Journal of Water Resource and Protection
Vol.5 No.6(2013), Article ID:33034,17 pages DOI:10.4236/jwarp.2013.56058
Reclaiming Biologically Pretreated Greywater for Reuse by Photocatalytic Oxidation: Qualitative Study on the Removal of Trace Organics
1Institute of Wastewater Management and Water Protection, Hamburg University of Technology, Eissendorfer Str. 42, Hamburg, Germany
2Central Laboratory of Analytical Chemistry, Hamburg University of Technology, Eissendorfer Str. 38, Hamburg, Germany
Email: holli@tuhh.de
Copyright © 2013 Holger Gulyas et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 13, 2013; revised April 15, 2013; accepted May 9, 2013
Keywords: Flame Retardants; Greywater; Photocatalytic Oxidation; Reuse; Trace Organics
ABSTRACT
This study was carried out for gathering qualitative information about the potential of photocatalytic oxidation for the removal of trace organics (analysed by gas chromatography coupled to mass spectrometry, GC/MS) from biologically pretreated greywater to make it suitable for high quality reuse applications like groundwater recharge. Additionally, fractions of bulk organics (humic substances, building blocks, and low molecular weight organic acids) were quantified by liquid chromatography with organic carbon detection. Biologically pretreated greywater was subjected to photocatalytic oxidation in open stirred vessel reactors with UV lamps positioned over the reactors. UV doses of 0, 5, and 15 Wh∙L−1 and TiO2 P25 photocatalyst concentrations of 1, 5, 10, and 20 g∙L−1 were investigated. Photocatalysis experiments with a 15 Wh∙L−1 UV dose were also conducted in the presence of 1 g∙L−1 powdered activated carbon. Subsequent to mere contact of the photocatalyst to biologically pretreated greywater without UV, GC/MS did not indicate a substantial removal of trace organics, while humic substances were increasingly adsorbed by increasing photocatalyst concentration. A UV dose of 15 Wh∙L−1 and TiO2 concentrations > 5 g∙L−1 were favorable conditions for photocatalytic oxidation resulting in the removal of most of the trace organics, especially chlorinated phosphate flame retardants. Also humic substances were efficiently removed under these conditions. Photocatalytic oxidation is thus a promising process for advanced greywater treatment prior to groundwater recharge. Addition of powdered activated carbon did not improve trace and bulk organics removal by photocatalysis with a UV dose of 15 Wh∙L−1 and with photocatalyst concentrations > 5 g∙L−1.
1. Introduction
Compared to municipal wastewater, greywater is a more suitable resource for high quality reuse applications such as groundwater recharge, because it is safely segregated from industrial wastewaters (potentially containing hazardous contaminants) and less polluted with nutrients and pathogens. Organics can be removed from greywater by aerobic biological treatment. TOC concentrations were reported to be in the range of 12 - 48 mg∙L−1 when bathroom greywater was treated by multistage rotating biological contactors [1]. Subsequent to treatment of the entire greywater (including kitchen greywater) of the ecosettlement Luebeck-Flintenbreite (Germany) in an intermittently fed vertical-flow constructed wetland, it exhibited TOC concentrations of 5 - 15 mg∙L−1 [2].
Purity of aerobically treated greywater disinfected by UV irradiation is sufficient for reuse purposes such as toilet flushing and even laundry [3]. However, for higher quality reuse applications, biological treatment of greywater might not suffice. It has been suggested that for groundwater recharge the TOC of the infiltrated wastewater should be reduced to a level similar to the natural background of the local aquifer [4], i.e. to a concentration of 3 mg∙L−1 or even less. In California, the TOC guideline for direct groundwater recharge is as low as 0.5 mg∙L−1 unless the reclaimed wastewater is diluted with clean water [5]. When treated wastewaters have to be chlorinated before reuse, the TOC should be lower than 2 mg∙L−1. Otherwise, carcinogenic trihalomethanes are formed in concentrations above the German drinking water guideline of 10 µg∙L−1 [6].
Besides aggregate organic compounds measured as TOC or COD, the concentration of particular recalcitrant organic micropollutants is relevant when reclaimed greywater is reused for groundwater recharge. Hundreds of trace organics have been identified or at least tentatively identified in raw greywater [7-9], many of them related to personal care products. Also priority pollutants listed in the EU Water Framework Directive have been detected in bathroom greywater [1]. Among the organic micropollutants found in greywater samples, there were also recalcitrant compounds detectable subsequent to biological treatment [1,9,10].
As reverse osmosis is no complete barrier against organic micropollutants [11-14], the inclusion of oxidative processes such as ozonation or advanced oxidation processes (AOPs) in wastewater reclamation treatment trains containing a reverse osmosis stage was recommended [15]. With an ozone dose of 15 mg∙L−1, efficient removal of eight organic micropollutants from a biologically treated greywater spiked with the trace organics in the 50 - 1700 ng∙L−1 range was demonstrated [16].
Heterogeneous photocatalytic oxidation (PCO) using the stable catalyst TiO2, is a favorable AOP, because TiO2 is an economically feasible industrial mass product and the process can be powered by sunlight. Boyjoo et al. [17] have investigated photocatalytic oxidation of shower water (TOC: 25 mg∙L−1) in 31 L batches using an 800 W medium pressure mercury lamp as UV source. After 6 hours illumination, only slightly more than half of the initial TOC was removed, however. Sanchez et al. [18] subjected hotel greywater with a DOC of 29 mg∙L−1 to heterogeneous photocatalytic oxidation also using a medium pressure mercury lamp (150 W) for UV illumination. DOC reduction after 2.5 h was only slightly above 65%. This data suggests that photocatalytic oxidation of greywater without any pretreatment requires a high energy input.
Unfortunately, also biologically pretreated greywater is a difficult candidate for heterogeneous PCO when compared to other pretreated wastewaters because of its inorganic matrix [19,20]. Reactors for solar heterogeneous PCO have been recently reviewed [21] and were evaluated as not feasible for treating large volumes of wastewater because of the considerable area demand. The combination of PCO at TiO2 concentrations of 1 and 5 g∙L−1 with powdered activated carbon (PAC) adsorption using 1 g∙L−1 PAC was promising for the advanced treatment of biologically pretreated greywater with an initial nonpurgeable dissolved organic carbon (np-DOC) concentration of 10 mg∙L−1; even when the photocatalyst/PAC mixtures were reused ten times, the np-DOC concentration could be kept below 2 mg∙L−1 [22].
In this study, the influence of an unusually wide range of TiO2 concentrations (1 to 20 g/L) on the elimination of trace and bulk organics from biologically treated greywater by PCO with UV doses of 5 and 15 Wh∙L−1 (UV illumination periods of one and three days, respectively) was evaluated. Additionally, trace and bulk organics removal by PCO with the different TiO2 concentrations was tested in the presence of 1 g/L PAC at a UV dose of 15 Wh∙L−1. Trace organics were tentatively identified by GC/MS in non-target analyses subsequent to solid phase extraction (SPE) of the aqueous phase. Bulk organic fractions such as humic substances, building blocks and low molecular weight (LMW) organic acids were studied by liquid chromatography with organic carbon detection (LCOCD).
2. Materials and Methods
2.1. Biologically Pretreated Greywater
An effluent grab sample of a constructed wetland exclusively treating separately collected greywater of the ecosettlement Luebeck-Flintenbreite, Germany, was used for the PCO experiments. The subsurface vertical flow constructed wetland with intermittent feeding (ensuring aerobic conditions) is described in more detail in [9]. At the time of sampling, greywater of about 120 residents was treated in the 280 m2 constructed wetland. The treated greywater was allowed to flow directly from the effluent pipe into 5-L and 10-L borosilicate glass bottles which had been rinsed with analytical grade methanol (Merck Eurolab, Darmstadt, Germany) and dried prior to sampling. Subsequently, the bottles were transported to the laboratory and stored in the dark until PCO experiments.
2.2. Photocatalytic Oxidation Experiments
One liter suspensions of different concentrations (1, 5, 10, and 20 g∙L−1) of the TiO2 photocatalyst “Aeroxideâ P25” (Evonik Industries AG, Hanau-Wolfgang, Germany) in biologically pretreated greywater were stirred (magnetic stirring bar length 7 cm; stirrer speed 300 min−1) in slim 2-L beakers (inner diameter 10.8 cm) without UV (stirring time 24 h) and with 24 or 72 h UV irradiation by a face tanner (HD 172, Philips, Hamburg, Germany) with an emission maximum at 352 nm located 30 cm above the liquid surface. At the liquid surface, UV intensity was 25 W∙m−2 as measured with a pyranometer (CM6B Kipp & Zonen, Delft, Netherlands) resulting in UV doses of 0, 5 and 15 Wh∙L−1 in the different experiments. Moreover, three days irradiation experiments were performed adding 1 g∙L−1 PAC “Hydraffin WG” (Lurgi GmbH, Frankfurt/M., Germany) to the photocatalyst. In order to remove any trace organics from the TiO2, the photocatalyst (and also the PAC in the respective experiments) was suspended in one liter of deionized water and UV-irradiated for 3 days prior to separating it by centrifugation and resuspending it in one liter of biologically pretreated greywater for the actual PCO experiment.
Subsequent to irradiation of TiO2 suspensions in greywater, evaporation losses from the reactors (which were quantified by weighing the reactors before and after irradiation) were replenished by addition of deionized water which was mixed with the reactor content by vigorous stirring.
At the end of each experiment, the photocatalyst was separated from the greywater by centrifugation and subsequent membrane filtration (Magna Nylon filters, pore width 0.45 µm, Carl Roth GmbH, Karlsruhe, Germany) of the centrifugation supernatant using stainless steel filter holders and pressurized air. Filtrates were collected in borosilicate glass bottles and immediately subjected to np-DOC analysis and solid phase extraction.
All items having contact with the biologically pretreated greywater (reactors, stirring bars, filter apparatus, bottles receiving the membrane filtrate) were rinsed with analytical grade methanol and dried before being used in order to remove any organic trace contamination.
2.3. General Greywater Characterization
Aggregate organic compounds were analyzed as nonpurgeable DOC (np-DOC) in a multi N/C 3000 TOC analyzer (analytic Jena AG, Jena, Germany) according to German standard procedures. The TOC analyzer’s furnace contained CeO2 catalyst and was operated at 850˚C with CO2-free air as incineration gas. The samples were acidified to pH 1 - 2 with concentrated HCl and purged for 20 min with CO2-free air before TOC analysis. Conductivity and pH were analyzed with probes (WTW, Weilheim, Germany). Alkalinity was determined by titration of 100 mL of the biologically pretreated greywater with hydrochloric acid (0.1 mol∙L−1, Titrisolâ, Merck Eurolab, Darmstadt, Germany) to pH 4.3 as indicated by a pH probe.
2.4. Determination of Organic Fractions in Biologically Treated Greywater
Humic substances, building blocks and low molecular weight (LMW) organic acids were quantified by means of an LC-OCD analyzer (DOC-Labor Dr. Huber, Karlsruhe, Germany) [23] equipped with a size exclusion column HW-55S, a 190 nm irradiation thin film reactor organic carbon detector, a UV detector and FIFFIKUS software for quantification.
2.5. Solid Phase Extraction and Gas Chromatography Coupled to Mass Spectrometry
Membrane filtrates of all UV-irradiated samples were spiked with dihydrocarbamazepine as internal standard. Volumes of 400 mL of the membrane-filtered differently treated greywater samples were sucked through 500 mg Abselut NEXUS SPE cartridges (Varian, Darmstadt, Germany) pre-conditioned with methanol and water. Subsequently, cartridges were rinsed with 10 mL deionized water and dried by gently sucking air through the cartridge for 30 min. Then the organics were eluted with 5 mL of analytical grade methanol (Merck, Darmstadt, Germany). The eluates were concentrated to 1 mL by vacuum evaporation and further to 100 µL with a gentle stream of nitrogen.
Concentrated eluates were analyzed with a gas chromatograph 6890 N coupled to a mass-selective detector 5975 B (Agilent Technologies Deutschland GmbH, Böblingen, Germany). Conditions for gas chromatography were as follows; injector (cold injection system KAS 4, Gerstel GmbH & Co KG, Mülheim/Ruhr, Germany): 300˚C; column (30 m HP-5 ms, Agilent, I.D. 0.25 mm, film thickness 0.25 µm) conditions: temperature program 70˚C (2 min), 8˚C min−1, 290˚C (15 min); carrier gas: helium (1 mL∙min−1). Conditions for mass spectrometry: MS source 230˚C, EI 70 eV, scan mode. Recorded mass spectra were compared to mass spectra of the computerized library NIST 05a.
As a blank, also deionized water was extracted by SPE and the methanol eluates subjected to GC/MS analysis.
3. Results and Discussion
The investigated biologically pretreated greywater exhibited an electric conductivity of 1060 µS∙cm−1, an alkalinity of 400 mg CaCO3 L−1 and a pH of 8.3. The np-DOC was 6 mg∙L−1. A great deal of the organics were well adsorbable to the photocatalyst, as np-DOC concentrations were decreasing with increasing photocatalyst concentrationin the absence of UV irradiation (open triangles in Figure 1). Organic greywater constituents were not markedly more removed by PCO than by adsorption to photocatalyst when UV irradiation period was one day (equivalent to a UV dose of 5 Wh∙L−1; open diamonds in Figure 1) while a three days UV irradiation (equivalent to a UV dose of 15 Wh∙L−1) led to efficient np-DOC removal (open squares in Figure 1).
The addition of PAC to 3 days PCO did not increase np-DOC removal (filled squares in Figure 1) except for PCO with the smallest photocatalyst concentration of 1 g∙L−1. So, a UV dose of 15 Wh∙L−1 which was achieved within 3 days irradiation under the experimental conditions, seems to be sufficient for nearly complete mineralization of organics contained in biologically pretreated greywater. In a previous study on advanced treatment of biologically pretreated greywater by PCO combined with PAC dosage [22], it was shown that PAC addition contributed very well to np-DOC removal when the UV dose

Figure 1. Relative concentrations of np-DOC in biologically pretreated greywater without UV irradiation and after 1 and 3 d UV irradiation in the presence of different TiO2 “P25” concentrations as well as after 3 d PCO in the presence of 1 g∙L−1 PAC.
was only about 10 Wh∙L−1.
Results of liquid chromatography with organic carbon detection are shown in Figure 2. Organics of greywater subsequent to biological treatment are predominantly represented by humic substances. The analyses showed that humic substances, building blocks and LMW organic acids were present in concentrations equivalent to 3530 µg DOC L−1, 880 µg DOC L−1 and 790 µg DOC L−1, respectively. Polysaccharides detected by LC-OCD were present only in a very low concentration range of around 100 µg DOC L−1 and disappeared nearly completely after contact of the biologically pretreated greywater with the photocatalyst (data not shown).
Figure 2(a) indicates that humic substances were also well adsorbable to TiO2. Their concentration decreased by 20% when the greywater was stirred for 24 h with 1 g∙L−1 TiO2 without illumination (“no UV”), but by nearly 50%, when photocatalyst concentration was 20 g∙L−1. Building blocks (Figure 2(b)) were reduced only by less than 10% by adsorption to 1 g∙L−1 photocatalyst, but by

Figure 2. Concentrations of humic substances (a); building blocks (b) and LMW organic acids (c) in biologically pretreated greywater without UV irradiation and after 1 and 3 d UV irradiation in the presence of different TiO2 “P25” concentrations as well as after 3 d PCO in the presence of 1 g∙L−1 PAC.
more than 50% with 20 g∙L−1 TiO2. About 27% of lowmolecular weight organic acids were adsorbed to the photocatalyst present in a concentration of 1 g∙L−1, while a TiO2 concentration of 20 g∙L−1 led to a removal of 43% (Figure 2(c)). From these data obtained without illumination, it can be suggested that the application of photocatalyst concentrations far above 1 g∙L−1 is beneficial for PCO of biologically treated greywater, because only organics adsorbed to the photocatalyst are efficiently oxidized during PCO.
Accordingly, with TiO2 concentrations of 5, 10 and 20 g∙L−1, humic substances were removed by more than 85% within 24 h UV illumination (equivalent to a UV dose of 5 Wh∙L−1; open diamonds in Figure 2(a)). So, humic substances were more efficiently removed by PCO than np-DOC; under the same conditions, np-DOC removal was only about 70% (open diamonds in Figure 1). After three days irradiation, humic substances were nearly completely removed in the presence of TiO2 concentrations of 10 and 20 g∙L−1, regardless of PAC addition (Figure 2(a)).
The time dependent behavior of building blocks elimination by PCO (Figure 2(b)) was different from that of humic substances. For 1 and 10 g∙L−1 TiO2, the building block concentrations were even enhanced when the UV dose was 5 Wh∙L−1. LMW organic acids (Figure 2(c)) showed slightly increased concentrations when the UV dose was increased from 5 to 15 Wh∙L−1 for a TiO2 concentration of 1 g∙L−1. The observed temporary increase of these TOC fractions during PCO might be an artifact due to analytical errors or to slightly deviating conditions among the experiments with different irradiation time. On the other hand, it has been reported that building blocks and LMW organic acid concentrations increase in the initial phase of river water PCO as a consequence of humic substances oxidation [24]. Moreover, it was demonstrated by model calculations on heterogeneous PCO of natural organic matter from lake water that larger molecules (represented by humic substances) were oxidized earlier than the smaller molecules (building blocks and LMW organic acids) [25]. This resulted in increased concentrations of smaller molecules, because oxidation of larger mother compounds usually yields molecules with lower molecular weights. On the other hand, also the formation of high molecular weight humic substances by the reaction of lower molecular size fractions of photocatalytic oxidation products has been discussed [26].
Figures 2(b) and (c) still show concentrations of building blocks and LMW organic acids which are equivalent to about 0.1 mg∙L−1 TOC for PCO experiments with three days UV irradiation and TiO2 concentrations of 5, 10 and 20 g∙L−1, although np-DOC concentrations after the same experiments were nearly 0. This contradiction might be explained by volatility of many representatives of the two groups of wastewater bulk organics “building blocks” and “LMW organic acids”. So, they were probably lost during purging prior to the actual np-DOC analysis.
In order to discriminate the organic micropollutants contained in biologically pretreated greywater from any artifacts caused by solid phase extraction as well as by replenishment of water evaporated from the reactors with deionized water, also deionized water was subjected to solid phase extraction and GC/MS. The five organics listed in Table 1 were found in these extracts.
Figure 3 shows the total ion current chromatogram of the 1:10 diluted eluate of the original biologically pretreated greywater. Some predominant peaks (among them the largest peak at 22.54 min) are artifacts as discussed above and indicated in Table 1. The other 16 substances tentatively identified in biologically treated greywater by means of mass spectra are displayed in Table 2 in the column “OG”.
These organics may occur in foodstuffs, beverages, personal care products or other items related to households as published in other studies. Acetic acid (retention time 3.71 min) is used in kitchens as well as for cleaning purposes and was also present in raw hotel and laundry greywater [18]. As it is biodegradable, its occurrence in biologically treated greywater was not expected. The volatile methoxyphenyloxime (retention time 5.16 min) was detected in some rice cultivars [27], in several cabernet sauvignon wines [28] and was also shown to be emitted during the operation of smoothing irons [29].
Besides these two volatile compounds, there were two not exactly identified alcohols representing rather prominent peaks in the chromatogram of the original biologically treated greywater (Figure 2) at 6.86 and 7.25 min retention time. The relatively low retention times suggest that these alcohols do not exhibit very high molecular weights. Alcohols such as iso-butanol, n-butanol and 2- ethylhexanol are used as solvents in many cosmetics and personal care ingredients. Tert.-butyl-methylphenol (retention time 11.84 min) is referred as a synthetic food

Table 1. Retention times of organic contaminations tentatively identified by GC/MS in methanolic eluatesof cartridges after SPE of deionized water; these substances have to be looked at as artifacts in greywater solid phase extracts.
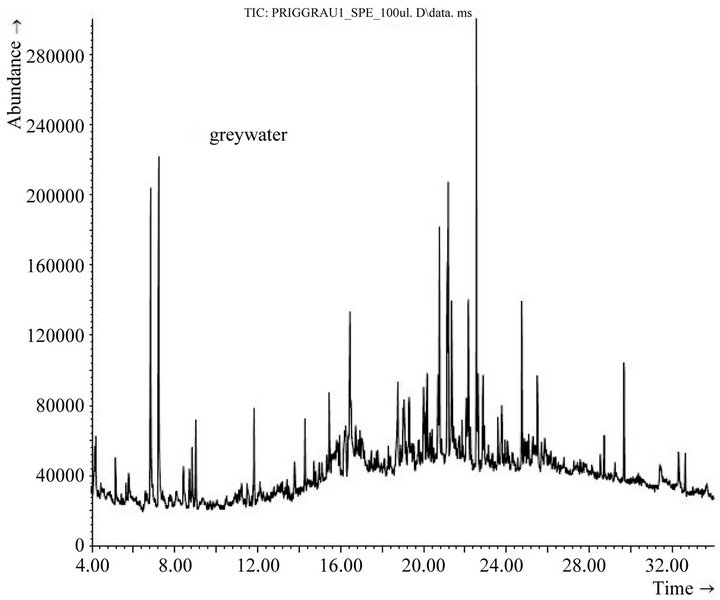
Figure 3. Total ion current chromatogram of 1:10 diluted solid phase extract of biologically pretreated greywater without photocatalyst and UV irradiation; for tentatively identified substances, see Table 2, column “OG”.
additive [30]. Whether the detected phenylamide derivative (retention time 13.78 min) is a phenylamide fungicide or not, cannot be answered with the present data. Phenylamide fungicides such as Metalaxyl-M were found in traded cut roses in Germany [31].
Detection of tetradecanoic acid (retention time 19.73 min), a constituent of soaps, Camembert and mould cheese, in the biologically treated greywater was contrasting to a previous study [9] where it was only found in the raw greywater from the same settlement but not subsequent to treatment in the constructed wetland. Hexadecanoic acid (retention time 22.68 min), a constituent of soaps, emulsifiers for facial creams and lotions, shaving cream formulations and also mould cheese, was probably the predominant trace organic presented by the largest peak in the chromatogram in Figure 3 (although coeluting with a phthalic acid ester, an artifact listed in Table 1 with the retention time 22.54 min). This is in accordance with a previous study on organic micropollutants in raw and biologically treated greywater sampled from the same constructed wetland [9].
Phenyl-iso-quinoline (retention time 21.35 min) was tentatively identified as a representative of azaarenes. Substituted iso-quinolines were also found in atmospheric particulate matter [32,33], although no phenyl-iso-quinolines were among the azaarenes detected in those studies. Nevertheless, it is hypothesized that phenyl-iso-quinoline can be a constituent of airborne dusts and washed out by wet deposition on to the constructed wetland surface thus reaching the biologically treated greywater, inasmuch as small quantities of phenyl-iso-quinoline are formed during combustion of materials containing elemental carbon, e.g. carbon fiber composites [34]. Accordingly, phenyl-iso-quinoline has been tentatively identified in the off-gas of a ring furnace for baking graphite electrodes [35]. Bubnov et al. [36] have detected phenyliso-quinoline in surface runoff.
As representatives of hazardous emerging pollutants, two chlorinated phosphoric acid esters were tentatively identified in the biologically treated greywater: tris (2- chloroethyl) phosphate (TCEP, retention time 19.73 min) and tris (2-chloro-1-methylethyl) phosphate (trischloroiso-propylphosphate, TCIPP, retention time 20.18 min). Both are used as flame retardants; TCEP in PVC, furniture foam, home electronics, upholstery, carpet backings, paints and wallpapers, and TCIPP in rigid and flexible polyurethane foams used in construction and furniture. Both TCEP and TCIPP were shown to alter sex hormone balance; they increased 17-ß-estradiol and testosterone concentrations in cultivated H295R cells, but did not act as estrogen receptor agonists or antagonists in MVLN cell line, on the other hand [37]. In other in vitro systems (recombinant yeast reporter gene assay and human endometrial cancer Ishikawa cells), no estrogenic or antiestrogenic effects were detected [38]. Because of its carcinogenicity [39], neurotoxicity [39] and reproductive toxicity [40], TCEP has been replaced in some consumer products [41].
Bank filtration of Lake Wannsee and Lake Tegel water both containing TCEP and TCIPP in the concentration ranges of 0.5 and 2 µg∙L−1, respectively, was shown to result in TCEP and TCIPP levels of several ten and 100
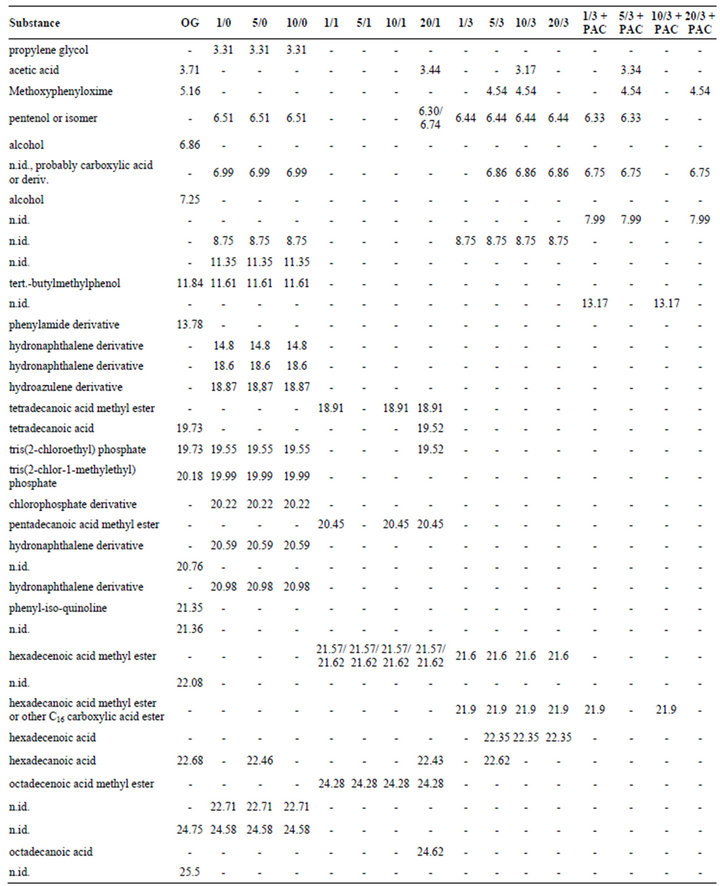
Table 2. Retention times of tentatively identified organic compounds (referring to total ion current gas chromatograms in Figures 3-7) in biologically pretreated greywater before and after photocatalytic oxidation; OG: original biologically treated greywater; numbers in front of a slash indicate concentration of TiO2 in g∙L−1; numbers following slash indicate UV illumination time in days; not detectable: -; artifacts displayed in Table 1 are not contained in this table.
ng∙L−1, respectively, in the groundwater receiving the bank filtrate, although they were attenuated not only with the groundwater dilution, but also by degradation on their passage [42]. In a study on soil aquifer treatment (SAT) of secondary municipal effluent, TCEP was biodegraded during long-term passage, while TCIPP was removed to a much lower extent [43]. In another SAT study [44], TCEP and TCIPP removal rates were 89 and 54%, respectively, when travel time was 60 days. Travel times <3 days led to negligible removal of both substances.
Because of their adverse health effects and their recalcitrance during soil passage, it is of concern that organophosphate flame retardants, among them TCEP and TCIPP, are common pollutants of secondary municipal effluents and consequently found in surface waters [45- 47]. Due to their application in furniture, upholstery and home electronics, halogenated organophosphates were frequently detected in indoor air as well as in indoor dust [45,48]. Proesch and Puchert [49] have interpreted the presence of these flame retardants in effluents of washing machines by their transfer from air and indoor dust to garment. Thus, the occurrence of this group of chemicals in greywater also shown in previous studies [8,9] is selfevident. It is therefore inevitable that biologically pretreated greywater is treated further by suitable processes before it is reused for groundwater aquifer recharge targeting for indirect potable reuse.
Furthermore, five major peaks were visible in the chromatogram of the original biologically pretreated greywater at retention times 20.76, 21.36, 22.08, 24.75 and 25.5 min (Table 2). However, these substances could not be characterized.
The chromatograms displayed in Figure 4 are total ion current chromatograms of SPE eluates of biologically treated greywater which had been stirred with 1, 5 and 10 g∙L−1 TiO2, respectively, for 24 h without UV irradiation. It is plausible that the chromatograms in general show peaks which are decreasing with increasing photocatalyst concentration. This indicates that the trace organics are partly removed by adsorption to the photocatalyst.
The following substances were tentatively identified in the original biologically treated greywater (see above) before and also after contact to different TiO2 concentrations without UV irradiation (see Table 2): tert.-butylmethylphenol (11.84 and 11.61 min), TCEP (19.73 and 19.55 min), TCIPP (20.18 and 19.99 min), and hexadecanoic acid (22.68 and 22.46 min). They were thus not completely removed from the aqueous phase by adsorption to the photocatalyst. Different retention times for the same substances in different sample extracts can be explained by the fact that the chromatograms were not recorded on the same day. Thus, slight differences in the flow of the mobile phase resulted in slightly different retention times.
Some substances tentatively identified in the original constructed wetland effluent were obviously removed to concentrations below limits of detection by adsorption to TiO2, because they were not detected after TiO2 addition (“-” in columns 1/0, 5/0 and 10/10 in Table 2): acetic acid, methoxyphenyloxime, a phenylamide derivative, tetradecanoic acid, and five compounds which could not be characterized at all (n.id.).
An unexpected result was that some substances were identified in the constructed wetland effluent after contact with TiO2 (columns “1/0”, 5/0” and 10/0” in Table 2), although not detected in the original biologically pretreated greywater (column “OG” in Table 2). As these samples were not subjected to UV irradiation, an oxidative transformation of other compounds into these substances can be excluded. However, they might be derived from oxidative transformation of organic contaminants of the photocatalyst or deionized water during the irradiation period of TiO2/deionized water suspensions prior to utilization of the photocatalyst with biologically treated greywater. Some of the organics might also originate from the biologically treated greywater, but were not detected in the greywater without TiO2 addition because of coelution with other greywater constituents. By TiO2 adsorption, some of the coeluting organics might have been removed to an extent, that now some substances were no longer covered by coeluted compounds and thus became detectable by GC/MS. Among these compounds were propylene glycol (retention time 3.31 min), pentenol (6.51 min), and four substances which were not identified (6.99, 8.75, 11.35 and 22.71 min). The mass spectrum of the substance eluted at 6.99 min suggests that it was a carboxylic acid or derivative. Additionally, four different not exactly characterized hydronaphthalene derivatives (retention times 14.8, 18.6, 20.59 and 20.98 min) as well as a hydroazulene derivative (retention time 18.87 min) were found in the biologically treated greywater only subsequent to contact with different doses of TiO2.
There are probable origins of the substances tentatively identified in the greywater subsequent to stirring with TiO2: Propylene glycol is one of the most widely used ingredients in cosmetics and personal care products (e.g. fragrances, creams, aftershave lotions, sprays, hair dyes and deodorants). Pentenol is a volatile reported to be released from leaves subsequent to wounding [50]. Very likely, one of the not exactly identified hydronaphthalene derivatives is tonalide (7-acetyl-1,1,3,4,4,6-hexamethyl-1,2,3,4-tetrahydronaphthalene, AHTN), a synthetic musk fragrance in cosmetics, detergents, and cigarettes, which has been analyzed in municipal effluents [51]. Hydroazulene derivatives were found in essential oil from the leaves of Pseudopanax lessonii [52], and hydroazulene-type sesquiterpene lactones are widespread
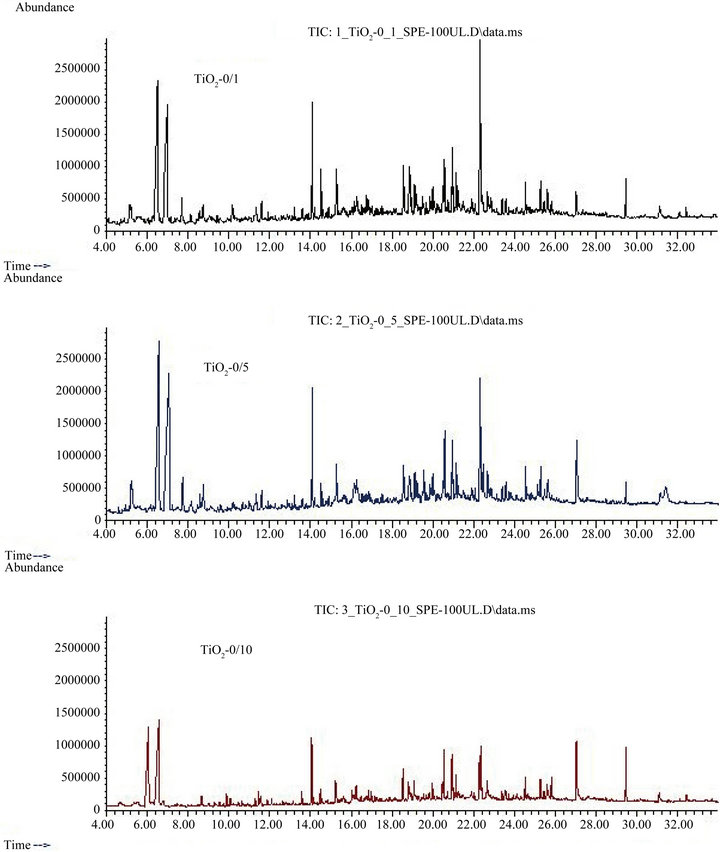
Figure 4. Total ion current chromatograms of solid phase extracts of biologically pretreated greywater subsequent to stirring with 1 g∙L−1 (top), 5 g∙L−1 (middle) and 10 g∙L−1 (bottom) TiO2 “P25” without UV irradiation; no internal standard added; for tentatively identified substances, see Table 2, columns “1/0”, “5/0” and “10/0”.
in natural sources although in minute quantities [53].
Moreover, another chlorinated phosphoric acid ester derivative (20.22 min) was found subsequent to TiO2 contact, but not in the original biologically treated greywater. The third chlorinated phosphoric acid ester derivative (retention time 20.22 min) detected in the greywater after contact to the photocatalyst was not exactly identified. It might be tris (1,3-dichloro-2-propyl) phosphate (TDCP), another common flame retardant.
In Figure 5, the chromatograms of biologically pretreated greywater are shown which has been subjected to one day PCO (UV dose: 5 Wh∙L−1) with different photo-

Figure 5. Total ion current chromatograms of solid phase extracts of biologically pretreated greywater subsequent to 1 day UV irradiation in the presence of 1 g∙L−1 (“TiO2—1/1”), 5 g∙L−1 (“TiO2—1/5”), 10 g∙L−1 (“TiO2—1/10”) and 20 g∙L−1 (“TiO2— 1/20”) TiO2 “P25”; *: internal standard dihydrocarbamazepine; for tentatively identified substances, see Table 2, columns “1/1”, “5/1”, “10/1”, and “20/1”.
catalyst concentrations. Comparing Figure 5 to Figure 4 indicates that one day UV illumination of the greywater/ TiO2 suspensions led to further removal of gas chromatographically detectable trace organics, because the chromatograms of samples irradiated in the presence of 1 to 10 g∙L−1 TiO2 (Figure 5) show more smooth baselines than those of the samples only stirred with the same TiO2 concentrations, but without UV light (Figure 4). The large peaks at retention times 6.5 and 6.9 min (representing pentenol and one not exactly identified alcohol) and many peaks in the retention time range from 18 to 21 min disappeared when the suspensions were irradiated in the presence of TiO2 concentrations of 1, 5, and 10 g∙L−1. Contrasting to the TiO2 contact experiments displayed in Figure 4, UV illumination was also executed with a photocatalyst concentration of 20 g∙L−1 (“TiO2—1/20” in Figure 5). A striking result of that experiment was that larger peaks can be observed after photocatalytic oxidation with this very high photocatalyst concentration than after UV irradiation in the presence of smaller TiO2 concentrations. This is coherent with the finding that among the samples irradiated for one day, the sample irradiated in the presence of 20 g∙L−1 exhibited more tentatively identified organics (Table 2, column “20/1”) than those with lower photocatalyst concentrations (Table 2, columns “1/1”, “5/1”, and “10/1”). The impaired efficiency of 1 d UV irradiation in the presence of 20 g∙L−1 TiO2 can be explained by shading of photocatalyst particles at too high TiO2 concentrations [54].
Table 2 indicates that many organic trace compounds which were identified after mere contact of the biologically pretreated greywater with different concentrations of TiO2 (columns “1/0”, “5/0”, and “10/0”) could no longer be identified after one day UV irradiation of the greywater/TiO2 suspensions (columns “1/1”, “5/1”, and “10/ 1”). According to the hypothesis of photocatalyst shading at very high TiO2 concentrations, acetic acid, pentenol, tetradecanoic acid, TCEP, and hexadecanoic acid were tentatively identified after 24 h UV irradiation in the presence of 20 g∙L−1 TiO2 (column “20/1” in Table 2), but not at lower photocatalyst concentrations. These compounds were also tentatively identified either in TiO2 suspensions without UV or in the original biologically treated greywater. The occurrence of acetic acid in the samples subjected to photocatalytic oxidation can be interpreted with the formation of this substance as oxidation intermediate from other organic mother compounds as observed during PCO of laundry low strength greywater [18]. Tentative identification in UV-irradiated samples is in accordance with LC-OCD results showing low molecular weight organic acids being present in all greywater samples subjected to photocatalytic oxidation (Figure 2(c)).
Octadecanoic acid (retention time 24.62 min) was detected in the biologically pretreated greywater subjected to 24 h photocatalytic oxidation in the presence of 20 g∙L−1 TiO2, but in no other sample. A further unexpected result was the occurrence of some long chain carboxylic acid methyl esters in greywater/TiO2 suspensions which were UV-irradiated for one day, but not in greywater/ TiO2 suspensions without illumination: tetradecanoic acid methyl ester (18.91 min), pentadecanoic acid methyl ester (20.54 min), hexadecenoic acid methyl esters (21.57 and 21.62 min), and octadecenoic acid methyl ester (24.28 min). All these compounds were also found by Eriksson et al. [8] in raw greywater. Additionally, hexadecanoic acid methyl ester and octadecenoic acid methyl ester were tentatively identified in raw greywater from the same source as investigated in this study, but not subsequent to biological treatment [9]. Long chain carboxylic acid methyl (and also other) esters are widely used as emollients. Emollient esters may represent 3% to 20% of skin care formulations ingredients and form semi-occlusive films on the skin maintaining its moisture. Besides “hiding” of these substances by coelution with other higher concentrated compounds with very similar retention times resulting in mass spectra which cannot be related to known compounds, another reason for the occurrence of substances in wastewaters purified to a higher extent which were not identified in the less purified wastewater was discussed elsewhere [9]; in case of separating the organics by solid phase extraction from the aqueous phase of the samples, lowly concentrated organic substances might be replaced from adsorption sites on the solid phase extractant by more highly concentrated organics and thus not occur in the methanol extract.
Also a three days UV illumination period (UV dose: 15 Wh∙L−1) of the biologically pretreated greywater in the presence of different TiO2 concentrations did not lead to complete removal of trace organics as can be seen in Figure 6. However, there are less and smaller peaks in all chromatograms displayed in Figure 6 compared to chromatograms contained in Figure 5. This means that threefold UV doses led to further removal of gas chromatographically detectable trace organics. It seems that for a three days UV illumination, the very high photocatalyst concentration of 20 g∙L−1 was the most efficient. Only a peak at a retention time of about 29.5 min was increasing with increasing photocatalyst concentration.
Table 2 indicates that the trace organics tentatively identified as pentenol (retention time 6.44 min), hexadecenoic acid methyl ester (21.6 min) and hexadecanoic acid methyl ester or another C16-carboxylic acid ester (21.9 min) were still present in the samples irradiated with a UV dose of 15 Wh∙L−1 irrespective of the photocatalyst concentration. Also organics with retention times of 6.86 and 8.75 min which could not be identified were

Figure 6. Total ion current chromatograms of solid phase extracts of biologically pretreated greywater subsequent to 3 days UV irradiation in the presence of 1 g∙L−1 (“TiO2—3/1”), 5 g∙L−1 (“TiO2—3/5”), 10 g∙L−1 (“TiO2—3/10”) and 20 g∙L−1 (“TiO2— 3/20”) TiO2 “P25”; *: internal standard dihydrocarbamazepine; for tentatively identified substances, see Table 2, columns “1/3”, “5/3”, “10/3”, and “20/3”.
detected in all samples treated by 3 days photocatalytic oxidation. These organics were the same as those detected in biologically treated greywater contacted with TiO2 for 24 hours without UV illumination (retention times 6.99 and 8.75 min) as indicated by identical mass spectra. The substance eluted at 6.86 min was a carboxylic acid or derivative as discussed above. Hexadecanoic acid (22.62 min) was found subsequent to three days photocatalytic oxidation only in the presence of 5 g∙L−1 TiO2, methoxyphenyloxime (4.54 min) for 5 and 10 g∙L−1 TiO2, and hexadecenoic acid (22.35 min) for 5, 10 and 20 g∙L−1 TiO2.
As the organophosphate flame retardants are probably the most hazardous organic trace organics contained in greywater with respect to toxicity and recalcitrance, the most important result visible in Table 2 is that these flame retardants were no longer found when the UV illumination was lasting three days. This means that at a UV dose of 15 Wh∙L−1, the flame retardants were removed to below limits of detection, which were not determined, however. A UV dose of 15 Wh∙L−1 is realized in solar photocatalytic oxidation when 1 m3 of the greywater/TiO2 suspension is spread on an area of 75 to 100 m2 and irradiated for one day at solar and sky radiations of 4 to as low as 3 kWh∙m−2∙d−1 (assuming 5% being represented by UV photons). This would result in very thin suspension layers of about 1 cm, however.
TCEP and TCIPP showed the lowest removal rate constants among 32 organics (with the exception of perfluorooctane sulfonic acid) in heterogeneous photocatalytic oxidation experiments with spiked river water [55]. In the same study, these compounds also showed the lowest rate constants for removal by the advanced oxidation process H2O2/UV. The flame retardant TCIPP was also reported to be fairly refractory to ozonation [56]. That investigation also revealed that addition of hydrogen peroxide to the ozonation process was only successful at high ozone doses, because at low ozone doses, wastewater organics substantially competed with hydrogen peroxide for reaction with ozone resulting in low formation rates of hydroxyl radicals. Therefore, it would be worth to investigate TCEP and TCIPP removal from non-spiked biologically pretreated greywater by heterogeneous photocatalytic oxidation in a more targeted way by GC/MS or HPLC/MS determination of the analytes using single ion monitoring or multi reaction monitoring thus enabeling lower limits of detection than non-target GC/MS analyses.
The chromatograms of biologically pretreated greywater subjected to 3 days PCO in the presence of 1 g∙L−1 PAC are shown in Figure 7. Comparison of Figure 7 to Figure 6 indicates, that the addition of 1 g∙L−1 powdered activated carbon did not improve trace organics removal by three days photocatalytic oxidation; there were still a couple of peaks observable in all chromatograms displayed in Figure 7. For a photocatalyst concentration of 20 g∙L−1, the presence of PAC obviously led to a slightly lower PCO efficiency, as peaks in the respective chromatogram (“TiO2/1-3/20”) in Figure 7 were larger than in the corresponding chromatogram in Figure 6. This might be attributed to additional shading of photocatalyst by PAC particles.
Table 2 indicates that acetic acid (retention time 3.34 min) methylphenyloxime (4.54 min), pentenol (6.33 min) and a not exactly characterized substance, probably a carboxylic acid ester (6.75 min), were tentatively identified in some of the samples subjected to three days photocatalytic oxidation in the presence of powdered activated carbon. Acetic acid in the “5/3 + PAC” sample might be an oxidation intermediate derived from other organics. Hexadecanoic acid methyl ester (21.9 min) was tentatively identified in two of the samples subjected to PCO in the presence of PAC, but also in the absence of PAC. Contrasting to samples subjected to three days photocatalytic oxidation without activated carbon, hexadecenoic acid methyl ester, hexadecanoic and hexadecenoic acid were no longer found in samples oxidized under similar conditions but in the presence of 1 g∙L−1 activated carbon. Also the organophosphate flame retardants were not identified in these samples.
Additionally, two organics were detected which could not be identified. Interestingly, these unidentified substances were compounds with retention times (7.99 and 13.17 min) not observed in the other samples. It can be assumed that these compounds were oxidation intermediates either of mother compounds present in the original biologically pretreated greywater or of activated carbon surface structures which had been separated from the activated carbon grain subsequent to oxidation.
Overall, the removal of trace organics by PCO equalled the removal of bulk organics; the chromatograms of biologically pretreated greywater subjected to PCO with a UV dose of 15 Wh∙L−1 in the absence (Figure 6) as well as in the presence of PAC (Figure 7) exhibited only few peaks. At the same time, the np-DOC concentrations of these samples (open and filled squares in Figure 1) were also very small and not largely influenced by PAC addition.
4. Conclusions
In the biologically pretreated greywater, eleven trace organics were tentatively identified. All of them could be related to household activities, food stuffs, beverages and personal care products or to wet precipitation from the atmosphere to the constructed wetland surface. The occurrence of chlorinated phosphoric acid ester flame retardants is of concern. Also after stirring the biologically
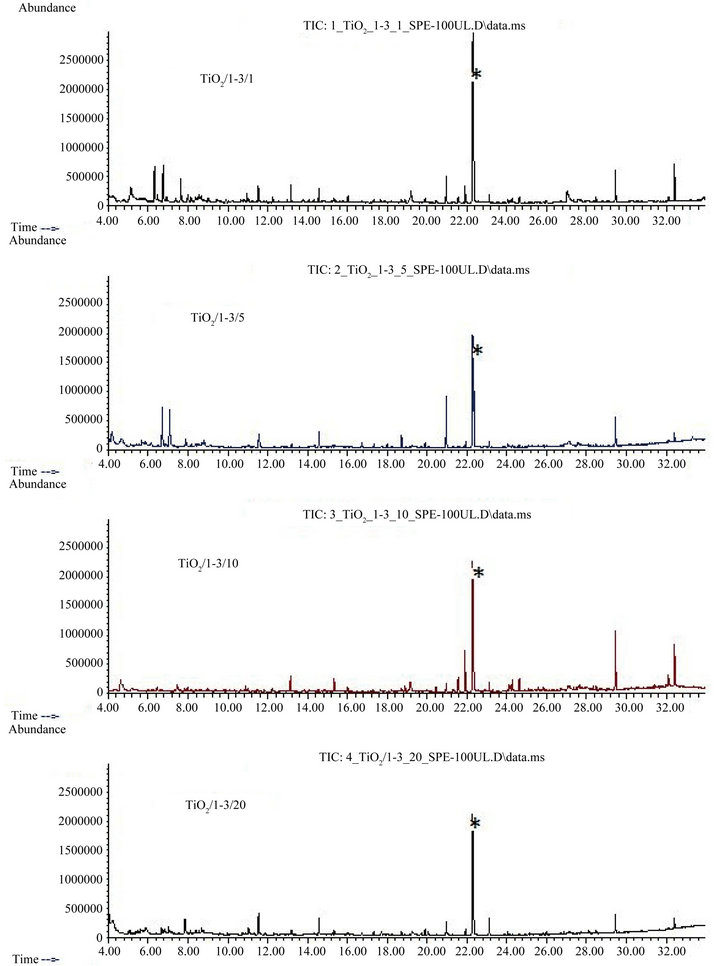
Figure 7. Total ion current chromatograms of solid phase extracts of biologically pretreated greywater subsequent to 3 days UV irradiation in the presence of powdered activated carbon “Hydraffin WG” (1 g∙L−1) and additionally 1 g∙L−1 (“TiO2/ 1-3/1”), 5 g∙L−1 (“TiO2/1-3/5”), 10 g∙L−1 (“TiO2/1-3/10”) and 20 g∙L−1 (“TiO2/1-3/20”) TiO2 “P25”; *: internal standard dihydrocarbamazepine; for tentatively identified substances, see Table 2, columns “1/3 + PAC”, “5/3 + PAC”, “10/3 + PAC”, and “20/3 + PAC”.
treated greywater with different concentrations of photocatalyst without UV illumination, several trace organics were tentatively identified in the liquid phase, among them chlorinated phosphoric acid ester flame retardants. Humic substances were well adsorbable to the photocatalyst.
PCO with a UV dose of 5 Wh∙L−1 resulted in insufficient np-DOC removal from biologically pretreated greywater (about 70% with 5, 10 and 20 g∙L−1 TiO2 and only about 45% with 1 g∙L−1 TiO2), although the fraction of humic substances was more efficiently removed under these conditions (>85% removal with TiO2 concentrations > 5 g∙L−1 and about 60% with 1 g∙L−1 TiO2), but not building blocks and LMW organic acids. GC/MS total ion current chromatograms of solid phase extracts of these samples qualitatively indicated that trace organics disappeared to some extent.
With a UV dose of 15 Wh∙L−1 and TiO2 concentrations of 5 to 20 g∙L−1, np-DOC was nearly completely removed, while PCO with the same UV dose, but with 1 g∙L−1 TiO2 led to an np-DOC removal of only about 80%. Building blocks and LMW organic acids showed residues of about 100 to 200 µg∙L−1 with TiO2 concentrations of > 5 g L-1 and even about 650 and 450 µg∙L−1, respectively, when the TiO2 concentration was 1 g∙L−1. Trace organics were removed by PCO with a UV dose of 15 Wh∙L−1 more efficiently than with 5 Wh∙L−1 as shown by GC/MS; chlorinated phosphoric acid ester flame retardants which were assumed to be the most hazardous trace organics in biologically treated greywater were no longer detectable subsequent to PCO with UV doses of 15 Wh∙L−1 irrespective of the photocatalyst concentration. However, some peaks were still visible in the total ion current chromatograms.They represented compounds tentatively identified as acetic acid, methoxyphenyloxime, pentenol, hexadecenoic and hexadecanoic acid, and hexadecenoic and hexadecanoic acid methyl esters.
Presence of 1 g∙L−1 PAC had no marked influence on bulk organics removal by PCO with UV doses of 15 Wh∙L−1 and photocatalyst concentrations of 5, 10, and 20 g∙L−1. Only at the lowest tested photocatalyst concentration of 1 g∙L−1, PAC addition led to improved removal of bulk organics. Trace organics removal seemed to be slightly impaired by PAC.
5. Acknowledgements
The authors greatly acknowledge Andreas Albers and Jeldrik Moritz for LC-OCD analyses and Ms. Susanne Eggers for her assistance in TOC analyses. This work was supported by a European Union scholarship for C.F. Liriano Jorge within the “Joint European Master Program in Environmental Studies” (JEMES).
REFERENCES
- E. Eriksson, E. Donner and A. Ledin, “Presence of Selected Priority and Personal Care Substances in an Onsite Bathroom Greywater Treatment Facility,” Water Science and Technology, Vol. 62, No. 12, 2010, pp. 2889-2898. doi:10.2166/wst.2010.988
- Z. Li, H. Gulyas, M. Jahn, D. R. Gajurel and R. Otterpohl, “Greywater Treatment by Constructed Wetlands in COMBINATIon with TiO2-Based Photocatalytic Oxidation for Suburban and Rural Areas without Sewer System,” Water Science and Technology, Vol. 48, No. 11, 2003, pp. 101- 106.
- E. Nolde, “Greywater Recycling Systems in Germany— Results, Experiences and Guidelines,” Water Science and Technology, Vol. 51, No. 10, 2005, pp. 203-210.
- M. Ernst and M. Jekel, “Advanced Treatment Combination for Groundwater Recharge of Municipal Wastewater by Nanofiltration and Ozonation,” Water Science and Technology, Vol. 40, No. 4-5, 1999, pp. 277-284. doi:10.1016/S0273-1223(99)00509-0
- California Department of Public Health, “Groundwater Recharge Reuse DRAFT Regulation,” Division of Drinking Water and Environmental Management, Sacramento, 2008. http://www.cdph.ca.gov/certlic/drinkingwater/Documents/Recharge/DraftRechargeReg2008.pdf
- W. Kuehn and B. Wricke, “Trinkwasseraufbereitung im Mitteldeutschen Raum unter Besonderer Berücksichtigung der Haloformbildung,” GWF Wasser/Abwasser, Vol. 136, No. 14, 1995, pp. S92-S98.
- E. Donner, E. Eriksson, D. M. Revitt, L. Scholes, H.-C. Lützhøft and A. Ledin, “Presence and Fate of Priority Substances in Domestic Greywater Treatment and Reuse Systems,” Science of the Total Environment, Vol. 408, No. 12, 2010, pp. 2444-2451. doi:10.1016/j.scitotenv.2010.02.033
- E. Eriksson, K. Auffarth, A. M. Eilersen, M. Henze and A. Ledin, “Household Chemicals and Personal Care Products as Sources for Xenobiotic Organic Compounds in Grey Wastewater,” Water SA, Vol. 29, No. 2, 2003, pp. 135-146. doi:10.4314/wsa.v29i2.4848
- H. Gulyas, M. Reich and R. Otterpohl, “Organic Micropollutants in Raw and Treated Greywater: A Preliminary Investigation,” Urban Water Journal, Vol. 8, No. 1, 2011, pp. 29-39. doi:10.1080/1573062X.2010.528435
- L. Hernandez Leal, “Removal of Micropollutants from Greywater. Combining Biological and Physical/Chemical Processes,” Ph.D. Dissertation, Wageningen University, Wageningen, 2010.
- B. Levine, K. Reich, P. Sheilds, I. H. Suffet and V. Lazarova, “Water Quality Assessment for Indirect Potable Reuse: A New Methodology for Controlling Trace Organic Compounds at the West Basin Water Recycling Plant (California, USA),” Water Science and Technology, Vol. 43, No. 10, 2001, pp. 249-257.
- L. D. Nghiem, J. McCutcheon, A. I. Schäferand M. Elimelech, “The Role of Endocrine Disrupters in Water Recycling: Risk or Mania?” Water Science and Technology, Vol. 50, No. 2, 2004, pp. 215-220.
- H. Ozaki, N. Ikejima, Y. Shimizu, K. Fukami, S. Taniguchi, R. Takanami, R. R. Giri and S. Matsui, “Rejection of Pharmaceuticals and Personal Care Products PPCPs) and Endocrine Disrupting Chemicals (EDCs) by Low Pressure Reverse Osmosis Membranes,” Water Science and Technology, Vol. 58, No. 1, 2008, pp. 73-81. doi:10.2166/wst.2008.607
- J. Radjenovic, M. Petrovic, F. Ventura and D. Barcelo, “Rejection of Pharmaceuticals in Nanofiltration and Reverse Osmosis Membrane Drinking Water Treatment,” Water Research, Vol. 42, No. 14, 2008, pp. 3601-3610. doi:10.1016/j.watres.2008.05.020
- Y. Poussade, A. Roux, T. Walker and V. Zavlanos, “Advanced Oxidation for Indirect Potable Reuse: A Practical Application in Australia,” Water Science and Technology, Vol. 60, No. 9, 2009, pp. 2419-2424. doi:10.2166/wst.2009.665
- L. Hernández-Leal, H. Temmink, G. Zeeman and C. J. N. Buisman, “Removal of Micropollutants from Aerobically Treated Grey Water via Ozone and Activated Carbon,” Water Research, Vol. 45, No. 9, 2011, pp. 2887-2896. doi:10.1016/j.watres.2011.03.009
- Y. Boyjoo, M. Ang and V. Pareek, “Photocatalytic Treatment of Shower Water Using a Pilot Scale Reactor,” International Journal of Photoenergy, 2012, Article ID 578916. http://www.hindawi.com/journals/ijp/2012/578916/
- M. Sanchez, M. J. Rivero and I. Orti, “Photocatalytic Oxidation of Grey Water over Titanium Dioxide Suspensions,” Desalination, Vol. 262, No. 1-3, 2010, pp. 141- 146. doi:10.1016/j.desal.2010.05.060
- A. Armanious, A. Özkan, U. Sohmen and H. Gulyas, “Inorganic Greywater Matrix Impact on Photocatalytic Oxidation: Does Flocculation of TiO2 Nanoparticles Impair Process Efficiency?” Water Science and Technology, Vol. 63, No. 12, 2011, pp. 2808-2813. doi:10.2166/wst.2011.614
- H. Gulyas, H. B. Jain, A. L. Susanto, M. Malekpur, K. Harasiuk, I. Krawczyk, P. Choromanski and M. Furmanska, “Solar Photocatalytic Oxidation of Pretreated Wastewaters: Laboratory Scale Generation of Design Data for Technical-Scale Double-Skin Sheet Reactors,” Environmental Technology, Vol. 26, No. 5, 2005, pp. 501-514. doi:10.1080/09593332608618540
- R. J. Braham and A. T. Harris, “Review of Major Design and Scale-Up Considerations for Solar Photocatalytic Reactors,” Industrial and Engineering Chemistry Research, Vol. 48, No. 19, 2009, pp. 8890-8905. doi:10.1021/ie900859z
- H. Gulyas, P. Choromanski, N. Muelling and M. Furmanska, “Toward Chemical-Free Reclamation of Biologically Pretreated Greywater: Solar Photocatalytic Oxidation with Powdered Activated Carbon,” Journal of Cleaner Production, Vol. 17, No. 13, 2009, pp. 1223-1227. doi:10.1016/j.jclepro.2009.03.008
- M. Abert, “LC-OCD-OND Analysis of Sewage Treatment Steps,” DOC-Labor Dr. Huber, Analytical Services and LC-OCD Systems, Karlsruhe, 2008. http://www.doc-labor.de/Example_Report_WASTE.pdf
- S. Liu, M. Lim, R. Fabris, C. W. K. Chow, M. Drikas, G. Korshin and R. Amal, “Multi-Wavelength Spectroscopic and Chromatography Study on the Photocatalytic Oxidation of Natural Organic Matter,” Water Research, Vol. 44, No. 8, 2010, pp. 2525-2532. doi:10.1016/j.watres.2010.01.036
- L. A. Tercero Espinoza and F. H. Frimmel, “A Simple Simulation of the Degradation of Natural Organic Matter in Homogeneous and Heterogeneous Advanced Oxidation Processes,” Water Research, Vol. 43, No. 16, 2009, pp. 3902-3909. doi:10.1016/j.watres.2009.04.028
- C. S. Uyguner and M. Bekbolet, “A Comparative Study on the Photocatalytic Degradation of Humic Substances of Various Origins,” Desalination, Vol. 176, No. 1-3, 2006, pp. 167-176. doi:10.1016/j.desal.2004.11.006
- R. J. Bryant and A. M. McClung, “Volatile Profiles of Aromatic and Non-Aromatic Rice Cultivars Using SPME/ GC-MS,” Food Chemistry, Vol. 124, No. 2, 2011, pp. 501- 513. doi:10.1016/j.foodchem.2010.06.061
- J. Meng, Y. Fang, J. Gao, A. Zhang, J. Liu, Z. Guo, Z. Zhang and H. Li, “Changes in Aromatic Compounds of Cabernet Sauvignon Wines during Ageing in Stainless Steel Tanks,” African Journal of Biotechnology, Vol. 10, No. 55, 2011, pp. 11640-11647.
- Miljøstyrelsen, “Afgivelse og Vurdering af Kemiske Stoffer fra Udvalgte Elektriske og Elektroniske Produkter— del 2,” Kortlægning af Kemiske Stoffer i Forbrugerprodukter nr. 66, Danish Ministry of the Environment, Copenhagen, 2005. http://www2.mst.dk/common/Udgivramme/Frame.asp?http://www2.mst.dk/udgiv/publikationer/2005/87-7614-825-4/html/helepubl.htm
- J. E. N. Dolatabadi and S. Kashanian, “A Review on DNA Interaction with Synthetic Phenolic Food Additives,” Food Research International, Vol. 43, No. 5, 2010, pp. 1223-1230. doi:10.1016/j.foodres.2010.03.026
- BUND, “Rosen mit Pestiziden Belastet—Analyse des BUND,” Flyer of Bund für Umweltund Naturschutz Deutschland e.V., Berlin, 2012. http://www.bund.net/fileadmin/bundnet/publikationen/chemie/20120214_chemie_rosen_pestizide_hintergrund.pdf
- O. Delhomme and M. Millet, “Azaarenes in Atmospheric Particulate Matter Samples of Three Different Urban Sites in East of France,” Atmospheric Environment, Vol. 47, 2012, pp. 541-545. doi:10.1016/j.atmosenv.2011.06.044
- J. Leniček, M. Sekyra, K. Bednárkova, I. Beneš and F. Šipek, “Fractionation and Chemical Analysis of Urban Air Particulate Extracts,” International Journal of Environmental Analytical Chemistry, Vol. 77, No. 4, 2000, pp. 269-288.
- D. L. Courson, C. D. Flemming, K. J. Kuhlmann, J. W. Lane, J. H. Grabau, C. R. Miller, J. M. Cline, B. J. Larcom and J. C. Lipscomb, “Smoke Production and Thermal Decomposition Products from Advanced Composite Materials,” In: D. E. Dodd, Ed., 1996 Toxic Hazards Research Annual Report, United States Air Force Armstrong Laboratory, Dayton, 1997, pp. 133-147.
- M. Meyer zu Reckendorf, “Identification of Phenyl-Substituted Polycyclic Aromatic Compounds in Ring Furnace Gases Using GC-MS and GC-AED,” Chromatographia, Vol. 45, No. 1, 1997, pp. 173-182. doi:10.1007/BF02505557
- A. G. Bubnov, V. I. Grinevich and O. N. Maslova, “BarrierDischarge Plasma Treatment of Surface Water to Remove Organic Compounds,” Russian Journal of Applied Chemistry, Vol. 79, No. 6, 2006, pp. 934-940. doi:10.1134/S1070427206060139
- X. Liu, K. Ji and K. Choi, “Endocrine Disruption Potentials of Organophosphate Flame Retardants and Related Mechanisms in H295R and MVLN Cell Lines and in Zebrafish,” Aquatic Toxicology, Vol. 114, 2012, pp. 173- 181. doi:10.1016/j.aquatox.2012.02.019
- W. Föllmann and J. Wober, “Investigation of Cytotoxic, Genotoxic, Mutagenic, and Estrogenic Effects of the Flame Retardants Tris-(2-chloroethyl)-phosphate (TCEP) and Tris-2-chloropropyl)-phosphate (TCPP) in Vitro,” Toxicology Letters, Vol. 161, No. 2, 2006, pp. 124-134. doi:10.1016/j.toxlet.2005.08.008
- WHO, “Flame Retardants: Tris(chloropropyl)phosphate and Tris(2-chloroethyl)phosphate,” Environmental Health Criteria 209. World Health Organization, Geneva, 1998. http://whqlibdoc.who.int/ehc/WHO_EHC_209.pdf
- R. Chapin, D. Gulati and L. Barnes, “Tris(2-chloroethyl)phosphate,” Environmental Health Perspectives, Vol. 105, No. S1, 1997, pp. 365-366.
- C. Bergh, “Organophosphates and Phthalates in Air and Dust from Indoor Environments—Method Development and Applied Measurements,” Ph.D. Dissertation, Stockholm University, Stockholm, 2011.
- T. Heberer, A. Mechlinski, B. Franck, A. Knappe, G. Massmann, A. Pekdeger and B. Fritz, “Field Studies on the Fate and Transport of Pharmaceutical Residues in Bank Filtration,” Ground Water Monitoring and Remediation, Vol. 24, No. 2, 2004, pp. 70-77. doi:10.1111/j.1745-6592.2004.tb00714.x
- G. Amy and J. Drewes, “Soil Aquifer Treatment (SAT) as a Natural and Sustainable Wastewater Reclamation/Reuse Technology: Fate of Wastewater Effluent Organic Matter (EfOM) and Trace Organic Compounds,” Environmental Monitoring and Assessment, Vol. 129, No. 1-3, 2007, pp. 19-26. doi:10.1007/s10661-006-9421-4
- B. V. Laws, E. R. V. Dickenson, T. A. Johnson, S. A. Snyder and J. E. Drewes, “Attenuation of Contaminants of Emerging Concern during Surface-Spreading Aquifer Recharge,” Science of the Total Environment, Vol. 409, No. 6, 2011, pp. 1087-1094. doi:10.1016/j.scitotenv.2010.11.021
- T. Reemtsma, J. B. Quintana, R. Rodil, M. Garcia-Lopez and I. Rodriguez, “Organophosphorus Flame Retardants and Plasticizers in Water and Air. I. Occurrence and Fate,” Trends in Analytical Chemistry, Vol. 27, No. 9, 2008, pp. 727-737. doi:10.1016/j.trac.2008.07.002
- A. Marklund, B. Andersson and P. Haglund, “Organophosphorus Flame Retardants and Plasticizers in Swedish Sewage Treatment Plants,” Environmental Science and Technology, Vol. 39, No. 19, 2005, pp. 7423-7429. doi:10.1021/es051013l
- J. Meyer and K. Bester, “Organophosphate Flame Retardants and Plasticisers in Wastewater Treatment Plants,” Journal of Environmental Monitoring, Vol. 6, No. 7, 2004, pp. 599-605. doi:10.1039/b403206c
- C. Bergh, R. Torgrip, G. Emenius and C. Östtran, “Organophosphate and Phthalate Esters in Air and Settled Dust —A Multi-Location Indoor Study,” Indoor Air, Vol. 21, No. 1, 2011, pp. 67-76. doi:10.1111/j.1600-0668.2010.00684.x
- J. Proesch and W. Puchert, “Kontaminierte TextilienEine Ursache für die TCPP-Belastung Kommunaler Abwässer,” Vom Wasser, Vol. 100, 2003, pp. 1-8.
- R. Fall, T. Karl, A. Jordan and W. Lindinger, “Biogenic C5 VOCs: Release from Leaves after Freeze-Thaw Wounding and Occurrence in Air at a High Mountain Observatory,” Atmospheric Environment, Vol. 35, No. 22, 2001, pp. 3905-3916. doi:10.1016/S1352-2310(01)00141-8
- D. A. Chase, A. Karnjanapiboonwong, Y. Fang, G. P. Cobb, A. N. Morse and T. A. Anderson, “Occurrence of Synthetic Musk Fragrances in Effluent and Non-Effluent Impacted Environments,” Science of the Total Environment, Vol. 416, 2012, pp. 253-260. doi:10.1016/j.scitotenv.2011.11.067
- R. J. Weston, “Essential Oils from the Leaves of Three New Zealand Species of Pseudopanax (Araliaceae),” Zeitschrift für Naturforschung, Vol. 59c, 2004, pp. 39-42.
- H. J. Schäfer and K. H. Baringhaus, “Enantioselective Synthesis of a Highly Functionalized Perhydroazulene,” Liebigs Annalen der Chemie, Vol. 1990, No. 4, 1990, pp. 355-360. doi:10.1002/jlac.199019900168
- L. Mansouri, L. Bousselmi and A. Ghrabi, “Degradation of Recalcitrant Organic Contaminants by Solar Photocatalysis,” Water Science and Technology, Vol. 55, No. 12, 2007, pp. 119-125.
- M. J. Benotti, B. D. Stanford, E. C. Wert and S. A. Snyder, “Evaluation of a Photocatalytic Reactor Membrane Pilot System for the Removal of Pharmaceuticals and Endocrine Disrupting Compounds from Water,” Water Research, Vol. 43, No. 6, 2009, pp. 1513-1522.
- J. P. Pocostales, M. M. Sein, W. Knolle, C. von Sonntag and T. C. Schmidt, “Degradation of Ozone-Refractory Organic Phosphates in Wastewater by Ozone and Ozone/ Hydogen Peroxide (Peroxone): The Role of Ozone Consumption by Dissolved Organic Matter,” Environmental Science & Technology, Vol. 44, No. 21, 2010, pp. 8248- 8253.