strongly demonstrated that prostate cancer contained a subpopulation of cancer stem cells which lack expression of androgen receptors. During transformation to androgen independence state, these subpopulation of cells may on the other hands serve as a protection from the effect of androgen deprivation during therapy. An inherent genetic instability in this subpopulation may lead to the generation of cells with androgen independence in the later stage [2].
Furthermore, another variant of prostate tumour subpopulation have been identified; which tend to promote tumour progression and resistance to hormone therapy. They were described as neuroendocrine cells. Androgen refractory prostate cancer cells contain about 40 - 100 percent neuroendocrine cells [37]. The neuroendocrine cells secrete serotonins and b ombestins which have paracrine effects on the nearby cells and induce an increased in cell proliferation index [37]. Evidence in support of this mechanism was that tumour growth continued in Nu Mice bearing LNCap and neuroendocrine cells —these cells continued to grow even after the tumour bearing mice were castrated. It was also noted that the tumour size decreased in the mice that bore LNCaPs tumour xenograft without neuroendocrine cells [37] this strongly suggests that the presence of neuroendocrine cells in LNCaP tumours or prostate cancer tumour models may contribute in part in the promotion of cancer progression.
8. The Hypoxic Tumour Microenvironment
The peculiar nature of microenvironment of prostate tumour is characterised by irregular supply of oxygen, PH, and nutrients [39]. The consequences of this alteration are development of irregular vasculature, genetic and epigenetic changes, alterations in gene expression and cellular functions, see Figure 4 [40]. Tumour hypoxia has been recognised as one of the pivots of development and progression of prostate cancer through the activation of three major oxygen sensitive pathways; including hypoxia inducible factor-1 (HIF-1), Mammalian target of Rapamycin mTOR kinase, and the Unfolded protein response (UPR), see Figure 4, [40]. These three pathways are independent but may also act in an integrated manner, with some level of influence on each other and leading to a common cumulative effects on gene expression, cell survival and tumourigenesis [40]. Hence, long time androgen deprivation has been suggested to induce tumour hypoxia and the consequent transition from androgen dependence to independence state, culminating in resistance to therapy.
Additionally, tumour hypoxia induces the generation of radical oxygen species (ROS), which beyond a certain threshold may cause adaptive responses and loss of: 1) PTEN, the function of PTEN in a normal cell is induction of apoptosis and its loss in androgen refractory prostate cancer causes cell survival [37]; 2) Glutathione S-transferase GST-Pi; 3) androgen regulated specific home box NKX3, 4) over expression of bcl-2 and loss of apoptosis, 5) activation of hypoxia inducible factor-1 HIF-1, see Figure 4, and activation of nuclear factor (NFkB). NFkB is an innate molecule which has a central role in the activation of both innate and adaptive immunity. In this case, its activation results in production of pro-inflammatory cytokines. Put together, these molecular signalling above may result in the increased level of inflamematory cytokines and oxidative damage to the DNA and epimutation; this may be responsible for acquisition of aggressive and more malignant phenotype, and invasive regional and distant spreading and resistance to treatment, see Figure 4.
9. Conclusion
In conclusion, development of androgen resistance is an established consequence of therapeutic intervention using androgen ablation approach. Various mechanisms involveing the alteration of gene expression, through yet unclear signalling pathways results in various molecular and cellular changes which may independently and synergistically induce transformation to androgen resistant phenoltypes. The perturbation of gene expression profile that drives development of androgen independence has been discussed in this study. A wide range of genomic perturbations in the gene expression profile of androgen independent prostate cancer tumours have been high-lighted. In addition, some specific mechanisms that may result in development of androgen independent state have been discussed. However, more studies involving perhaps functional genomics may be important—this may provide evidence for molecular targeting of some hit and lead genes that drive prostate cancers towards malignant progression. This in turn may serve as potent therapeutic targets for overcoming treatment failures in patients with prostate cancer.
10. Submission Declaration
The authors declare that no part of this work has been published else where or is awaiting publication anywhere.
11. Conflict of Interest
I declare no conflict of interest, and that no fund from any funding body or the University was made available for this work. This study was funded by the authors only.
REFERENCES
- A. Jemal, R. Siegel, E. Ward, Y. P. Hao, J. Q. Xu, T. Murray and M. J. Thun, “Cancer Statistics, 2008,” CA: A Cancer Journal for Clinicians, Vol. 58, No. 2, 2008, pp. 71-96. doi:10.3322/CA.2007.0010
- N. J. Maitland and A. T. Collins, “Prostate Cancer Stem Cells: A New Target for Therapy,” Journal of Clinical Oncology, Vol. 26, No. 17, 2008, pp. 2862-2870. doi:10.1200/JCO.2007.15.1472
- F. T. Odedina, T. O. Akinremi, F. Chinegwundoh, R. Roberts, D. Yu, R. R. Reams, M. L. Freedman, B. Rivers, B. L. Green and N. Kumar, “Prostate Cancer Disparities in Black Men of African Descent: A Comparative Literature Review of Prostate Cancer Burden among Black Men in the United States, Caribbean, United Kingdom, and West Africa,” Infectious Agents and Cancer, Vol. 4, Suppl. 1, 2009, p. S2. doi:10.1186/1750-9378-4-S1-S2
- W. G. Nelson, A. M. De Marzo and W. B. Isaacs, “Prostate Cancer,” The New England Journal of Medicine, Vol. 349, No. 4, 2003, pp. 366-381. doi:10.1056/NEJMra021562
- J. B. Feldman and D. Feldman, “The Development of Androgen-Independent Prostate Cancer,” Nature Reviews. Cancer, Vol. 1, No. 1, 2001, pp. 34-35. doi:10.1038/35094009
- S. Koutros, S. I. Berndt, R. Sinha, X. Ma, N. Chatterjee, M. C. Alavanja, T. Zheng, W. Y. Huang, R. B. Hayes and A. J. Cross, “Xenobiotic Metabolizing Gene Variants, Dietary Heterocyclic Amine Intake, and Risk of Prostate Cancer,” Cancer Research, Vol. 69, No. 5, 2009, pp. 1877-1884. doi:10.1158/0008-5472.CAN-08-2447
- G. R. Stuart, J. Holcroft, J. G. de Boer and B. W. Glickman, “Prostate Mutations in Rats Induced by the Suspected Human Carcinogen 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine,” Cancer Research, Vol. 60, No. 2, 2000, pp. 266-268.
- M. Shelley, C. Harrison, B. Coles, J. Staffurth, T. Wilt and M. Mason, “Chemotherapy for Hormone-Refractory Prostate Cancer,” Cochrane Database of Systematic Reviews, Vol. 18, No. 4, 2006, Article ID: CD005247.
- J. D. Debes and D. J. Tindall, “Mechanisms of Androgen-Refractory Prostate Cancer,” The New England Journal of Medicine, Vol. 351, No. 15, 2004, pp. 1488-1490. doi:10.1056/NEJMp048178
- M. J. Barry, D. S. Kaufman and C. L. Wu, “Case, Records of the Massachusetts General Hospital, Case 15- 2008, a 55-Year-Old Man with an Elevated ProstateSpecific Antigen Level and Early-Stage Prostate Cancer,” The New England Journal of Medicine, Vol. 358, No. 20, 2008, pp. 2161-2168. doi:10.1056/NEJMcpc0707057
- W P. alsh, T. L. DeWeese and M. A. Eisenberger, “Localized Prostate Cancer,” The New England Journal of Medicine, Vol. 357, No. 26, 2007, pp. 2696-2705. doi:10.1056/NEJMcp0706784
- T. J. Wilt, “Clarifying Uncertainty Regarding Detection and Treatment of Early-Stage Prostate Cancer,” Seminars in Urologic Oncology, Vol. 20, No. 1, 2002, pp. 10-17. doi:10.1053/suro.2002.30393
- A. Bill-Axelson, L. Holmberg, M. Ruutu, M. Häggman, S. O. Andersson, S. Bratell, A. Spångberg, C. Busch, S. Nordling, H. Garmo, J. Palmgren, H. O. Adami, B. J. Norlén and J. E. Johansson, “Scandinavian Prostate Cancer Group Study No. 4. Radical Prostatectomy versus Watchful Waiting in Early Prostate Cancer,” The New England Journal of Medicine, Vol. 352, No. 19, 2005, pp. 1977-1984. doi:10.1056/NEJMoa043739
- E. M. Horwitz, K. Bae, G. E. Hanks, A. Porter, D. J. Grignon, H. D. Brereton, V. Venkatesan, C. A. Lawton, S. A. Rosenthal, H. M. Sandler and W. U. Shipley, “TenYear Follow-Up of Radiation Therapy Oncology Group Protocol 92-02: A Phase III Trial of the Duration of Elective Androgen Deprivation in Locally Advanced Prostate Cancer,” Journal of Clinical Oncology, Vol. 26, No. 15, 2008, pp. 2497-2504. doi:10.1200/JCO.2007.14.9021
- E. M. Messing, J. Manola, M. Sarosdy, G. Wilding, E. D. Crawford and D. Trump, “Immediate Hormonal Therapy Compared with Observation after Radical Prostatectomy and Pelvic Lymphadenectomy in Men with Node-Positive Prostate Cancer,” The New England Journal of Medicine, Vol. 341, No. 24, 1999, pp. 1781-1788. doi:10.1056/NEJM199912093412401
- Y. K. Lau, M. K. Chadha, A. Litwin and D. L. Trump, “A Dramatic, Objective Antiandrogen Withdrawal Response: Case Report and Review of the Literature,” Journal of Hematology & Oncology, Vol. 1, No. 1, 2008, p. 21. doi:10.1186/1756-8722-1-21
- E. M. Messing, J. Manola, J. Yao, M. Kiernan, D. Crawford, G. Wilding, P. A. di’SantAgnese and D. Trump, “Eastern Cooperative Oncology Group Study EST Immediate versus Deferred Androgen Deprivation Treatment in Patients with Node-Positive Prostate Cancer after Radical Prostatectomy and Pelvic Lymphadenectomy,” The Lancet Oncology, Vol. 7, No. 6, 2006, pp. 472-479. doi:10.1016/S1470-2045(06)70700-8
- T. J. Wilt, R. MacDonald, I. Rutks, T. A. Shamliyan, B. C. Taylor and R. L. Kane, “Systematic Review: Comparative Effectiveness and Harms of Treatments for Clinically Localized Prostate Cancer,” Annals of Internal Medicine, Vol. 148, No. 6, 2008, pp. 435-448.
- M. Roach, K. Bae, J. Speight, H. B. Wolkov, P. Rubin, R. J. Lee, C. Lawton, R. Valicenti, D. Grignon and M. V. Pilepich, “Short-Term Neoadjuvant Androgen Deprivation Therapy and External-Beam Radiotherapy for Locally Advanced Prostate Cancer: Long-Term Results of RTOG 8610,” Journal of Clinical Oncology, Vol. 26, No. 4, 2008, pp. 585-591. doi:10.1200/JCO.2007.13.9881
- L. Holmberg, A. Bill-Axelson, F. Helgesen, J. O. Salo, P. Folmerz, M. Häggman, S. O. Andersson, A. Spångberg, C. Busch, S. Nordling, J. Palmgren, H. O. Adami, J. E. Johansson and B. J. Norlén, “Scandinavian Prostatic Cancer Group Study No. 4. A Randomized Trial Comparing Radical Prostatectomy with Watchful Waiting in Early Prostate Cancer,” The New England Journal of Medicine, Vol. 347, No. 11, 2002, pp. 781-789. doi:10.1056/NEJMoa012794
- S. Kumar, M. Shelley, C. Harrison, B. Coles, T. J. Wilt and M. Mason, “Neo-Adjuvant and Adjuvant Hormone Therapy for Localised and Locally Advanced Prostate Cancer,” Cochrane Database of Systematic Reviews, Vol. 18, No. 4, 2006, Article ID: CD006019.
- A. Pollack, D. A. Kuban and G. K. Zagars, “Impact of Androgen Deprivation Therapy on Survival in Men Treated with Radiation for Prostate Cancer,” Urology, Vol. 60, Suppl. 1, 2002, pp. 22-30. doi:10.1016/S0090-4295(02)01564-9
- B. J. Patel, A. J. Pantuck, A. Zisman, K. H. Tsui, S. H. Paik, R. Caliliw, S. Sheriff, L. Wu, J. B. de Kernion, C. L. Tso, and A. S. Belldegrun, “CL1-GFP: An Androgen Independent Metastatic Tumor Model for Prostate Cancer,” Journal of Urology, Vol. 164, No. 4, 2000, pp. 1420-1425. doi:10.1016/S0022-5347(05)67210-2
- C. L. Tso, W. H. McBride, J. Sun, B. Patel, K. H. Tsui, S. H. Paik, B. Gitlitz, R. Caliliw, A. van Ophoven, L. Wu, J. de Kernion and A. Belldegrun, “Androgen Deprivation Induces Selective Outgrowth of Aggressive HormoneRefractory Prostate Cancer Clones Expressing Distinct Cellular and Molecular Properties Not Present in Parental Androgen-Dependent Cancer Cells,” The Cancer Journal, Vol. 6, No. 4, 2000, pp. 220-233.
- S. J. Freedland, A. J. Pantuck, P S. H. aik, A. Zisman, T. G. Graeber, D. Eisenberg, W. H. McBride, D. Nguyen, C. L. Tso and A. S. Belldegrun, “Heterogeneity of Molecular Targets on Clonal Cancer Lines Derived from a Novel Hormone-Refractory Prostate Cancer Tumor System,” The Prostate, Vol. 55, No. 4, 2003, pp. 299-307. doi:10.1002/pros.10226
- J. R. Zhou, L. Yu, L. F. Zerbini, T. A. Libermann, G. L. Blackburn, “Progression to Androgen-Independent LNCaP Human Prostate Tumors: Cellular and Molecular Alterations,” International Journal of Cancer, Vol. 110, No. 6, 2004, pp. 800-806. doi:10.1002/ijc.20206
- J. K. Oosterhoff, J. A. Grootegoed and L. J. Blok, “Expression Profiling of Androgen-Dependent and -Independent LNCaP Cells: EGF versus Androgen Signalling,” Endocrine-Related Cancer, Vol. 12, No. 1, 2005, pp. 135- 148. doi:10.1515/IJDHD.2005.4.2.77
- N. J. Nesslinger, X. B. Shi and R. W.de Vere White, “Androgen-Independent Growth of LNCaP Prostate Cancer Cells Is Mediated by Gain-of-Function Mutant p53,” Cancer Research, Vol. 83, No. 9, 2003, pp. 2228-2233.
- C. Hughes, A. Murphy, C. Martin, O. Sheils and J. O’Leary, “Molecular Pathology of Prostate Cancer,” Journal of Clinical Pathology, Vol. 58, No. 7, 2005, pp. 673-684. doi:10.1136/jcp.2002.003954
- A. T. Collins, P. A. Berry, C. Hyde, M. J. Stower and N. J. Maitland, “Prospective Identification of Tumorigenic Prostate Cancer Stem Cells,” Cancer Research, Vol. 65, No. 23, 2005, pp. 10946-10951. doi:10.1158/0008-5472.CAN-05-2018
- M. Marcelli, M. Ittmann, S. Mariani, et al., “Androgen Receptor Mutations in Prostate Cancer,” Cancer Research, Vol. 60, No. 4, 2000, PP. 944-949.
- F. M. Sirotnak, Y. She, N. Z. Khokhar, P. Hayes, W. Gerald and H. I. Scher, “Microarray Analysis of Prostate Cancer Progression to Reduced Androgen Dependence: Studies in Unique Models Contrasts Early and Late Molecular Events,” Molecular Carcinogenesis, Vol. 41, No. 3, 2004, pp. 150-163. doi:10.1002/mc.20051
- A. P. Singh, S. Bafna, K. Chaudhary, G. Venkatraman, L. Smith, J. D. Eudy, S. L. Johansson, M. F. Lin and S. K. Batra, “Genome-Wide Expression Profiling Reveals Transcriptomic Variation and Perturbed Gene Networks in Androgen-Dependent and Androgen-Independent Prostate Cancer Cells,” Cancer Letters, Vol. 259, No. 1, 2008, pp. 28-38. doi:10.1016/j.canlet.2007.09.018
- K. J. Pienta and D. Bradley, “Mechanisms Underlying the Development of Androgen-Independent Prostate Cancer,” Clinical Cancer Research, Vol. 12, No. 6, 2006, pp. 1665-1671. doi:10.1158/1078-0432.CCR-06-0067
- H. K. Tsai, A. V. D’Amico, N. Sadetsky, M. H. Chenand and P. R. Carroll, “Androgen Deprivation Therapy for Localized Prostate Cancer and the Risk of Cardiovascular Mortality,” Journal of National Cancer Institute, Vol. 99, No. 20, 2007, pp. 1516-1524.doi:10.1093/jnci/djm168
- A. J. Raffo, H. Perlman, M. W. Chen, M. L. Day, J. S. Streitman and R. Buttyan, “Over Expression of bcl-2 Protects Prostate Cancer Cells from Apoptosis in Vitro and Confers Resistance to Androgen Depletion in Vivo,” Cancer Research, Vol. 55, No. 19, 1995, pp. 4438-4445.
- J. D. Debes and D. J. Tindall, “Mechanisms of Androgen-Refractory Prostate Cancer,” The New England Journal of Medicine, Vol. 351, No. 15, 2004, pp. 1488-1490. doi:10.1056/NEJMp048178
- R. J. Jin, Y. Wang, N. Masumori, K. Ishii, T. Tsukamoto, S. B. Shappell, S. W. Hayward, S. Kasper and R. J. Matusik, “NE-10 Neuroendocrine Cancer Promotes the LNCaP Xenograft Growth in Castrated Mice,” Cancer Research, Vol. 64, No. 15, 2004, pp. 5489-5495. doi:10.1158/0008-5472.CAN-03-3117
- M. W. Dewhirst, Y. Cao and B. Moeller, “Cycling Hypoxia and Free Radicals Regulate Angiogenesis and Radiotherapy Response,” Nature Reviews Cancer, Vol. 8, No. 8, 2008, pp. 425-437. doi:10.1038/nrc2397
- B. G. Wouters and M. Koritzinsky, “Hypoxia Signalling through mTOR and the Unfolded Protein Response in Cancer,” Nature Reviews Cancer, Vol. 8, No. 11, 2008, pp. 851-864. doi:10.1038/nrc2501
- G. L. Semenza, “Targeting HIF-1 for Cancer Therapy,” Nature Reviews Cancer, Vol. 3, No. 10, 2003, pp. 721- 732. doi:10.1038/nrc1187
NOTES
*Corresponding author.