Open Journal of Anesthesiology
Vol.2 No.4(2012), Article ID:22247,7 pages DOI:10.4236/ojanes.2012.24043
Pseudocholinesterase Deficiency: A Case Report and Literature Review
![]()
Department of Anesthesiology, University of Pittsburgh School of Medicine, Pittsburgh, USA.
Email: sakait@upmc.edu
Received July 1st, 2012; revised August 9th, 2012; accepted August 27th, 2012
Keywords: Prolonged Paralysis; Succinylcholine; Pseudocholinesterase Deficiency; Dibucaine Number; Case Review
ABSTRACT
A 72-year-old male underwent neck dissection and parotidectomy with facial nerve preservation. Endotracheal intubation was facilitated with succinylcholine. Prolonged muscle paralysis which was first detected after failure to stimulate the facial nerve with electrocautery, lasted five hours. Laboratory tests indicated pseudocholinesterase (PChE) deficiency. A thyroidectomy one month later was performed uneventfully using rocuronium as a muscle relaxant. Literature review revealed a total of 40 PChE deficiency cases being reported since 1956.
1. Introduction
Pseudocholinesterase (PChE), also referred to as butyrylcholinesterase, serum cholinesterase, plasma cholinesterase, and false cholinesterase, is a liver-derived plasma protein capable of hydrolyzing esters including muscle relaxants (i.e. succinylcholine, atracurium, mivacurium) and ester type local anesthetics (i.e. procaine, chloroprocaine, tetracaine, cocaine) with a serum half-life of eight to 12 days. A deficiency of this enzyme, either inherited and/or acquired, can lead to significantly pro-longed activity of these medications.
We encountered a patient with prolonged paralysis after exposure to an intubation dose of succinylcholine for parotid gland resection. Succinylcholine was indicated to facilitate an endotracheal insertion with a laryngoscope with the expectation that muscle tone should quickly be regained for the surgeons to preserve the facial nerve during neck dissection. The importance of early recognition of prolonged paralysis is stressed and its management, as well as a literature review of reported PChE cases, is discussed.
2. Case Report
A 72-year-old patient presented for resection of a left parotid gland lesion. His past medical history was notable for benign prostatic hypertrophy, acid reflux, and basal cell cancer of the face. There were no prior surgeries or anesthetics. Family history was noncontributory. He was married with four children and six grandchildren. He denied use of tobacco, alcohol, or illicit drugs. There were no allergies at that time. His medications included only 0.5 mg dutasteride daily. Present illness at the time of surgery included a painful skin lesion overlying the angle of the left aspect of the mandible with an accompanied parotid mass. He was scheduled for an excision of the left facial skin lesion, left partial parotidectomy with facial nerve dissection, and left lymph node biopsy. His height and weight were 188 cm and 110 kg, respectively. On physical exam, his heart sounds were regular without murmurs, rubs, or gallops; lungs were clear bilaterally; abdomen was soft and non-tender; no lower extremity edema.
After premedication with 2 mg midazolam and 100 mcg fentanyl intravenously (iv) and preoxygenation, general anesthesia was induced with 200 mg propofol with 100 mg lidocaine iv. Succinylcholine 200 mg iv was administered to facilitate the placement of an endotracheal tube. General anesthesia was maintained with desflurane and 50% oxygen. Initial vital signs were stable with an SaO2 of 100%, heart rate at 85/min and regular, blood pressure of 145/75 mmHg, and a temperature of 36.3˚C. Volume controlled ventilation was used with a tidal volume of 750 mL, a respiratory rate of 10/min, and peak inspiratory pressure of 20 cm H2O. The skin lesion, actinic keratosis, was excised using an elliptical incision and the tissue was submitted to pathology. The anterior aspect of the sternocleidomastoid muscle was mobilized and the anterior aspect of the external auditory canal was identified along with the posterior belly of the digastrics muscle, revealing the left facial nerve structure. At this point, approximately one hour into the procedure, the surgeon was unable to create muscle contractions by direct stimulation of the facial nerve with an electrocautery. The anesthesiologist confirmed muscle paralysis with an ulnar nerve stimulator via a train-of-four (TOF) technique without any twitch.
Given the use of succinylcholine and the patient’s otherwise unremarkable preoperative condition, PChE deficiency was suspected. A blood sample was submitted to measure serum PChE level. The surgical team continued to carefully identify and protect the facial nerve, and the lateral aspect of the parotid gland was removed and submitted to pathology. The wound was irrigated and closed in layers. The patient’s paralysis continued at the end of the two-hour and eleven-minute operation. He was transported to the post-anesthesia care unit with a propofol infusion (100 mcg/kg/min) under mechanical ventilation. At five hours after induction of general anesthesia, an adequate TOF stimulation was obtained. No fresh frozen plasma was used. The propofol infusion was then terminated and the endotracheal tube was removed after the patient was able to follow commands, sustain a head lift, and register adequate tidal volumes.
The serum PChE level submitted during surgery was 1100 IU/L (normal 3300 - 7600 IU/L), supporting the diagnosis of PChE deficiency. The patient was counseled in regards to the function of PChE and what it means to be deficient. The patient was instructed to wear a Medic Alert bracelet and to obtain further testing for him and his family.
He was discharged home on post-operative day one, and returned approximately one month later for an uneventful total thyroidectomy using rocuronium instead of succinylcholine for muscle paralysis. Before the second surgery, a repeated plasma PChE test revealed a level of 1100 IU/L, effectively confirming his PChE deficiency. He declined additional testing, including dibucaine number and genetic testing.
3. Discussion
PChE deficiency is one of many causes of prolonged paralysis during general anesthesia. In the current case, failure to induce muscle contraction with direct electrostimulation of the facial nerve led to the identification of the prolonged paralysis. The differential diagnoses for prolonged paralysis can be seen in Table 1. While most are common, treatable, and avoidable causes of pro-longed muscle blockade, PChE deficiency after the exposure of potential offending muscle relaxants should also be suspected.
PChE deficiency can be either inherited or acquired. The inherited etiologies of PChE deficiency is attributed to genetic mutations at a single autosomal location on the long arm of chromosome 3. The genetics of PChE deficiency were first documented in the 1950’s; five autosomal alleles on locus E1 are now known to be responsible for synthesizing PChE. The five alleles are: usual (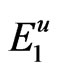 ), atypical (
), atypical (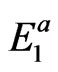 ), fluoride resistant (
), fluoride resistant (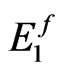 ), K variant (
), K variant (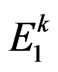 ) and silent (
) and silent (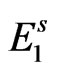 ). The usual allele codes for a normal PChE quantity and quality while the others to a changeable degree do not. These five alleles give rise to 15 phenotypes that produce varying degrees of PChE deficiency. To further illustrate the commonality of this disorder, it has been reported that 96% of the population is homozygous for the usual enzyme while 4% posses at least one abnormal allele. This translates to 1:3000 in the general population possessing two abnormal alleles and 1:2400 belonging to those with a clinically significant prolonged paralysis after exposure to PChE dependent muscle relaxants [1]. On the other hand, there are many acquired etiologies for having low PChE levels (Table 1). However, as common as these modifiers may be, more often a patient possessing any of these risk factors will have a normal response to paralytics. Imerman et al. points out that a reduction to 5% of normal PChE activity would be required to increase paralysis by one hour after administration of succinylcholine [2]. Furthermore, Foldes and colleagues documented that liver disease alone, resulting in PChE levels 20% of normal, merely increases neuromuscular blockade threefold [3]. It is unlikely that any of the above acquired etiologies could produce a clinically significant prolongation of paralysis, illustrating that a genetic basis of PChE deficiency should be considered most likely.
). The usual allele codes for a normal PChE quantity and quality while the others to a changeable degree do not. These five alleles give rise to 15 phenotypes that produce varying degrees of PChE deficiency. To further illustrate the commonality of this disorder, it has been reported that 96% of the population is homozygous for the usual enzyme while 4% posses at least one abnormal allele. This translates to 1:3000 in the general population possessing two abnormal alleles and 1:2400 belonging to those with a clinically significant prolonged paralysis after exposure to PChE dependent muscle relaxants [1]. On the other hand, there are many acquired etiologies for having low PChE levels (Table 1). However, as common as these modifiers may be, more often a patient possessing any of these risk factors will have a normal response to paralytics. Imerman et al. points out that a reduction to 5% of normal PChE activity would be required to increase paralysis by one hour after administration of succinylcholine [2]. Furthermore, Foldes and colleagues documented that liver disease alone, resulting in PChE levels 20% of normal, merely increases neuromuscular blockade threefold [3]. It is unlikely that any of the above acquired etiologies could produce a clinically significant prolongation of paralysis, illustrating that a genetic basis of PChE deficiency should be considered most likely.
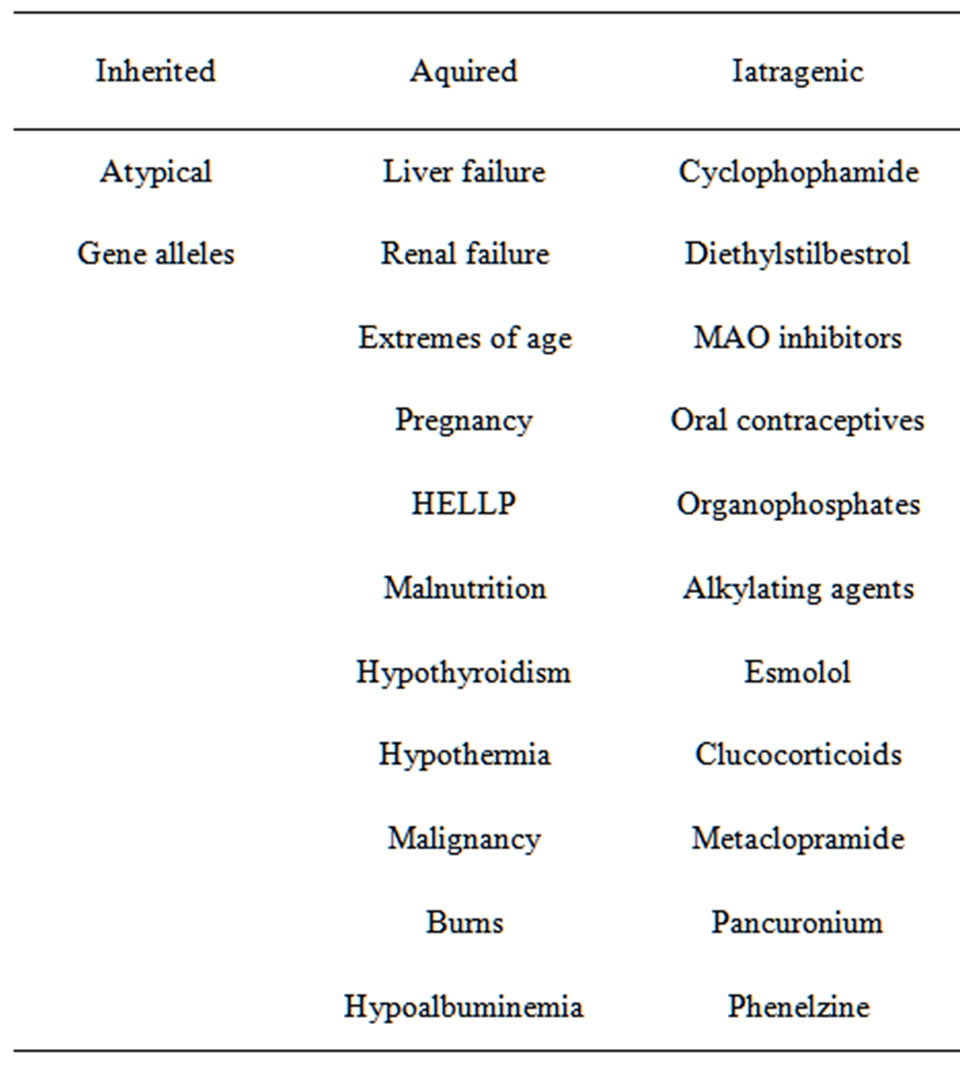
Table 1. Causes of pseudocholinesterase deficiency [2,7-11, 14,17,18].
To verify a suspected PChE deficiency, one should first obtain a serum PChE level, the normal reference range being 3300 - 7600 IU/L. Of note, a repeated test approximately one week or more after exposure to the offending agents is warranted for confirmation, because PChE level can be artificially decreased initially [4]. Our patient’s PChE level immediately after the exposure of succinylcholine was 1080 IU/L and was confirmed low (1100 IU/L) one month later. Also, a dibucaine number (DN) helps to decipher whether the PChE deficiency is genetic in origin, and can imply heteroor homozygosity. Dibucaine is a local anesthetic which inhibits PChE activity when mixed with a blood sample. The percentage of PChE inhibited will yield a DN; 80 - 100 indicates normal PChE function, 40 - 70 indicates heterozygous, and less than 30 indicates homozygous for atypical genotype. From a practical standpoint, PChE level and DN have been shown to be sufficient in determining a genetic origin of PChE deficiency. However, molecular testing is also available, including gel-electrophoresis and immunoelectrophoresis, and this method has been shown to improve biological diagnosis in one-third of patients; however, the clinical implications are lacking [5].
The management of patients with prolonged paralysis due to PChE deficiency is mainly conservative. Patients are maintained with mechanical ventilation under sedation while the effect of the paralytic is cleared [3]. Theoretically, if urgent reversal of paralysis becomes necessary, for example to assess neurologic function, fresh frozen plasma (FFP) or packed red blood cells can be utilized to provide exogenous PChE [6,7]. Of note, anticholinesterase agents (i.e. neostigmine and physostigmine) inhibit PChE activity and can lead to paradoxical worsening of paralysis [7]. If PChE deficiency is suspected, the patient and family should be counseled with recommendation of further diagnostic tests and a Medic Alert bracelet should be issued [4].
In a literature review searching for the key words “pseudocholinesterase deficiency” in PubMed (http://www.ncbi.nlm.nih.gov/pubmed/:lastaccessedMay5, 2012), a total of 39 articles featuring 40 case reports on PChE deficiency were found and are summarized in Tables 2 and 3 [1-39]. The earliest description of PChE deficiency was reported in 1956 [3] and the latest case report was published in 2011 [20]. The length of paralysis ranged from 50 minutes [28] to more than ten hours [34]. Only one case reported the use of FFP to facilitate recovery from paralysis [6]. No mortality or lasting complications were reported. PChE levels were checked in all but six cases, ranging from 5 - 2765 IU/L. DN was checked in only 12 of the 40 cases ranging from 19 - 81 [1,4,8,9,12,14,20,24,26,33,38]. Two cases reported a “normal” DN [8,9] and one with a “very low” value [1]. In terms of etiology, 20 cases were considered genetic [1, 4,7,12,14,19,20,23,24,26,27,29,34,35,37,38]. Six were due to acquired causes such as liver disease [28], pregnancy [33], HELLP syndrome [17], malnutrition [8], or from administration of cyclophosphamide [10] or diethylstilbestrol [9]. The remaining 15 cases were from an unknown etiology [2,6,13,21,22,25,27,30,32,36,39]. Medical staff in only 18 cases inquired about family history; eight reported a family member with a similar experience [1,4,7, 29,37]. None of these case reports performed genetic testing on family members.
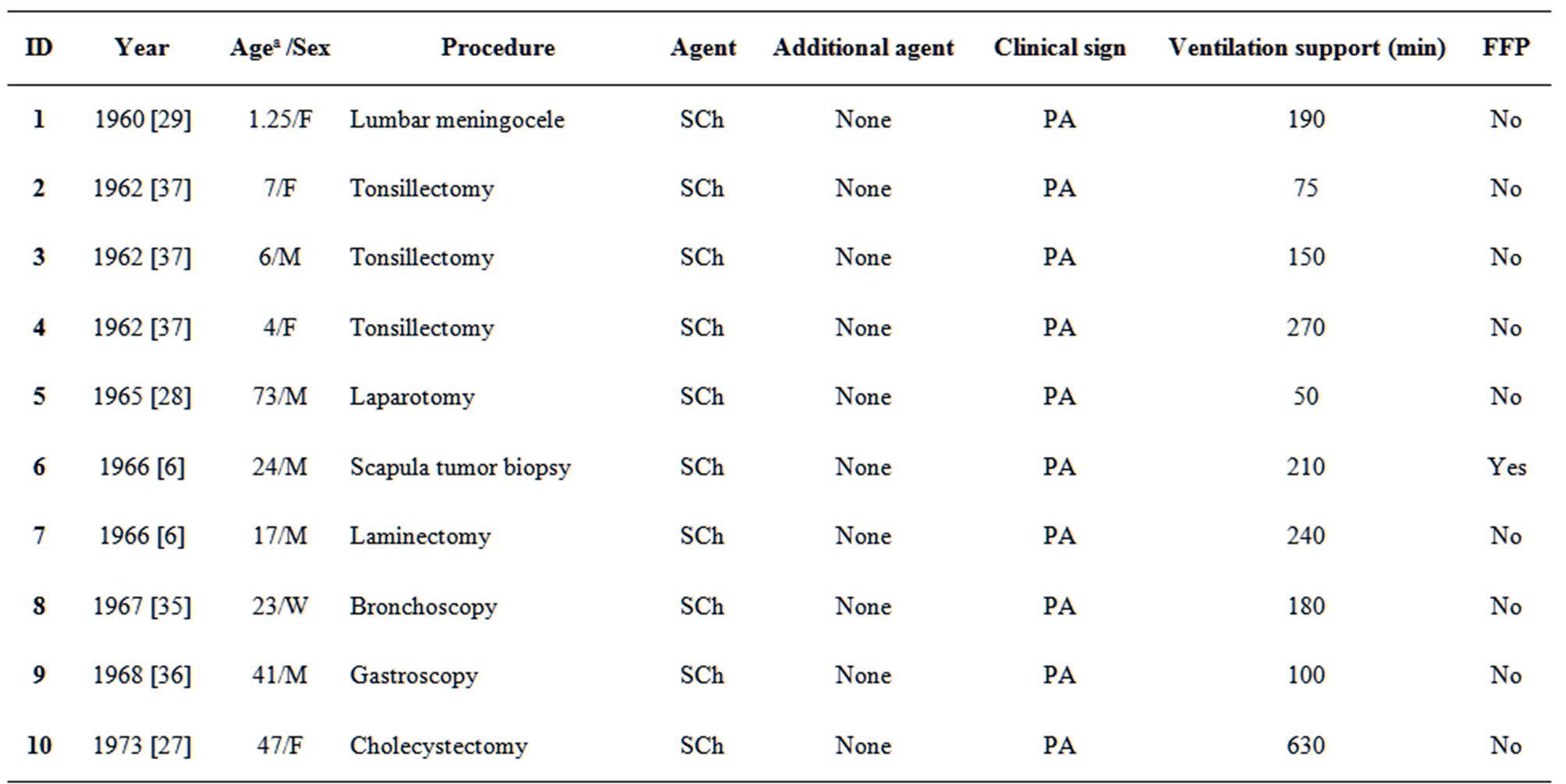
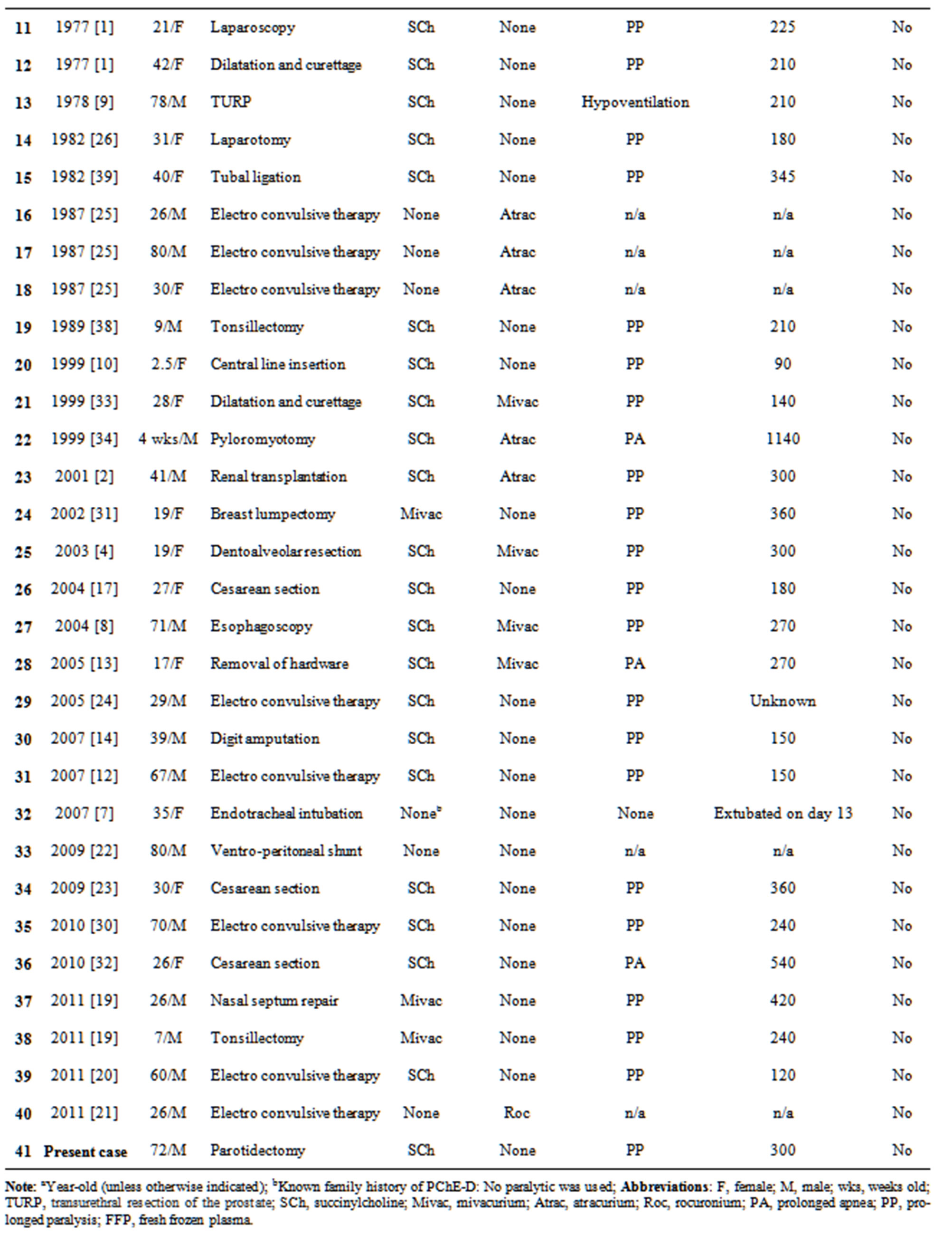
Table 2. Summary of the reported cases of pseudocholinesterase deficiency.
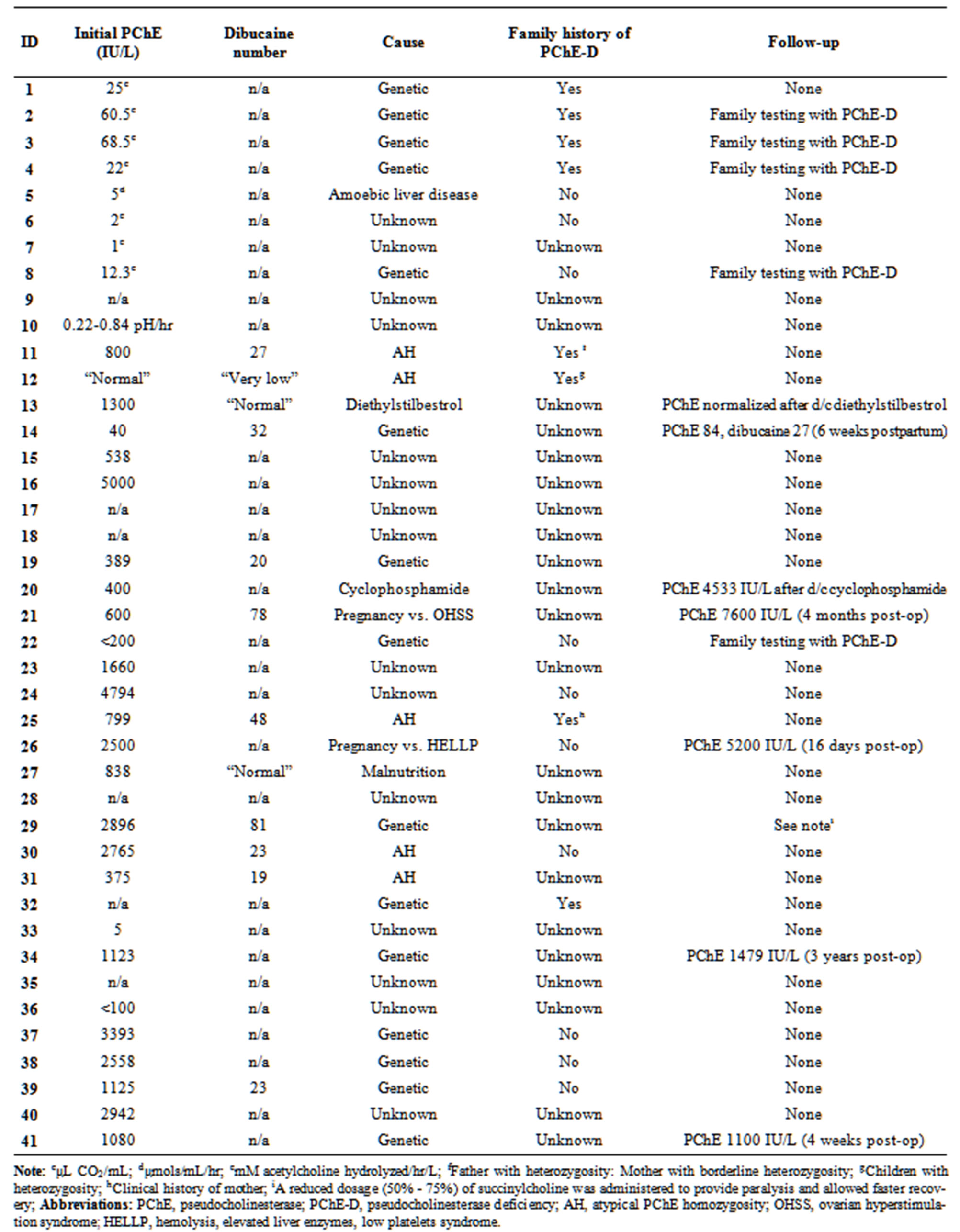
Table 3. Summary of the reported cases of pseudocholinesterase deficiency (continued).
Note: cμL CO2/mL; dμmols/mL/hr; emM acetylcholine hydrolyzed/hr/L; fFather with heterozygosity: Mother with borderline heterozygosity; gChildren with
4. Conclusion
PChE deficiency is a rare cause of prolonged paralysis, regarding which any clinician utilizing paralytics should be educated. When suspecting the diagnosis, supportive care should be given to ensure patient safety, a PChE level and DN should be obtained, and genetic testing should be offered if indicated. Genetic counseling should be offered not only to the patient but also to members of family.
5. Acknowledgements
The authors thank Christine M. Heiner, BA (Scientific Writer, Department of Anesthesiology/Department of Surgery, University of Pittsburgh School of Medicine) for her editorial assistance with the manuscript.
REFERENCES
- L. Putnam, “Pseudocholinesterase Deficiency: An Additional Preoperative Consideration in Outpatient Diagnostic Procedures,” Southern Medical Journal, Vol. 70, No. 7, 1977, pp. 831-833. doi:10.1097/00007611-197707000-00021
- B. Imerman and L. J. Caruso, “Prolonged Neuromuscular Block in a Patient Undergoing Renal Transplantation,” Journal of Clinical Anesthesia, Vol. 13, No. 7, 2001, pp. 540-544. doi:10.1016/S0952-8180(01)00321-X
- F. F. Foldes, L. Rendell-Baker and J. H. Birch, “Causes and Prevention of Prolonged Apnea with Succinylcholine,” Anesthesia and Analgesia, Vol. 35, No. 6, 1956, pp. 609-633. doi:10.1213/00000539-195611000-00005
- A. Maiorana, “Heterozygous Pseudocholinesterase Deficiency: A Case Report and Review of the Literature,” Journal of Oral Maxillofacial Surgery, Vol. 61, No. 7, 2003, pp. 845-847. doi:10.1016/S0278-2391(03)00163-0
- C. Cerf, M. Mesguish, I. Gabriel, S. Amselem and P. Duvaldestin, “Screening Patient with Prolonged Neuromuscular Blockade after Succinylcholine and Mivacurium,” International Anesthesia Research Society, Vol. 94, No. 2, 2001, pp. 461-466.
- A. M. Karahasanoglu and P. Ozand, “Pseudocholinesterase: Two Cases of Pseudocholinesterase Deficiency,” The Turkish Journal of Pediatrics, Vol. 8, No. 1, 1966, pp. 10-19.
- A. J. Whyte and H. E. Wang, “Prehospital Airway Management Complicated by Reported Pseudocholinesterase Deficiency,” Prehospital Emergency Care, Vol. 11, No. 3, 2007, pp. 343-345. doi:10.1080/10903120701347984
- A. Niazi, I. E. Leonard and B. O’Kelly, “Prolonged Neuromuscular Blockade as a Result of Malnutrition-Induced Pseudocholinesterase Deficiency,” Journal of Clinical Anesthesia, Vol. 16, No. 1, 2004, pp. 40-42. doi:10.1016/j.jclinane.2003.02.010
- T. Archer and E. C. Janowsky, “Plasma Pseudocholinesterase Deficiency Associated with Diethylstilbestrol Therapy,” Anesthesia and Analgesia, Vol. 57, No. 6, 1978, pp. 726-729.
- V. Koseoglu, J. Chiang and K. W. Chan, “Acquired Pseudocholinesterase Deficiency after High-Dose Cyclophosphamide,” Bone Marrow Transplant, Vol. 24, No. 12, 1999, pp. 1367-1368. doi:10.1038/sj.bmt.1702097
- F. K. Soliday, Y. P. Conley and R. Henker, “Pseudocholinesterase Deficiency: A Comprehensive Review of Genetic, Acquired, and Drug Influences,” American Association of Nurse Anesthetists Journal, Vol. 78, No. 4, 2010, pp. 313-320.
- J. Williams, P. Rosenquist, L. Arias and W. V. McCall, “Pseudocholinesterase Deficiency and Electroconvulsive Therapy,” Journal of ECT, Vol. 23, No. 3, 2007, pp. 198- 200. doi:10.1097/YCT.0b013e318070c686
- K. Kendrick, “Prolonged Paralysis Related to Mivacurium: A Case Study,” Journal of Perianesthesia Nursing, Vol. 20, No. 1, 2005, pp. 7-12. doi:10.1016/j.jopan.2004.11.006
- C. A. Leadingham, “A Case of Pseudocholinesterase Deficiency in the PACU,” Journal of Perianesthesia Nursing, Vol. 22, No. 4, 2007, pp. 265-274. doi:10.1016/j.jopan.2007.05.005
- J. G. Ramirez, J. Sprung, M. T. Keegan and B. A. Hall, “Neostigmine-Induced Prolonged Neuromuscular Blockade in a Patient with Atypical Pseudocholinesterase,” Journal of Clinical Anesthesia, Vol. 17, No. 3, 2005, pp. 221-224. doi:10.1016/j.jclinane.2004.06.012
- W. E. Hodgkin, E. R. Giblett, H. Levine, W. Bauer and A. G. Motulsky, “Complete Pseudocholinesterase Deficiency: Genetic and Immunologic Characterization,” Journal of Clinical Investigation, Vol. 44, No. 3, 1965, pp. 486-493. doi:10.1172/JCI105162
- S. Lurie, O. Sadan, M. Glezerman and T. Ezri, “Pseudocholinesterase Deficiency Associated with HELLP Syndrome,” American Journal of Perinatology, Vol. 21, No. 6, 2004, pp. 315-317. doi:10.1055/s-2004-831881
- M. J. Lovely, S. K. Patteson, F. J. Beuerlein and J. T. Chesney, “Perioperative Blood Transfusion May Conceal Atypical Pseudocholinesterase,” Anesthesia and Analgesia, Vol. 70, No. 3, 1990, pp. 326-327. doi:10.1213/00000539-199003000-00017
- R. Gelo, A. Jurek-Gelo, R. Wardaszko and A. Kanski, “Congenital Pseudocholinesterase Deficiency,” Anesthesiology Intensive Therapy, Vol. 1, 2011, pp. 31-33.
- I. M. Reti, J. Torres, A. Morad and G. Javaram, “Pseudocholinesterase Deficiency in an ECT Patient: A Case Report,” Psychosomatics, Vol. 52, No. 4, 2011, pp. 392- 397. doi:10.1016/j.psym.2011.01.029
- C. Batistaki, K. Kesidis and S. Apostolaki, “Rocuronium Antagonized by Sugammadex for Series of Electroconvulsive Therapy (ECT) in a Patient with Pseudocholinesterase Deficiency,” Journal of ECT, Vol. 27, No. 1, 2011, pp. 47-48. doi:10.1097/YCT.0b013e318205e1d3
- J. Shiotsuka, M. Sanui and A. Lefor, “Safe Use of LanDiolol Hydrochloride in a Patient with Marked Pseudocholinesterase Deficiency,” Journal of Anesthesia, Vol. 24, No. 2, 2010, pp. 309-310. doi:10.1007/s00540-010-0889-2
- R. Somers, Y. Jacquemyn, L. SerMeus and M. Vercauteren, “Corrected Scoliosis, Cholinesterase Deficiency, and Cesarean Section: A Case Report,” Case Reports in Medecine, 2009, Article ID: 957479. doi:10.1155/2009/957479
- J. G. Augoustides, H. H. Hosalkar, J. P. O’Reardon, W. A. Kofke and C. J. Datto, “Customized Anesthetic Preservation of Ictal Threshold in Electroconvulsive Therapy: Role of Adjunctive Remifentanil with Etomidate,” Journal of ECT, Vol. 21, No. 2, 2005, pp. 128-131. doi:10.1097/01.yct.0000167463.71704.52
- F. G. Hicks, “ECT Modified by Atracurium,” Convulsive Therapy, Vol. 3, No. 1, 1987, pp. 54-59.
- B. R. Kuhnert, E. H. Philipson, R. Pimental and P. M. Kuhnert, “A Prolonged Chloroprocaine Epidural Block in a Postpartum Patient with Abnormal Pseudocholinesterase,” Anesthesiology, Vol. 56, No. 6, 1982, pp. 477-478. doi:10.1097/00000542-198206000-00017
- M. Cherington and G. Lasater, “Prolonged Paralysis in Pseudocholinesterase Deficiency,” Archives of Neurology, Vol. 28, No. 4, 1973, pp. 274-275. doi:10.1001/archneur.1973.00490220082014
- D. P. Mullan and N. E. Williams, “Left-Sided Amoebic Liver Abscess with Pulmonary Involvement, Pericarditis, and Pseudocholinesterase Deficiency,” British Medical Journal, Vol. 1, 1965, pp. 235-236. doi:10.1136/bmj.1.5429.235
- L. Kaufman, H. Lehmann and E. Silk, “Suxamethonium Apnoea in an Infant: Expression of Familial Pseudocholinesterase Deficiency in Three Generations,” British Medical Journal, Vol. 1, 1960, pp. 166-167. doi:10.1136/bmj.1.5167.166
- E. Martinez-Amoros, E. Real, P. A. Barrado, M. Carulla, M. Urretavizcaya and N. Cardoner, “Optimizing Electroconvulsive Therapy in Non-Suspected Pseudocholinesterase Deficiency: Laryngeal Mask Use and Neuromuscular Selection,” Psychosomatics, Vol. 51, No. 6, 2010, pp. 537- 538.
- M. Hemadi, M. Purva and V. Traykova, “Unexpected Prolonged Neuromuscular Block after Mivacurium: A Case Report,” Medical Principles and Practice, Vol. 11, No. 1, 2002, pp. 50-52. doi:10.1159/000048662
- K. J. Grim, K. W. Arendt, A. K. Jacob, C. H. Rose and M. T. Keegan, “Urgent Cesarean Delivery and Prolonged Ventilatory Support in a Parturient with Fontan Circulation and Undiagnosed Pseudocholinesterase Deficiency,” International Journal of Obstetric Anesthesia, Vol. 10, No. 6, 2010, pp. 184-188.
- H. J. Southgate, S. K. Anderson, N. G. Lavies and M. J. Rymer, “Pseudocholinesterase Deficiency: A Dangerous, Unrecognized Complication of Ovarian Hyperstimulation Syndrome,” Annals of Clinical Biochemistry, Vol. 36, No. 2, 1999, pp. 256-258.
- V. Ho and H. Osiovich, “A Case of Pseudocholinesterase Deficiency in the Neonate,” American Journal of Perinatology, Vol. 16, No. 7, 1999, pp. 351-353. doi:10.1055/s-2007-993883
- J. Theodore, J. E. Millen, H. V. Murdaugh and E. D. Robin, “Prolonged Postoperative Apnea with Pseudocholinesterase Deficiency,” The American Review of Respiratory Disease, Vol. 96, No. 3, 1967, pp. 508-511.
- J. F. Glasgow and J. H. Elwood, “Pseudocholinesterase Deficiency,” Journal of the Irish Medical Association, Vol. 61, No. 374, 1968, pp. 268-272.
- J. S. Ruddell, “Apnea Due to Suxemathonium,” Lancet, Vol. 1, No. 7234, 1962, pp. 832-833. doi:10.1016/S0140-6736(62)91842-1
- P. S. Myles and R. N. Westhorpe, “A Patient with Sanfilippo Syndrome and Pseudocholinesterase Deficiency, Further Complicated by Post-Tonsillectomy Hemorrhage,” Anesthesia and Intensive Care, Vol. 17, No. 1, 1989, pp. 86-88.
- S. J. Chung and D. Andrews, “Prolonged Postoperative Succinylcholine-Induced Apnea with Pseudocholinesterase Deficiency,” Journal of Tennessee Medical Association, Vol. 75, No. 8, 1982, pp. 535-536.