Case Reports in Clinical Medicine
Vol.2 No.1(2013), Article ID:28927,5 pages DOI:10.4236/crcm.2013.21021
Lymphoid interstitial pneumonia—Another consideration for recurrent bacterial pneumonia in an HIV positive patient (A case report)
![]()
1Howard University College of Medicine, Washington DC, USA
2Department of Internal Medicine, Division of Hospital Medicine, Johns Hopkins School of Medicine, Baltimore, USA
3Department of Internal Medicine, Division of Infectious Diseases, University of Illinois College of Medicine at Peoria, Peoria, USA; *Corresponding Author: shwetaramsahai@gmail.com
Received 24 January 2013; revised 27 February 2013; accepted 14 March 2013
Keywords: HIV; AIDS; Lymphoid Interstitialpneumonia (LIP); Lymphocytic Interstitial Pneumonitis; Recurrent Pneumonia
ABSTRACT
We present a 39-year-old female with HIV, medication non-compliance, and recurrences of bacterial pneumonia. She presented with a two-day bout of worsening shortness of breath, dyspnea and productive cough. A CT of the chest was notable for multi-lobular pneumonia. She underwent flexible bronchoscopy and biopsy which revealed lymphocytic interstitial pneumonitis. Conclusion: Recurrent bacterial pneumonia is a rare complication of lymphoid interstitial pneumonia (LIP) in HIV positive patients, and should be considered in the differential as a possible precipitator of recurrent pneumonia inan HIV positive patient.
1. INTRODUCTION
There are two idiopathic interstitial pneumonias associated with human immunodeficiency syndrome (HIV): Lymphoid Interstitial Pneumonia (LIP) and Nonspecific Interstitial Pneumonia (NSIP) according to the American Thoracic Society/European Respiratory Society (ATS/ ERS) Consensus Statement published in 2002 [1]. LIP is characterized by interstitial infiltration of the alveolar septae with mature lymphocytes and plasma cells. Many cases have been associated with lymphoproliferative, autoimmune and infectious disorders [1,2]. And it has been reported that patients with LIP may be more susceptible to bacterial pneumonias [3,4]. There has only been one reported case of recurrent pneumococcal pneumonia in an HIV patient with LIP [4]. We describe an African American female with a history of Acquired Immunodeficiency Syndrome (AIDS) and biopsy-proven Lymphoid Interstitial Pneumonia who was found to have streptococcal bacteremia after multiple hospitalizations for pneumonia.
2. CASE PRESENTATION
A 39-year-old African American female presented to the emergency room March 3rd, 2009 with a two dayhistory of worsening dyspnea, productive cough and chest tightness. Her past medical history was significant for AIDS diagnosed in 1994 with CD4 T-cell count of 131, childhood asthma and emphysema. She was not taking antiretroviral therapy or any other regular medications. She admitted to a history of chronic tobacco use (40 pack/ years) but denied intravenous drug abuse. She had three previous hospitalizations for confirmed bacterial pneumonia over the past year with her most recent hospitalization one month prior to this current case presentation.
On physical examination, her temperature was 98.7˚F, heart rate 116 beats/minute, blood pressure 119/57 mmHg and respiratory rate 19 breaths/minute. The patient was using accessory muscles of respiration and had decreased breath sounds with diffuse wheezes bilaterally. She had left lower lobe crackles and the rest of the examination was normal.
Her initial arterial blood gas showed: pH 7.42, pCO2 41 mmHg, HCO3 27, pO2 68 mmHg and saturation of 94% while breathing two liters of oxygen. The calculated alveolar-arterial oxygen difference (P(A-a)O2) was 30 mmHg. The white blood cell count was 23.9 th/cm, hemoglobin 13.1 g/dl and platelet count 171 th/cm. The CD 4 T-cell count was 131 cells/ul, CD 8 T-cell count was 778 cells/ul and CD 4/CD 8 ratio was 0.16. Chest roentgenogram showed hyperaeration with bilateral upper lobe bullae and bibasilar interstitial disease (Figure 1).
She was empirically treated for hospital acquired pneumonia with piperacillin-tazobactam, vancomycin and le-
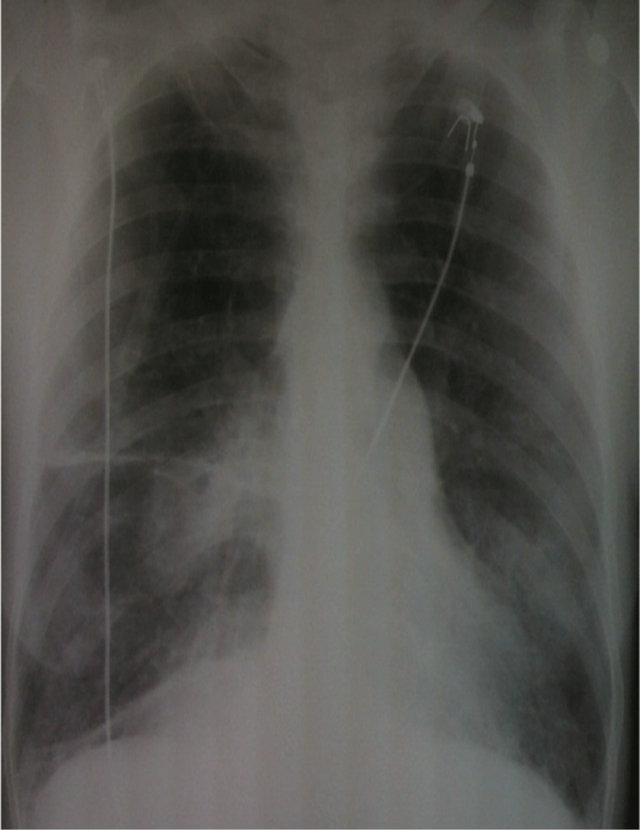
Figure 1. Chest roentgenogram showing hyper inflated lung fields along with bilateral upper lobe bullae and bibasilar infiltrates.
vaquin. She was initiated on steroids and nebulizers for COPD. The patient demonstrated minimal respiratory improvement over the next 24 hours. A follow up thoracic computer topography (CT) scan without contrast showed a 1 cm irregular border nodular density in the apical posterior segment of the left upper lobe and patchy opacities in the middle lobe, lingular and lower lobes (Figure 2). Given that the patient had Mycobacterium avium Complex (MAC), which was positive from sputum cultures during a previous, recent hospitalization this prompted the need for flexible bronchoscopy for further evaluation. In the interim, admission blood cultures returned positive for Streptococcus pneumonia. Piperacillin-tazobactam and vancomycin were stopped, and the patient was continued on levaquin.
Transbronchial biopsy showed evidence of lymphocytic interstitial pneumonitis with diffuse interstitial infiltration, mature lymphocytes, plasma cells, macrophages, neutrophils and moderate benign squamous cells (Figure 3).
Stains and cultures for acid fast bacilli, Pneumocystis jiroveci and fungi were negative. Repeat blood cultures were negative.
The patient clinically improved with peak flows back to baseline and pulse oxygen saturations in the mid to
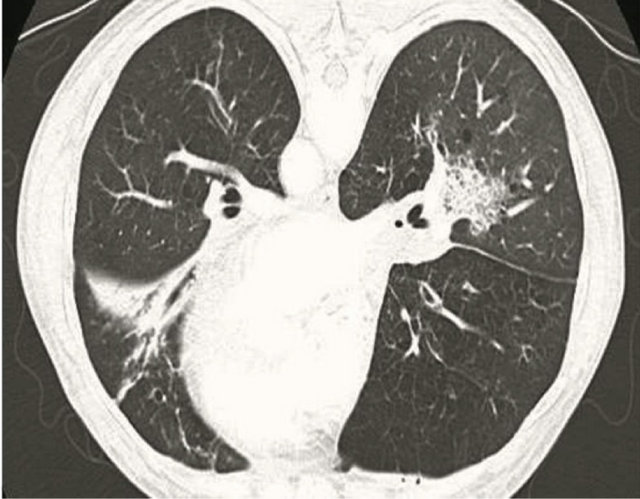
Figure 2. Chest CT showing a 1 cm irregular border nodular density in the apical posterior segment of the left upper lobe and patchy opacities in the middle lobe, lingular and lower lobes.
upper 90’s on room air. The patient was discharged with a steroid taper, and oral levaquin (for 10 days) as she appeared clinically stable. However, the patient was lost toimmediate outpatient follow-up.
3. DISCUSSION
This case highlights LIP as a rare condition (affecting only about 3% of HIV positive patients) that has previously been implicated to rarely cause the complication of recurrent bacterial pneumonias [4]. As such, it is quite possible that our patient’s repeated bouts of pneumonia may have been precipitated by an undiagnosed case of LIP, as she was not on HAART at the time of diagnosis. The previous case report noted a patient with recurrent pneumonia in the setting of LIP in the pre-HAART era, which may explain why the presentation is not common [4]. However, there remain cases of late HIV diagnosis and medication non-adherence, which makes knowledge of our patient’s case pertinent.
In patients with HIV not on HAART, who present with inexplicable recurrent bouts of bacterial pneumonia, the astute clinician should consider LIP in the differential underlying diagnosis for increased susceptibility. This in turn will reduce morbidity and mortality for these patients as LIP remains an often overlooked etiology.
LIP, itself, was first described by Liebow and Carrington in 1969 [5]. It is characterized by infiltration of alveolar interstitium predominantly with lymphocytes [1]. Usually there is also involvement of subpleural areas and intralobular septae [6]. Overall there is a mixture of polyclonal lymphocytes, immunoblasts, plasma cells, histiocytes, epithelioid cells, giant cells and macrophages. Also, reactive lymphoid follicles along peribronchial regions, noncaseating granulomas, interstitial fibrosis and honeycomb changes (the latter two in advanced stages)
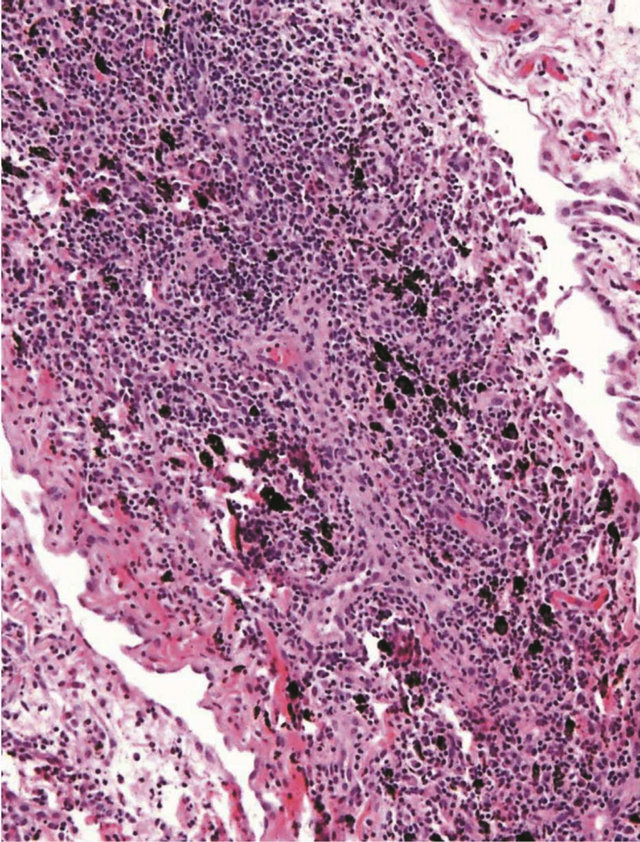
Figure 3. Trans bronchial biopsy showing evidence of lymphocytic interstitial pneumonitis with diffuse interstitial infiltration, mature lymphocytes, plasma cells, macrophages, neutrophils and moderate benign squamous cells.
have all been described on pathology [2]. LIP is delineated as the diffuse hyperplasia of bronchus-associated lymphoid tissue (BALT) [7].
The differential diagnosis of predominantly lymphocytic interstitial infiltrates on lung biopsy specimens of HIV-positive patients ranges from infectious etiologies, for instance, Pneumocystis jiroveci pneumonia, viral pneumonia, EBV-associated infections (infectious mononucleosis or post-transplant lymphoproliferative disorders) to non-infectious lymphoproliferative disorders such as hypersensitivity pneumonitis, sarcoidosis, Langerhans’ cell histiocytosis, malignant lymphomas and drug-induced interstitial pneumonitis, for example, caused by diphenylhydantoin use [1,8].
LIP occurs more commonly in females, particularly in their fifth decade of life, however the disease can occur at any age [1,9]. In HIV-positive children younger than 13 years of age, LIP is an AIDS defining illness [10]. This is not the case in adults where it occurs rarely and is frequently diagnosed at autopsy. The majority of HIVpositive adults with LIP are black (black Africans and Afro-Caribbean patients) whereas there is no race predilection in children [2,6].
LIP was first reported to be associated with HIV in case reports published in the 1980’s [9]. The pathogenesis of LIP in HIV patients remains unclear but has been postulated as a pulmonary process that may involve an immune response to the direct presence of HIV in lymphoreticular cells [8]. Serum and lung tissue specimens had increased HIV reverse transcriptase in patients with both AIDS and LIP in some studies, though absent in others [9,10]. Previous case reports have demonstrated by in situ hybridization large amounts of HIV RNA in germinal centers of lymphoid tissue; p24 antigen in macrophages and the interstitium; and HIV antigen and antibody in bronchoalveolar lavage fluid of HIV-positive patients with LIP [6]. In mice transgenic for HIV-1 genes, the severity of LIP-associated clinical manifestations and histological lesions correlated with the level of tissular transgene expression [11].
The altered trafficking of lymphocytes in LIP leading to increased epithelial permeability is mediated predominantly by CD 8+ cytotoxic lymphocytes which can destroy HIV-infected macrophages in vitro, leading to release of cytokines [8]. Increased human leukocyte antigen (HLA)-DR5, HLA-DR6 in black patients, and HLADR7 in white patients which predispose to increased circulating CD 8+ lymphocytes have been documented in salivary glands and lungs in diffuse infiltrative lymphocytosis syndrome (DILS) [6,12]. This syndrome is associated with lymphocytic interstitial pneumonitis, myopathy, glandular enlargement and sicca symptoms [12]. Approximately 50% of patients with DILS acquire LIP [2].
In addition to HIV, LIP has been associated with other infectious diseases including Epstein Barr Virus (EBV), human T-cell leukemia virus type I and III (HTLV-I and III), legionella, mycoplasma, chlamydia, tuberculosis, and chronic active hepatitis [2,9,13,14]. It has previously been reported that patients dually infected with HIV and EBV have a higher likelihood of developing LIP [2,9]. However, a review article concluded that though some patients with HIV may have EBV antigen and antibody positivity in lung biopsy, it is quite possible that another condition could have contributed to the disease process in their LIP [2].
LIP is associated with many lymphoproliferative and autoimmune disorders, including Sjogren’s syndrome, rheumatoid arthritis, autoimmune hemolytic anemia, systemic lupus erythematosus, dysproteinemias (agammaglobulinemias, common variable immune deficiency and hypergammaglobulinemias due to elevation of IgM and IgG), myasthenia gravis, autoerythrocyte sensitization syndrome, chronic active hepatitis with renal tubular acidosis, primary biliary cirrhosis, pernicious anemia, celiac sprue, autoimmune thyroid disease, multicentric Castleman’s disease and allogenic bone marrow transplantation [1,2,7,9,15].
Adults with LIP usually present with moderate to severe respiratory symptoms [8]. The diagnosis of LIP usually ranges from 1 - 12 months after the onset of clinical symptoms [11]. These include progressive nonproductive cough and exertional dyspnea, occasionally accompanied by fever, night sweats, chest pain, weight loss and arthralgias. On physical examination, there may be bilateral basal crackles. Wheezes and diminished breath sounds may also be present. In many instances, patients may be asymptomatic with a normal physical examination [6]. Laboratory abnormalities include mild anemia, polyclonal increase in gammaglobulin or a monoclonal increase in IgG or IgM [1]. Extrapulmonary involvement, such as lymphadenopathy (peripheral and mediastinal), parotid enlargement, splenomegaly, lymphocytic infiltration of the kidney, nasopharynx, liver, bone marrow and gastrointestinal tract is rare, as well as, clubbing is usually absent [2,8].
Chest imaging studies are imperative in working up the diagnosis of LIP. Chest x-rays have been reported to appear in several forms predominantly affecting the lower lung zones. These are listed as follows: 1) normal; 2) fine reticular nodular infiltrates; 3) larger reticular nodular infiltrates (features may mimic miliary tuberculosis); 4) combination of #1 and #2 with superimposed alveolar opacities; and 5) focal consolidations [2,6]. Of note, LIP may also lead to atelectasis, fibrosis or bronchiectasis that may be detected on chest imaging [6]. Das and Miller reported that high resolution computer tomography assists in distinguishing fibrosis from reversible inflammatory processes, and also helps in determining the severity of pulmonary involvement [6]. However, nonspecific infiltrations and ground glass appearance may also be present. Chest CT scan also may detect interlobular septal and peribronchovascular interstitium thickening [16]. It must be emphasized that imaging studies, in general, may be indistinguishable from Pneumocystis jiroveci and many other pulmonary diseases [6].
Pulmonary function testing has mainly demonstrated restrictive disease with a reduced or normal total lung diffusion capacity, though obstructive disease and normal findings have also been reported in LIP [6,17]. Arterial blood gas measurements are unhelpful as they may reveal a normal PaO2, or mild to profound hypoxaemia with an increased alveolar to arterial oxygen gradient [6]. Previous case reports suggest chest x-ray infiltrate resolution with declining CD4 lymphocyte counts in the absence of corticosteroids or HAART predicts worsening immunosuppression [2].
As in our patient’s case, the diagnosis of LIP can be made via transbronchial biopsy, but the gold standard of diagnosis is thoracoscopic or open-lung biopsy [6,9]. This is because transbronchial biopsy has a lower diagnostic yield (small sample size, which often leads to multiple biopsy attempts) and increased risks of complications such as pneumothorax [6].
Due to LIP’s rarity, no controlled trials have been carried out to offer a standard recommended treatment regimen. However, immuno-suppression with steroids appears to be key in the initiation of therapy based on prior reports [2]. Before the advent of highly active antiretroviral therapy (HAART), it was noticed that HIV affected patients with LIP clinically improved with steroids. However, treatment duration ranged from weeks to chronic steroid therapy, and this appeared to be influenced by patients’ overall HIV status. Treatment with antiretroviral therapy in HIV affected patients with LIP (either with zidovudinemonotherapy or combination nucleoside analog therapy) has been shown to be effective in improving clinical outcomes. However, therapeutic responses may be influenced by the stage of a patient’s HIV (where lower CD4 counts and high viral loads may reflect poorer outcomes) [6].
No studies limited to individual non-nucleoside reverse transcriptase inhibitors have been recorded in the management of LIP, but HAART has been shown to suppress LIP in several reports [6,18,19]. However, one case report showed manifestation of LIP as a complication of immune reconstitution syndrome shortly after the initiation of HAART [20]. Considering the risks of pulmonary fibrosis, progressive respiratory failure or transformation to malignant lymphoma, we strongly feel as if treatment is warranted [3,9]. However, some patients have been reported to improve without treatment [9]. LIP is often misdiagnosed as Pneumocystis jiroveci in affected patients, which results in inappropriate management with cotrimoxazole [16].
4. CONCLUSION
The patient described in this case exhibited recurrent bouts of bacterial pneumonia, as well as clinical, radiographic and pathological features commonly found in lymphoid interstitial pneumonia (LIP). The complication of repeated bouts of bacterial pneumonia is a rare feature of LIP in HIV positive patients with LIP. To our knowledge, this is one of the first reported cases since the widespread availability and use of HAART.
REFERENCES
- American Thoracic Society/European Respiratory Society (2002) American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. American Journal of Respiratory and Critical Care Medicine, 165, 277-304.
- Swigris, J.J., Berry, G.J., Raffin, T.A. and Kuschner, W.G. (2002) Lymphoid interstitial pneumonia: A narrative review. CHEST, 122, 2150-2164. doi:10.1378/chest.122.6.2150
- Lin, R.Y., Gruber, P.J., Saunders, R. and Perla, E.N. (1988) Lymphocytic interstitial pneumonitis in adult HIV infection. New York State Journal of Medicine, 88, 273- 276.
- Harley, W., Horton, J.M. and Corbin, R.P. (1992) Recurrent pneumococcal pneumonia in an HIV-positive patient with lymphoid interstitial pneumonitis. Southern Medical Journal, 85, 1015-1016. doi:10.1097/00007611-199210000-00022
- Carrington, C.B. and Liebow, A.A. (1966). Lymphocytic interstitial pneumonia (abstract). American Journal of Pathology, 48, 36a.
- Das, S. and Miller, R.F. (2003) Lymphocytic interstitial pneumonitis in HIV infected adults. Sexually Transmitted Infections, 79, 88-93. doi:10.1136/sti.79.2.88
- Davies, C.W.H., Juniper, M.C., Gray, W., Gleeson, F.V., Chapel, H. and Davies, R.J.O. (2000) Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinemia treated with cyclosporin A. Thorax, 55, 88-90. doi:10.1136/thorax.55.1.88
- Travis, W.D., Fox, C.H., Devaney, K.O., Weiss, L.M., O’Leary, T.J., Ognibene, F.P., Suffredini, A.F., Rosen, M.J., Cohen, M.B. and Shelhamer, J. (1992) Lymphoid pneumonitis in 50 adult patients infected with the human immunodeficiency virus: Lymphocytic interstitial pneumonitis versus nonspecific interstitial pneumonitis. Human Pathology, 23, 529-541. doi:10.1016/0046-8177(92)90130-U
- Najjar, N., Gamal, A.E., Halabi, S. and Leyenson, V. (2005) A 38-year-old man with HIV Infection and sub acute onset of cough and dyspnea. CHEST, 128, 4008- 4012. doi:10.1378/chest.128.6.4008
- Dean, N.C., Golden, J.A., Evans, L.A., Warnock, M.L., Addison, T.E., Hopewell, P.C. and Levy, P.A. (1988) Human immunodeficiency virus recovery from bronchialveolar lavage fluid in patients with AIDS. CHEST, 93, 1176-1179. doi:10.1378/chest.93.6.1176
- Dufour, V., Wislez, M., Bergot, E., Mayaud, C. and Cadranel, J. (2003) Improvement of symptomatic human immunodeficiency virus-related lymphoid interstitial pneumonia in patients receiving highly active antiretroviral therapy. Clinical Infectious Disease, 36, e127-e130. doi:10.1086/374665
- Kazi, S., Cohen, P.R., Williams, F., Schempp, R. and Reveille, J.D. (1996) The diffuse infiltrative lymphocytosis syndrome: Clinical and immunogenetic features in 35 patients. AIDS, 10, 385-391. doi:10.1097/00002030-199604000-00006
- Ishikawa, N., Awaya, Y., Maeda, H., Miyazaki, M., Fujitaka, K., Yamasaki, M., Kondo, K., Oguri, T., Isobe, T. and kohno, N. (2002) KL-6 as an indicator for lymphocytic interstitial pneumonia (LIP) in a human T-lymphotrophic virus type 1 (HTLV-1) carrier. Annals of Hematology, 81, 474-477. doi:10.1007/s00277-002-0465-4
- Freemer, M.M. and King Jr., T.W. (2003) Lymphoplasmocytic infiltration of the lung. In: Schwarz, M.I. and King Jr., T.W., Eds, Interstitial Lung Disease, B. C. Decker, Hamilton, 827.
- Macfarlane, A. and Davies, D. (1973) Diffuse lymphoid interstitial pneumonia. Thorax, 28, 768-776. doi:10.1136/thx.28.6.768
- Rosewarne, D., Reynolds, J.H., Trotter, S.E. and Burge, P.S. (2008) The idiopathic interstitial pneumonias—A survival guide. Imaging, 20, 289-302.
- Balestro, E., Pastore, U., Dal Farra, F., Rossi, G., Calabro, S., Polverosi, R. and Vassallo, E. (2008) A 40-year-old man with hemolytic anemia, Ig deficiency, and bilateral pulmonary infiltrates. CHEST, 133, 1517-1523.
- Cao, M., Soogoor, M. and Vintch, J. (2005) Lymphocytic Interstitial Pneumonia (LIP) and Human Immunodeficiency Virus (HIV) in the modern era. CHEST, 128, 426S.
- Innes, A.L., Huang, L. and Nishimura, S.L. (2004) Resolution of lymphocytic interstitial pneumonitis in an HIV infected adult after treatment with HAART. Sexually Transmitted Infections, 80, 417-418. doi:10.1136/sti.2004.009886
- Ingiliz, P., Appenrodt, B., Gruenhage, F., Vogel, M., Tschampa, H., Tasci, S. and Rockstroh, J.K. (2006) Lymphoid pneumonitis as an immune reconstitution inflammatory syndrome in a patient with CD4 cell recovery after HAART initiation. HIV Medicine, 7, 411-414. doi:10.1111/j.1468-1293.2006.00389.x