Journal of Cancer Therapy
Vol. 4 No. 6A3 (2013) , Article ID: 34310 , 13 pages DOI:10.4236/jct.2013.46A3002
Synergistic Antitumor Activity of Vitamins C and K3 on Human Bladder Cancer Cell Lines
1The Apatone Development Center, St. Thomas Hospital, Summa Health System, Akron, USA; 2Department of Anatomical Sciences, St Georges’ University International School of Medicine, K B Taylor Scholar’s Programme, Newcastle upon Tyne, UK.
Email: *jamisonj@summahealth.org
Copyright © 2013 Karen McGuire et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 24th, 2013; revised May 26th, 2013; accepted June 3rd, 2013
Keywords: Bladder; Carcinoma; Vitamin C; Vitamin K3
ABSTRACT
Exponentially growing cultures of human bladder tumor cells (RT4 and T24) were treated with Vitamin C (VC) alone, Vitamin K3 (VK3) alone, or with a VC:VK3 combination continuously for 5 days or treated with vitamins for 1 h, washed with PBS and then incubated in culture medium for 5 days. Co-administration of the vitamins enhanced the antitumor activity 12- to 24-fold for the RT-4 cells and 6- to 41-fold for the T24 cells. Flow cytometry of RT4 cells exposed to the vitamins revealed a growth arrested population and a population undergoing cell death. Growth arrested cells were blocked near the G0/G1-S-phase interface, while cell death was due to autoschizis. Catalase treatment abrogated both cell cycle arrest and cell death which implicated hydrogen peroxide (H2O2) in these processes. The H2O2 production resulted in a moderate increase in lipid peroxidation and depletion of cell thiol levels. Analysis of cellular ATP levels revealed a transient increase in ATP production for VC and the VC:VK3 combination, but decreased ATP levels following VK3 treatment. Lipid peroxidation, thiol depletion and ATP modulation occurred at a 17-fold lower concentration in the vitamin combination than with either vitamin alone. These results suggested that the increased cytotoxicity of the vitamin combination was due to redox cycling and increased oxidative stress.
1. Introduction
Bladder cancer is the second most common urological malignancy in the United States of America with an estimated 72,570 new cases and 15,210 deaths in 2013 [1]. Unlike most epithelial tumors, divergent pathways of tumorigenesis are involved in urothelial carcinoma [2]. These separate mechanisms produce at least two distinct types of neoplasms: non-invasive, low-grade tumor and high-grade, often invasive, carcinoma [3]. Patients with low grade tumors usually undergo transurethral tumor resection (TUR). While the prognosis for these patients is usually good, they exhibit a lifelong risk of recurrence (50% - 70%) with occasional progression to invasion. Given the relatively high rates of recurrence and progression, it is necessary to consider adjuvant intravesical therapy in most patients [4,5]. Since the mid 1980s, the standard treatment for metastatic bladder cancer has been methotrexate, vinblastine, doxorubicin and cisplatin [6]. However, even with this regimen, the prognosis for patients with metastatic disease is poor with a median survival being approximately 12 - 14 months [7]. Furthermore, addition of new drugs, such as gemcitabine, to the standard cisplatin-based regimens has not improved clinical outcomes [8,9]. In addition, the use of several targeted agents, such as, antiangiogenics, anti-epidermal growth factor receptor agents, and immunomodulatory agents did not result in a major breakthrough [7]. Finally, because of the lifelong need for monitoring for recurrence, the typical cost incurred by a bladder tumor patient from diagnosis to death has been reported to be the highest among all cancers [4,10]. Taken together, these facts demonstrate the need for the development of agents that are more effective, less toxic, and more cost effective agents that are more effective, less toxic and affordable [7].
Due to their low systemic toxicity, several vitamins have been evaluated for their chemopreventive and therapeutic potential abilities against bladder cancer [11]. Vitamins A, B6, C, E, and K3 have all demonstrated activity in the prevention or treatment of bladder cancer [12]. In addition, Lamm et al. [12] performed a double blind, randomized trial in patients with bladder cancer who were treated with transurethral resection plus megadose vitamins daily vs the recommended daily allowance of multivitamins. The overall recurrence rate was 80% in the recommended daily allowance arm and 41% in the megadose vitamin arm (p = 0.0011). This vitamin treatment not only was nontoxic, but also produced a greater reduction in the rate of tumor recurrence than BCG immunotherapy which is the gold standard for the treatment of superficial bladder cancer. In addition, there is a growing body of evidence demonstrating the benefit of combining vitamins C and K3 for the treatment of: acute lymphoblastic leukemia [13], acute myelogenous leukemia [14-16], bladder [17-28], breast [29], glioblastoma [30], glioma [31], kidney [32], liver [33-37], lung [38], ovarian [39-42] and prostate cancers [43-55]. Unlike the majority of chemotherapeutic agents which target rapidly dividing cells, VC:VK3 appears to target tumor cells by inflammation [50]. Tumor cells possess a greater need for glucose than normal cells and express facilitative glucose transporters (GLUTs) to achieve this task. Because of the structural resemblance of dehydroascorbic acid (DHA, the oxidized form of vitamin C) to glucose, DHA can also enter the tumor cells and bio-accumulate. Epithelial tumors appear to rely on superoxide (inflammation) which is produced constitutively via NA DPH oxidase of non-neoplastic stromal cells to oxidize the ascorbic acid [56,57]. Once dehydroascorbic acid enters the cells, it is reduced and retained as ascorbic acid (AA) which is not transportable through the bidirectional GLUTs [58,59]. The purpose of the current study is to evaluate VC, VK3 and the VC:VK3 combination for their antitumor activity against two human bladder cancer cell lines and to make an initial attempt to elucidate the mechanism(s) of action of the VC:VK3 combination.
2. Materials and Methods
2.1. Culture Conditions
Human bladder cancer cell lines (T24 and RT4) were purchased from the American Type Culture Collection (ATTC, Rockville, MD, USA) and were grown in culture medium according to ATTC instructions. All media was supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY) and 50 µg/mL gentamicin sulfate (Sigma, St Louis, MO). All incubations were performed at 37˚C and with 5% CO2 unless other conditions are stated. Vitamin C (VC) and menadione bisulfite (VK3) were purchased from Sigma Chemical Company (St Louis, MO, USA) and were dissolved in phosphate-buffered saline (PBS) to create 8000 µM VC, 500 µM VK3 and 8000 µM VC:80 µM VK3 test solutions. To prevent photodegradation of the vitamins, all the vitamin solutions were prepared and experiments were performed in a darkened laminar flow hood.
2.2. Protein Concentration Assay
In all experiments total protein concentration was determined using the method of Bradford [60]. Sham-treated cells served as controls in all experiments.
2.3. Cytotoxicity Assay
The cytoxicity assay was performed using the microtetrazolium assay[MTT,3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-diphenyltetrazolium bromide] assay as described previously [23]. Corning 96-well titer plates were seeded with tumor cells (5 × 103 per well) and incubated for 24 hr. Vitamin test solutions were serially diluted with media in twelve 2-fold dilutions. Each dilution was added to seven wells of the titer plates and co-incubated with the tumor cells for 5 days. After vitamin treatment and the incubation period, cytotoxicity was evaluated using the MTT assay. Following linear regression, the line of best fit was determined and the CD50 was calculated. The fractional inhibitory concentration index (FIC) was employed to evaluate synergism.
2.4. Flow Cytometry
Determination of cell DNA content and ploidy were performed according to our previously published procedure [45]. Briefly, titer dishes were seeded with 1.0 × 106 RT4 cells suspended in MEM (10% FCS). Following 24 hours of incubation, the MEM was removed and the cells were washed twice with 3 ml of PBS. The cells in each titer dish were then overlaid with 2 ml of MEM containing the vitamins. Human foreskin fibroblast cells served as diploid internal standard cells in flow cytometric studies. After a one hour incubation period with vitamins, the cultures were washed free of vitamin and overlaid fresh MEM. Following a 24-hour of incubation period, the cells were harvested from the titer dishes and suspended in 0.1% NP-40 in a Tris-citrate solubilization buffer which contained propidium iodide (PI, 5 mg/ml) and 0.1% RNase A. Following a 30-minutes incubation, DNA ploidy and cell cycle analysis was performed on a Ortho Cytoron flow cytometer. The data from 2 × 104 cells were collected (when possible), stored, and analyzed using ModFit Cell Cycle Analysis.
2.5. Addition of Catalase
Titer plates were seeded and incubated as described in the cytotoxicity assay. After 24 h, the appropriate nontoxic concentrations of catalase and the vitamin test solutions were added to the wells and the titer plates were incubated at 37˚C in 5% CO2 for 1 h. The cells were subsequently washed with PBS, overlain with culture media and incubated for 5 days. Cytotoxicity was evaluated using the MTT assay.
2.6. Analysis of Lipid Peroxidation
Lipid peroxidation was evaluated using the thiobarbituric acid (TBA) method [61]. RT4 cells were treated and harvested as described in the thiol assay. After centrifugation, the cell pellets were resuspended in 6.0% TCA (trichloroacetic acid), mixed with 1 ml of 0.25 N HCl containing 0.375% TBA and 15% TCA, heated in a water bath for 15 min at 95˚C and then allowed to cool. Following centrifugation the supernatant was monitored fluorimetrically for malondialdehyde (MDA) production using an excitation wavelength of 532 nm and an emission wavelength of 555 nm. Data was expressed as nM MDA per mg of protein, calculated on the basis of an MDA standard curve generated using 1,1,3,3-tetramethoxypropane.
2.7. Analysis of ATP
RT4 cells (1.0 × 106) were seeded and then incubated at 37˚C and 5% CO2. After 24 h, the culture medium was removed and the cells were exposed for 1 h to culture media containing the vitamins at their CD90 concentrations. The cells were then washed with PBS, overlaid with vitamin-free culture media and solubilized in somatic cell ATP releasing reagent (Sigma Chemical Co, St Louis, USA) at 1-h intervals for 6 h and intracellular ATP content was assayed using an ATP bioluminescent assay kit (Sigma, St Louis, U.S.A.) [62]. Bioluminescence was measured using a Beckman LS 9000 scintillation counter set for single photon counting. Data was expressed as nM ATP per mg of protein, calculated on the basis of an ATP standard curve.
2.8. Analysis of Protein Thiols
Thiols were assayed using the method of Nagelkerke and co-workers [63]. RT4 cells were exposed for 1 h to culture media containing the vitamins at their CD90 concentrations. The cells were then washed with PBS, overlaid with vitamin-free culture media, trypsinized at 1-h intervals for 6 h and centrifuged for 5 min at 1000 rpm. The cell pellets were washed twice with 6.5% TCA and resuspended in 1 ml of 0.5 M Tris-HCl (pH 7.6). Subsequently, 50 µl of 10 mM methanolic Ellman’s Reagent was added and the solution was incubated at room temperature. After 20 min, the solution was centrifuged for 5 min at 1000 rpm and the absorbance of the supernatant was measured at 412 nm. Data was expressed as µM thiols per mg of protein, calculated on the basis of a reduced glutathione (GSH) standard curve.
2.9. Statistics
Three-way ANOVA was performed using BMDP statistical software. In the three-way ANOVA, the two-way interactions were tested at the 0.005 level of significance, while all other effects were tested at the 0.0022 level of significance. A summary of the experimental design is given in Figure 1.
3. Results
3.1. Cytotoxicity
VC, VK3 and the VC:VK3 combination with a VC:VK3 ratio of 100:1 have been evaluated for their cytotoxicity against two human bladder carcinoma cell lines following continuous 5-day vitamin exposure or 1-h vitamin exposure followed by a 5-day incubation in media (Table 1). A continuous 5-day exposure to VC:VK3 treatment of the RT-4 cells resulted in a 22-fold decrease of the CD50 of VC (2430 to 110 µM) and a 12-fold decrease in the CD50 of the VK3 (12.8 to 1.10 µM) and a 12-fold decrease in the CD50 of the VK3 (12.8 to 1.10 µM) with an FIC value of 0.136 indicating that the combination was synergistic. T24 cells treated continuous for 5 days with VC:VK3 resulted in a 41 fold decrease in VC (1,490 µM to 13.1 µM) and a 6-fold decrease in VK3 (212 µM to 2.13 µM) with an FIC value of 0.158 also demonstrating a significant synergism after only 1hr of VC:VK3 treatment.
Taper and his associates [34] have shown that the VC:VK3 combination exhibited antitumor activity with exposure times as short as 1 h. We sought to determine if the vitamins would exert significant antitumor activity against RT-4 and T24 cells following a 1 h exposure (Table 1). A 1 h VC:VK3 treatment of the RT-4 cells resulted in an 18-fold decrease in the CD50 value of VC (4740 µM reduced to 267 µM) and a 22-fold decrease in VK3 CD50 values (60.7 µM reduced to 2.68 µM) with a FIC value of 0.100. The same VC:VK3 1hr treatment on T24 cells resulted in a 41-fold decrease in the CD50 of VC (4970 µM reduced to 120 µM) and a 59-fold decrease in the CD50 values of VK3 (73.2 µM reduced to 1.21 µM) with an FIC of 0.093.
3.2. Flow Cytometry
Flow cytometry was also employed to determine whether vitamin treatment effects the cell cycle of RT4 cells. Human foreskin fibroblasts were mixed with RT4 cells and then analyzed by flow cytometry in an effort to determine the channel number of the true diploid G0 - G1 peak. The mean channel of the fibroblast G0 - G1 peak is 59 and the mean channel of the G2 - M peak is 118. The mean channel of the RT4 cell G0 - G1 is 108, and the mean channel of the G2 - M peak is 216. The DNA index (mean channel of RT4 G0 - G1/mean channel of fibroblast G0 - G1) is 1.83. This reading indicates that RT4 is an aneuploid cell line. In fact it is hypotetraploid. The
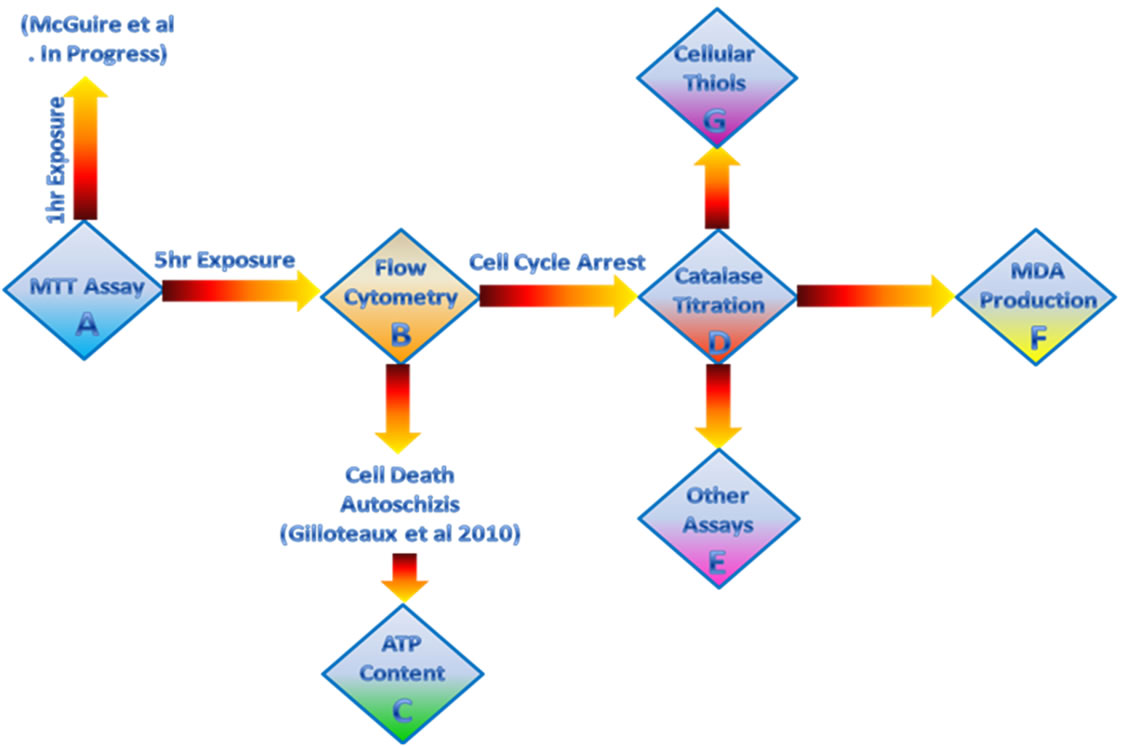
Figure 1. (A) An MTT assay was employed to measure VC:VK3 induced cell cytotoxicity. Since VC:VK3 was equally potent following a 1hr or 5 day exposure both values were reported. (B) Since the MTT value may be a composite measure of metabolic arrest, cell death and cell cycle arrest, cell cycle arrest was measured by flow cytometry and cell death by autoschizis was reported earlier. (C) Electron microscopy (Gilloteaux et al. 2010) showed that mitochondrial architecture was damaged by the VC:VK3 combination, therefore changes in ATP levels were used to evaluate cellular energy content and to determine if cell death was ATP-dependant or ATP-independent. (D) The role of hydrogen peroxide in the activity of VC:VK3 was determined by exogenous catalase titration. (E) If H2O2was not responsible for the antitumor activity other mechanistic activities were performed (not shown). (F) If H2O2 were involved in the mechanism of action lipid peroxidation should occur. Therefore, malondialderayde production was monitored. (G) VC:VK3 have been reported to form a redox pair. If redox was involved there should be a concomitant decrease in cellular thiol content.
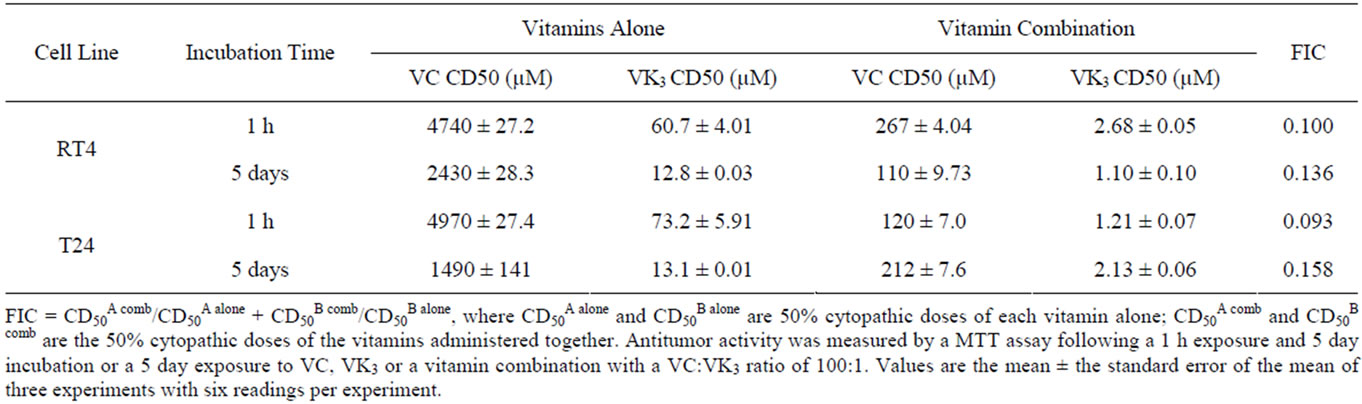
Table 1. Antitumor activity of vitamins against bladder carcinoma cells.
distribution of the cells within the phases of the cell cycle can be found in Table 2. Like untreated RT4 cells, VC-treated RT4 cells exhibited a G0 - G1 peak in channel 115 and a G2 - M peak in channel 231. An aneuploid shoulder was visible on the G0 - G1 peak in channel 130 and the trace also showed a small amount of sub-G0 - G1 “multi-cut debris”. When compared to control cells, the VC-treated cells exhibited 34% of the cells in G0 - G1 phase and 60% of cells in the S phase as opposed to 76% of the cells in G0 - G1 phase and 18% of the cells in S phase in control cells. VK3-treated cells showed a G0 - G1 peak in channel 121 and a G2 - M peak in channel 241. Sub-G0 - G1 multi-cut debris was also evident. As a consequence of this treatment, the proportion of RT4 cells in the G0 - G1 phase decreased to 30% while the number of cells in S phase increased to 64%. VC:VK3-treated cells
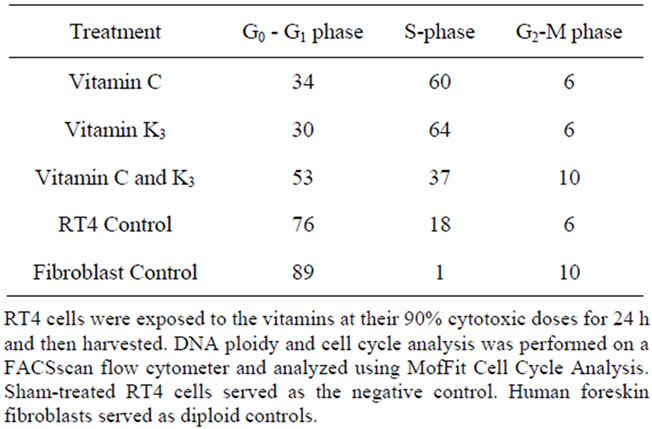
Table 2. Cell cycle distribution of the RT4 cells.
showed a G0 - G1 peak in channel 107 and a G2 - M peak in channel 214. Sub-G0 - G1 multi-cut debris was also present. As a consequence of this treatment, the proportion of RT4 cells in the S phase and G2-M phase were 37 and 10% respectively, compared with 18% and 6% for control cells.
3.3. Hydrogen Peroxide
In our previous studies, catalase administration to DU145 and T24 cells was shown to abrogate the antitumor activity of the vitamins at catalase doses as low as 100 µg/ml [17,45]. Therefore, administration of exogenous catalase has been employed to elucidate the role of hydrogen peroxide (H2O2) in the antitumor activity of vitamins. Catalase administration to RT4 cells abrogated the antitumor activity of VC at catalase doses as low as 100 µg/ml (Table 3). The majority of the antitumor activity of the vitamin combination was lost at a catalase concentration of 100 µg/ml. However, the antitumor activity of VK3 could not be completely neutralized by the administration of catalase even at concentrations as high as 1000 µg/ml. Conversely, the antitumor activity of VC:VK3 was lost following administration of as little as 300 µg/ml of catalase. These results demonstrated that H2O2 production was necessary for the antitumor activity of the vitamins.
3.4. Lipid Peroxidation
Exposure of tumor cells to VC, VK3 or the VC:VK3 combination has been shown to generate hydrogen peroxide (H2O2) and other reactive oxygen species (ROS) that may initiate membrane lipid peroxidation [17,45]. Therefore the effect of vitamin treatment on cellular lipid peroxidation (Table 4) was examined using the thiobarbituric acid method. The lipid peroxidation of shamtreated RT-4 cells displayed an average value of 3.17 nM(MDA)/mg of protein. However, this is only a measure of the lipid peroxidation that occurs during the heating
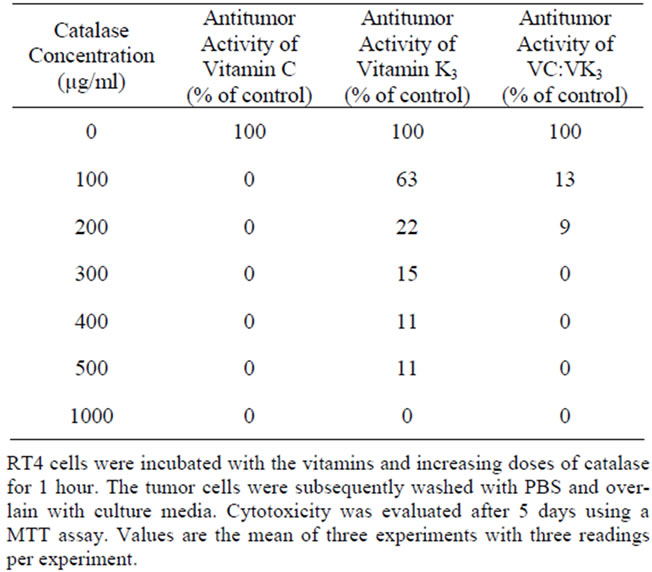
Table 3. Effect of catalase treatment of RT-4 cells antitumor activity.
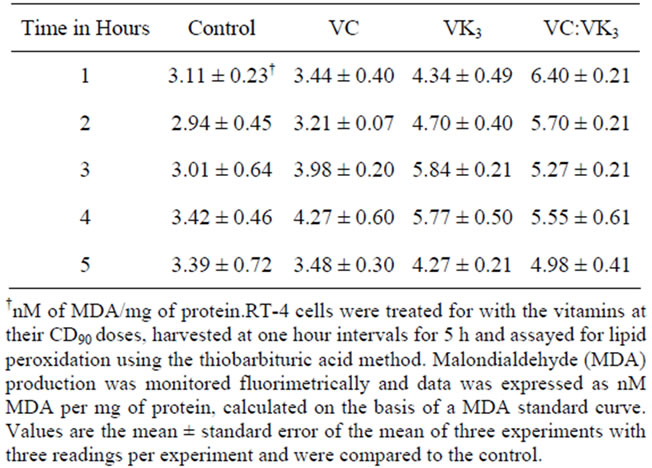
Table 4. Vitamin-induced lipid peroxidation in RT-4 cells.
of samples to 95˚C during the assay and can, therefore, be considered as a baseline for MDA production. Lipid peroxidation values following VC treatment peaked at 4.27 nM/mg with an average value of 3.67 nM/mg, while lipid peroxidation of VK3-treated cells was significantly higher at 5.84 nM/mg with an average of 4.98 nM/mg. Lipid peroxidation values for VC:VK3 peaked at 6.7 nM/mg with an average value of 5.58 nM/mg of protein. The treatment of the cells with the vitamins resulted in a statistically significant alteration in lipid peroxidation (p < 0.005). This lipid peroxidation was vitamin related because lipid peroxidation values rapidly returned to control levels when the vitamins were removed (data not shown).
3.5. ATP Production
Transmission electron microscopy has shown that mitochondrial architecture was altered by vitamin treatment [36,63]. In the following experiments, intracellular levels of ATP synthesis was measured to determine if vitamininduced cell death was related to mitochondrial damage and subsequent “ATP-less” cell death (Figure 2). The ATP content of sham treated RT4 cells displayed averaged of 59.64 nM ATP/mg of protein. VC exposure resulted in an increase in ATP levels to 147 ± 8.64 nM during the first hour. Subsequently, the ATP levels decreased to 86.0 ± 4.73 nM during the second hour and remained relatively constant during the third and fourth hours and then fell to 56.1 ± 4.09 nM during the final hour. VK3 treatment lowered ATP levels to 39.9 ± 0.99 nM during the first hour. ATP levels rose slightly to 48.8 ± 4.52 nM during the second hour, remained relatively constant for the next 3 hours and increased to near control levels during the final hour. The VC:VK3 combination produced a slight decrease in ATP concentration to 46.7 ± 2.13 nM during the first hour. ATP levels increased during the second and third hours to 134 ± 1.46 nM and decreased gradually to near control levels during the final two hours. These results demonstrate that pulse treatment of RT4 cells with VC alone or with the VC: VK3 combination resulted in a transient increase in intracellular ATP levels following vitamin treatment. The treatment of the cells with the vitamins resulted in a significant alteration in ATP levels (p < 0.005).
3.6. Thiols
Administration of VK3 to hepatocytes is known to induce a variety of effects including: depletion of GSH and oxidation of protein sulphydryl groups in cytoskeletal
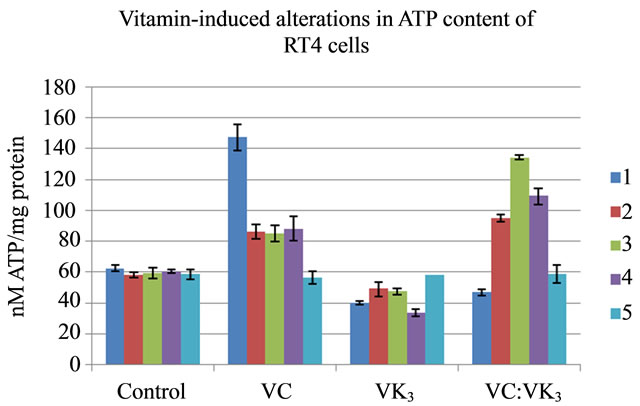
Figure 2. Cultures of exponentially growing RT4 cells were treated for 1 hour with the vitamins at their CD90 doses and then harvested at one hour intervals for 5 h. ATP content was assayed using a bioluminescence assay. Data has been expressed as nM ATP per mg of protein, calculated on the basis of an ATP standard curve. Values are the mean ± standard error of the mean of three experiments with three readings per experiment and were compared to the control (p << 0.05 between all groups relative to control).
proteins [35,65]. Therefore, the effect of vitamin treatment on cellular thiols has been examined (Table 5). The thiol content of sham-treated RT4 cells averaged 1.39 µM/mg of protein. VC treatment resulted in a decrease in cellular thiol levels to 0.92 ± 0.31 µM/mg during the first hour. Subsequently, the thiol level remained constant during the second hour, dropped precipitously to 0.47 ± 0.03 µM/mg during the third hour, rebounded to 0.73 ± 0.12 µM/mg of during the fourth hour and then returned to 0.45 ± 0.03 µM/mg during the final hour. VK3 treatment lowered thiol levels to 0.62 ± 0.5 µM/mg during the first hour. Thiol levels remained constant during the next three hours and then dropped slightly to 0.54 ± 0.1 µM/mg during the final hour. The VC:VK3 combination produced a decrease in thiol concentration to 0.63 ± 0.05 µM/mg during the first hour. Thiol levels gradually decreased to 0.45 ± 0.03 µM/mg during the second hour and then remained constant. VC:VK3 treated cells induced significant depletion of cellular thiols, The treatment of the cells with the vitamins resulted in a significant alteration in thiol levels (p < 0.005).
4. Discussion
VC exhibits selective toxicity against a plethora of tumor cell lines as well as experimental tumors [64,65]. In addition, VC is a chemosensitizing agent [66,67] and radiosensitizing agent [68]. The mechanism(s) responsible for the antitumour activity of VC appears to be related to the prooxidant properties of ascorbate and dehydroascorbate, the oxidative product of ascorbate, which generate intracellular H2O2 and other reactive oxygen species (ROS) which may deplete cellular thiol levels and initiate membrane lipid peroxidation [45,66].
Likewise, VK3 is a synthetic derivative of phylloquinone (VK1) that exhibits in vitro cytotoxic activity against a variety of tumor cell lines [69] as well as in
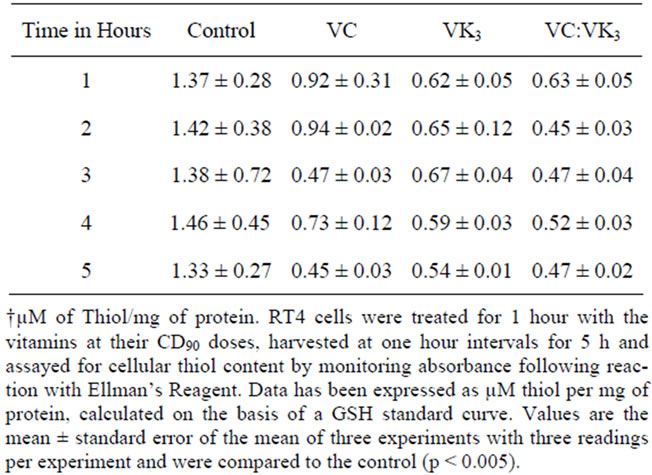
Table 5. Vitamin-induced alterations in the thiol content of RT4 cells.
vivo antitumor activity [70]. VK3 is also a chemosensitizer for most of traditional chemotherapeutic agents [70]. Administration of VK3 to tumor cells induces depletion of glutathione, reduction of nicotinamide adenine dinucleotide phosphate and adenosine triphosphate pools, oxidation of sulfhydryl groups in cytoskeletal proteins, and induction of single-stranded DNA breaks [71-73].
When VC and VK3 were combined in a ratio of 100:1 and administered to three human tumor cell lines, the vitamin combination exhibited a synergistic inhibition of cell growth at concentrations that were 10 to 50 times lower than for the individual vitamins [74]. The vitamin combination also potentiated the in vitro growth inhibitory activities of several chemotherapeutic agents 3- to 14-fold [75]. The VC:VK3 combination also inhibited tumor growth, increased lifespan and decreased metastasis of C3H mice implanted with a murine transplantable liver tumor (TLT) cells [34-37,49]. Additional studies indicated that the VC:VK3 combination was an effective chemosensitizer [34,35] and radiosensitizer that induced little systemic or major organ pathology [35,36]. The potentiation and specificity of the antitumor activity was attributed to the possible generation of hydrogen peroxide followed by membrane lipid alteration, DNase activation and DNA destruction by the VC:VK3 combination in the catalase-deficient cancer cells [35]. When VC and VK3 were combined in a 100:1 ratio, the VC:VK3 interaction not only fostered single-electron reduction to produce the long-lived semiquinone and ascorbyl radicals and increased the rate of redox cycling of the quinone to form H2O2 and other ROS, but also fostered two-electron transfer, which ensured that ascorbic acid (AA) and dehydroascorbic acid (DHA) were present at pharmacologic levels for a protracted period of time [24,50, 76,77]. Single electron redox cycling generated a moderate increase in oxidative stress [24,50,76] and induced a complex stress response that resulted in structural damage to the catalase-deficient cancer cells. A number of cellular processes were effected by the presence of AA and especially DHA, including: modulation of signal transduction, cell cycle arrest and inhibition of glycolytic respiration, inhibition of metastasis [15,23,24,26,35,49, 50,78,79]. Together the two redox cycles reactivated DNase I and II and induced autoschizic cell death [20,23, 46]. A number of other laboratories have subsequently have modified or added more components to the VC: VK3 combination. Modification of one of the constituents (i.e. brominated VK3) or replacement of the VK3 with any number of constituents (arsenic trioxide, benzoquinones, coenzyme Q10, doxorubicin, lipoic acid, resveratrol, vitamin B12, Vitamin E and others) [67,80-85].
In the current study, VC, VK3 and the VC:VK3 combination were evaluated for their antitumor activity against two human bladder cancer cell lines (RT4 and T24). An MTT assay was employed to demonstrate that a VC:VK3 combination with a VC:VK3 ratio of 100:1 exhibited synergistic antitumor activity against both the RT4 and the T24 cell lines following continuous 5-day vitamin exposure or a 1-h vitamin exposure followed by a 5-day incubation in culture media. The fact that there was a close agreement between the CD50 values of the VC:VK3 combination for the 1-h and 5-day vitamin exposures indicated that the antitumor activity generated during a 1-h vitamin treatment was almost as effective as the antitumor activity generated during a continuous 5-day vitamin treatment. These results were consistent with those found in both prostate cancer (DU145) and bladder cancer (T24) cell lines and suggested some of the events responsible for triggering tumor cell death occurred during the first hour of vitamin treatment [17,45]. Characterization of the 5 day vitamin treatment is presented in this report while characterization of the 1-h vitamin treatment will be presented in a future report. In a previous study with T24 cells, flow cytometry revealed that VC:VK3 treatment resulted in a growth arrested population of cells and a population of cells undergoing autoschizic cell death [24].
Likewise, in the current study, a cell cycle block was observed at G0 - G1/S. From previous studies,it is known that enhanced population of cells in early S phase represent both growth arrested and autoschizic cells [24]. The disposition of these autoschizic cells was described in a previous publication [22]. Similar results were seen in an in vivo study when UMUC-14 tumorigenic urothelial carcinoma cells were implanted into the subcutis of nude mice and the mice were treated with gemcitabine GEM), VC:VK3 or a GEM-VC:VK3 combination. In vivo analysis suggested that synergistic antitumor activity was due to antiproliferative effects rather than to enhanced apoptosis. However, there was significant necrosis in combination group tumors which was most likely autoschizis [28]. From our experience with VC:VK3, these effects can be attributed to the combined effects of H2O2 and the vitamins [26].
When RT4 cells were co-incubated with catalase and VC, VK3 or the VC:VK3 combination, the antitumor activity of the vitamins was negated. The fact that a greater amount of catalase was required to destroy the antitumor activity of VK3 than was required to destroy the antitumor activity of the vitamin combination, suggested that while H2O2 was involved in the mechanism of action of these vitamins, the enhanced antitumor activity of the vitamin combination was not simply due to an excessive increase in H2O2 production. Furthermore, the fact that the majority of VC is known to enter tumor cells through Glut channels as DHA coupled with the observation that antitumor activity of the vitamins was abrogated by exogenous catalase suggested that at least a portion of lost antitumor activity of VC and VC:VK3 in the presence of catalase may be a function of diminished extracellular conversion of VC to DHA and a decreased uptake of the vitamin by the tumor cells [86].
The observation that H2O2 generation was essential for the antitumor activity of the vitamin combination suggested that lipid peroxidation may have been responsible for the vitamin-induced cytotoxicity. When the RT4 cells were co-incubated with the vitamins for 1 - 5 h, the level of lipid peroxidation increased 1.1- to 1.3-fold for VC, 1.3- to 1.9-fold for VK3 and 1.5- to 2-fold for VC:VK3. These results suggested that, while lipid peroxidation increased, the modest increase in lipid peroxidation was not the primary cause of the cytotoxicity of the vitamin combination. These results were consistent with those of McGuire and co-workers [87] who demonstrated that when the vitamins were removed from the RT4 cells following a 1-h exposure, lipid peroxidation values returned to near control levels while the cells continued to undergo autoschizis.
Since VC:VK3 treatment of RT4 cells was shown to produce severely damaged mitochondria [26], intracellular levels of ATP were measured to determine if vitamin induced cell death was related to the depletion of ATP. Treatment of RT4 cells with VK3 decreased intracellular ATP levels to approximately 60% to 80% of control levels, while following exposure to VC, intracellular ATP levels increased 2.4-fold during the first hour, decreased to about 1.5-fold greater than control during the second hour and remained constant for the next two hours. Following VC:VK3 exposure, intracellular ATP levels decreased 25% during the first hour, rose to a maximum of about 2.3-fold greater than control by 3 hours and then began to decrease. Interestingly, the fact that the level of intracellular ATP levels for all treatments decreased to control levels by the fifth hour may be indicative of the duration of the pharmacological activity of the vitamins in RT4 cells. These results are consistent with those seen in prostate cancer cells (DU145) and another bladder cancer cell line (T24) and suggested that ATP depletion was not the proximal cause of VC:VK3-induced tumor cell death in RT4 cells [17,45]. While the cause of these ATP spikes has not yet been elucidated, they may reflect the ability of the VC:VK3 to form a shunt around a defective region of complex III of the electron transport chain by having menadione accept electrons from coenzyme Q (ubiquinone), shuttle them to ascorbate and then to cytochrome c. Impaired oxidative phosphorylation has been observed in a variety of cancers including: prostate tumors, and bladder (infiltrating bladder urothelial) carcinomas due to alterations in the protein complexes, especially in complex III [88,89]. Such a shuttle was observed in a patient with a defect in electron transport at complex III in skeletal muscle [91]. The shunt was able to bypass the antimycin-a-sensitive site in both forward and reversed electron transport (had two intact phosphorylation sites) and produced a shift from glycolytic activity to increased mitochondrial oxidative phosphorylation and a dimunition of lactic acidosis.
The effect of vitamin treatment on cellular thiols was examined because redox cycling was shown to draw down cell thiols [71-73,77,78,87]. Following a one hour VC treatment, the intracellular thiol levels of RT4 decreased to 67% of control levels. Thiol levels remained at this level for another hour and then decreased to 34% of control levels. Subsequently, thiol levels oscillated between 34% and 50% of control levels. Exposure of RT4 cells to VK3 led to a drop in intracellular levels to 45% of that of control levels. Following a one hour VC:VK3 treatment, the intracellular thiol levels of RT4 decreased to 46% of control levels. In the second hour, intracellular thiol levels decreased to approximately 35% of control levels and then remained constant for the remaining 3 hours. These results suggested that the cells were only affected by vitamin treatment when the oxidative stress of the vitamins surpassed the reducing ability of the cellular thiols and cellular or genetic damage occurred. Tumor cells appeared to be particularly susceptible because they have reduced levels of catalase, superoxide dismutase, and/or glutathione peroxidase as well as other ROS detoxifying enzymes. Therefore, they have difficulty in metabolizing hydrogen peroxide and other ROS that can accumulate, alter cellular processes, and induce cellular damage or cell death [74].
While the morphologic trends and defects observed following combined vitamin treatment were the composite of the biochemical and cytological damage induced by ascorbate or the menadione-treated alone, the damage induced by VC:VK3 was seen at a VC concentration of 520 µM and a VK3 concentration of 5.2 µM concentrations which were 17-fold less than the concentrations of 8750 and 90 µM when VC and VK3 were administered individually. The values indicated a synergistic interaction between VC and VK3. The vitamin combination was shown to be safe and effective against human prostate cancer cell lines in vitro [44,45], against androgen-independent prostate cancer in nude mice [47] and in two clinical trials with endstage prostate cancer patients [51,55]. Likewise, VC:VK3 was shown to be safe and effective against human bladder cancer cell lines [17-27] and in a murine bladder cancer model when administered alone or in conjunction with gemcitabine [28].
5. Conclusion
While the results described in the current study provide information concerning the effect of VC:VK3 administration on tumor cells death, cell cycle arrest, cellular ATP levels, the effect of redox cycling on cellular thiol levels and confirm the role of H2O2 in lipid peroxidation and tumor cell death, the major limitation of these techniques is that they provide no information concerning the genes and signal transduction mechanisms involved these processes. The authors concluded that, based on their results, a clinical trial was warranted to examine the efficacy and toxicity of VC:VK3. Because of these results, on July 31, 2007, the combination of sodium ascorbate and menadione sodlium bisulfite (tradename Apatone®, designation request #06-2366) was granted orphan drug status for the treatment of metastatic or locally advanced, inoperable transitional cell carcinoma of the urothelium (stage III and IV bladder cancer). Efforts are underway to conduct a phase II clinical trial for this indication.
6. Acknowledgements
This research was funded by grants from The Summa Health System Foundation and The Women’s Board at St. Thomas Hospital.
REFERENCES
- R. Siegel, D. Naishadham and A. Jemal, “Cancer Statistics, 2013,” CA: A Cancer Journal for Clinicians, Vol. 63, No. 1, 2013, pp. 11-30. doi:10.3322/caac.21166
- P. J. Goebell and M. A. Knowles, “Bladder Cancer or Bladder Cancers? Genetically Distinct Malignant Conditions of the Urothelium,” Urologic Oncology, Vol. 28, No. 4, 2010, pp. 409-428.
- Y. Li, K. Izumi and H. Miyamoto, “The Role of the Androgen Receptor in the Development and Progression of Bladder Cancer,” The Japanese Journal of Clinical Oncology, Vol. 42, No. 7, 2012, pp. 569-577. doi:10.1093/jjco/hys072
- C. Logan, M. Brown and D. Hayne, “Intravesical Therapies for Bladder Cancer-Indications and Limitations,” British Journal of Urology International, Vol. 110, No. S4, 2012, pp. 12-21.
- A. Pawinski, R. Sylvester, K. H. Kurth, et al., “A Combined Analysis of European Organization for Research and Treatment of Cancer, and Medical Research Council Randomized Clinical Trials for the Prophylactic Treatment of Stage TaT1 Bladder Cancer,” The Journal of Urology, Vol. 156, No. 6, 1996, pp. 1934-1941.
- C. N. Sternberg, A. Yagoda, H. I. Scher, et al., “Methotrexate, Vinblastine, Doxorubicin, and Cisplatin for Advanced Transitional Cell Carcinoma of the Urothelium. Efficacy and Patterns of Response and Relapse,” Cancer Vol. 64, No. 12, 1989, pp. 2448-2458. doi:10.1002/1097-0142(19891215)64:12<2448::AID-CNCR2820641209>3.0.CO;2-7
- F. Calabrò and C. N. Sternberg, “Metastatic Bladder Cancer: Anything New?” Current Opinion in Supportive and Palliative Care. Vol. 6, No. 3, 2012, pp. 304-309.
- H. von der Maase, S. W. Hansen, J. T. Roberts, et al., “Gemcitabine and Cisplatin versus Methotrexate, Vinblastine, Doxorubicin, and Cisplatin in Advanced or Metastatic Bladder Cancer: Results of a Large, Randomized, Multinational, Multicenter, Phase III Study,” Journal of Clinical Oncology, Vol. 17, No. 17, 2000, pp. 3068-3077.
- M. Hussain, U. Vaishampayan, W. Du, et al., “Combination Paclitaxel, Carboplatin, and Gemcitabine Is an Active Treatment for Advanced Urothelial Cancer,” Journal of Clinical Oncology, Vol. 19, No. 9, 2001, pp. 2527-2533.
- K. D. Sievert, B. Amend, U. Nagele, et al., “Economic Aspects of Bladder Cancer: What Are the Benefits and Costs?” World Journal of Urology, Vol. 27, No. 3, 2009, pp. 295-300. doi:10.1007/s00345-009-0395-z
- D. L. Lamm and M. Allaway, “Current Trends in Bladder Cancer Treatment,” Annales Chirurgiae et Gynaecologiae, Vol. 89, 2000, pp. 234-241.
- D. L. Lamm, D. R. Riggs, J. S. Shriver, et al., “Megadose Vitamins in Bladder Cancer: A Double-Blind Clinical Trial,” The Journal of Urology, Vol. 151, No. 1, 1994, pp. 21-26.
- R. Bonilla-Porras, M. Jimenez-Del-Rio and C. VelezPardo, ”Vitamin K3 and Vitamin C Alone or in Combination Induced Apoptosis in Leukemia Cells by a Similar Oxidative Stress Signalling Mechanism,” Cancer Cell International, Vol. 11, No. 10, 2011, p. 19. doi:10.1186/1475-2867-11-19
- J. Verrax, S. Vanbever, J. Stockis, et al., “Role of Glycolysis Inhibition and Poly(ADP-ribose) Polymerase Activation in Necrotic-Like Cell Death Caused by Ascorbate/ Menadione-Induced Oxidative Stress in K562 Human Chronic Myelogenous Leukemic Cells,” International Journal of Cancer, Vol. 120, No. 6, 2007, pp. 1192-1197. doi:10.1002/ijc.22439
- J. Verrax, J. Stockis, A. Tison, et al., “Oxidative Stress by Ascorbate/Menadione Association Kills K562 Human Chronic Myelogenous Leukaemia Cells and Inhibits Its Tumour Growth in Nude Mice,” Biochemical Pharmacology, Vol. 72, No. 6, 2006, pp. 671-680. doi:10.1016/j.bcp.2006.05.025
- J. Verrax, M. Delvaux, N. Beghein, et al., “Enhancement of Quinone Redox Cycling by Ascorbate Induces a Caspase-3 Independent Cell Death in Human Leukemia Cells. An in Vitro Comparative Study,” Free Radical Research, Vol. 39, No. 6, 2005, pp. 649-657. doi:10.1080/10715760500097906
- M. Venugopal, J. M. Jamison, J. Gilloteaux, et al., “Synergistic Antitumor Activity of Vitamins C and K3 on Human Urologic Tumor Cell Lines,” Life Sciences, Vol. 56, No. 17, 1996, pp. 1389‑1400. doi:10.1016/0024-3205(96)00466-3
- J. Gilloteaux, J. M. Jamison, E. Ervin, et al., “Scanning Electron Microscopy and Transmission Electron Microscopy Aspects of the Synergistic Antitumor Activity of Vitamin C/ Vitamin K3 Combinations against Human T24 Bladder Carcinoma: Another Kind of Cell Death?” Scanning, Vol. 20, No. 3, 1998, pp. 208‑209.
- E. Ervin, J. M. Jamison, J. Gilloteaux, et al., “Characterization of the Early Events in Vitamin C and K3‑Induced Death of Human Bladder Tumor Cells,” Scanning, Vol. 20, No. 3, 1998, pp. 210‑211.
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Cancer Cell Necrosis by Autoschizis: Synergism of Antitumor Activity of Vitamin C:Vitamin K3 on Human Bladder Carcinoma T24 Cells,” Scanning, Vol. 20, No. 3, 1998, pp. 564‑576.
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Autoschizis: Another Cell Death for Cancer Cells Induced by Oxidative Stress,” In Advances in Microanatomy of Cells and Tissues, Biophysical and Biochemical Correlates, Marcello Malphigi Series, Italian Journal of Anatomy and Embryology, Vol. 106, No. 2, 2001, pp. 79-91.
- J. Gilloteaux, J. M. Jamison, D. Arnold, H. S. Taper, J. L. Summers, “Ultrastructural Aspects of Autoschizis: A New Cancer Cell Death induced by the Synergistic Action of Ascorbate/Menadione on Human Bladder Carcinoma Cells,” Ultrastructural Pathology, Vol. 25, No. 3, 2001, pp. 183-192. doi:10.1080/019131201300343810
- J. M. Jamison, J. Gilloteaux, H. S. Taper, et al., “Autoschizis: A Novel Cell Death,” Biochemical Pharmacology, Vol. 63, No. 10, 2002, pp. 1773-1783. doi:10.1016/S0006-2952(02)00904-8
- J. M. Jamison, J. Gilloteaux, M. R. Nassiri, et al., “Induction of Cell Cycle Arrest and Autoschizis in a Human Bladder Carcinoma Cell Line by Vitamins C and K3,” Biochemical Pharmacology, Vol. 67, No. 2, 2004, pp. 337-351. doi:10.1016/j.bcp.2003.08.040
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Morphology and DNA Degeneration during Autoschizic Cell Death in Bladder Carcinoma T24 Cells Induced by Ascorbate and Menadione Treatment,” Anatomical Record, Part A: Discoveries in Molecular, Cellular, and Evolutionary Biology, Vol. 288, No. 1, 2006, pp. 58-83. doi:10.1002/ar.a.20276
- J. Gilloteaux, J. M. Jamison, D. R. Neal, et al., “Cell Damage and Death by Autoschizis in Human Bladder (RT4) Carcinoma Cells Resulting from Treatment by Ascorbate and Menadione,” Ultrastructural Pathology, Vol. 34, No. 3, 2010, pp. 140-160. doi:10.3109/01913121003662304
- J. M. Jamison, J. Gilloteaux, L. Perlaky, et al., “Nucleolar Changes and Fibrillarin Redistribution Following Apatone® Treatment of Human Bladder Carcinoma Cells,” Journal of Histochemistry and Cytochemistry, Vol. 58, No. 7, 2010, pp. 635-652. doi:10.1369/jhc.2010.956284
- W. Kassouf, R. Highshaw, G. M. Nelkin, et al., “Vitamins C and K3 Sensitize Human Urothelial Tumors to Gemcitabine,” The Journal of Urology, Vol. 176, No. 4, 2006, pp. 1642-1647.
- R. Beck, J. Verrax, N. Dejeans, et al., “Menadione Reduction by Pharmacological Doses of Ascorbate Induces an Oxidative Stress That Kills Breast Cancer cells,” International Journal of Toxicology, Vol. 28, No. 1, 2009, pp. 33-42. doi:10.1177/1091581809333139
- H. S. Taper, “Reversibility of Acid and Alkaline Deoxyribonuclease Deficiency in Malignant Tumor Cells,” Journal of Histochemistry and Cytochemistry, Vol. 29, No. 9, 1981, pp. 1053-1060.
- M. F. Vita, N. Nagachar, D. Avramidis, et al., “Pankiller Effect of Prolonged Exposure to Menadione on Glioma Cells: Potentiation by Vitamin C,” Investigational New Drugs, Vol. 82, No. 11, 2011, p. 1314. doi:10.1007/s10637-010-9489-0
- D. Arnold, J. Gilloteaux, J. Jamison, et al., “Synergistic Effect of Vitamin C and Vitamin K3 on Human Renal Adenocarcinoma Cell Line (Caki‑1): Scanning Electron Microscopy,” Scanning, Vol. 21, No. 2, 1999, pp. 109‑110.
- J. Verrax, R. Beck, N. Dejeans, et al., “Redox-Active Quinones and Ascorbate: An Innovative Cancer Therapy That Exploits the Vulnerability of Cancer Cells to Oxidative Stress,” Anti-Cancer Agents in Medicinal Chemistry, Vol. 11, No. 2, 2011, pp. 213-221. doi:10.2174/187152011795255902
- H. S. Taper, J. de Gerlache, M. Lans M, et al., “NonToxic Potentiation of Cancer Chemotherapy by Combined C and K3 Vitamin Pretreatment,” International Journal of Cancer, Vol. 40, No. 4, 1987, pp. 575-579. doi:10.1002/ijc.2910400424
- H. S. Taper, A. Keyeux and M. Roberfroid, “Potentiation of Radiotherapy by Non-Toxic Pretreatment with Combined Vitamin C and K3 in Mice Bearing Solid, Transplantable Tumor,” Anticancer Research Vol. 16, No. 1, 1996, pp. 499-503.
- H. S. Taper and M. Roberfroid, “Non-Toxic Sensitization of Cancer Chemotherapy by Combined C and K3 Vitamin Pretreatment in a Mouse Tumor Resistant to Oncovin,” Anticancer Research, Vol. 12, No. 5, 1992, pp. 1651-1654.
- H. S. Taper, J. M. Jamison, J. Gilloteaux, et al., “Inhibition of the Development of Metastases by Dietary Vitamin C: K3 Combination,” Life Sciences, Vol. 75, No. 8, 2004, pp. 955-967. doi:10.1016/j.lfs.2004.02.011
- M. F. Chen, C. M. Yang and C. M. Su, “Inhibitory Effect of Vitamin C in Combination with Vitamin K3 on Tumor Growth and Metastasis of Lewis Lung Carcinoma Xenografted in C57BL/6 Mice,” Nutrition and Cancer, Vol. 63, No. 7, 2011, pp. 1036-1043. doi:10.1080/01635581.2011.597537
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Microscopic Aspects of Autoschizic Cell Death in Human Ovarian Carcinoma (2774) Cells Following Vitamin C, Vitamin K3 or Vitamin C:K3 Treatment,” Microscopy and Microanalysis, Vol. 9, No. 4, 2003, pp. 311-329. doi:10.1017/S1431927603030125
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Autoschizis of Human Ovarian Carcinoma Cells: Scanning Electron and Light Microscopy of a New Cell Death Induced by Sodium Ascorbate: Menadione Treatment,” Scanning, Vol. 25, No. 3, 2003, pp. 137-149. doi:10.1002/sca.4950250306
- V. E. Von Gruenigen, J. M. Jamison, J. Gilloteaux, et al., “The in Vitro Antitumor Activity of Vitamins C and K3 against Ovarian Carcinoma,” Anticancer Research, Vol. 23, No. 4, 2003, pp. 3279-3287.
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Autoschizis: A New Form of Cell Death for Human Ovarian Carcinoma Cells Following Ascorbate: Menadione Treatment: Nuclear and DNA Degradation,” Tissue and Cell, Vol. 36, No. 3, 2004, pp. 197-209. doi:10.1016/j.tice.2004.01.006
- J. Gilloteaux, J. M. Jamison, M., Venugopal, et al., “Scanning Electron Microscopy and Transmission Electron Microscopy Aspects of Synergistic Antitumor Activity of Vitamin C‑Vitamin K3 Combinations against Human Prostatic Carcinoma Cells,” Scanning Microscopy, Vol. 9, No. 1, 1995, pp. 159‑173.
- J. M. Jamison, J. Gilloteaux, M. Venugopal, et al., “Flow Cytometric and Ultrastructural Aspects of the Synergistic Antitumor Activity of Vitamin C‑Vitamin K3 Combinations against Human Prostatic Carcinoma Cells,” Tissue and Cell, Vol. 28, No. 6, 1996, pp. 687‑701.
- M. Venugopal, J. M. Jamison, J. Gilloteaux, J. A. Koch, M. Summers, J. Hoke, C. Sowick and J. L. Summers, “Synergistic Antitumor Activity of Synergistic Antitumor Activity of Vitamin C and K3 against Human Prostate Carcinoma Cell Lines,” Cell Biology International Reports, Vol. 20, No. 12, 1996, pp. 787‑797.
- H. S. Taper, J. M. Jamison, J. Gilloteaux, et al., “In Vivo Reactivation of DNases in Implanted Human Prostate Tumors Following Administration of a Vitamin C/K3 Combination,” Journal of Histochemistry and Cytochemistry, Vol. 49, No. 1, 2001, pp. 109-120. doi:10.1177/002215540104900111
- J. M. Jamison, J. Gilloteaux, H. S. Taper, et al., “Evaluation of the In Vitro and In Vivo Antitumor Activities of Vitamin C and K3 Combinations against Human Prostate Cancer,” Journal of Nutrition, Vol. 131, No. 1, 2001, pp. 158S-160S.
- P. B. Calderon, J. Cadrobbi, C. Marques, et al., “Potential Therapeutic Application of the Association of Vitamins C and K3 in Cancer Treatment,” Current Medicinal Chemistry, Vol. 9, No. 24, 2003, pp. 2269-2285.
- J. Verrax, J. Cadrobbi, M. Delvaux, et al., “The Association of Vitamins C and K3 Kills Cancer Cells Mainly by Autoschizis, a Novel Form of Cell Death. Basis for Their Potential Use as Coadjuvants in Anticancer Therapy,” European Journal of Medicinal Chemistry, Vol. 38, No. 5, 2003, pp. 451-457. doi:10.1016/S0223-5234(03)00082-5
- J. M. Jamison, J. Gilloteaux, H. S. Taper, et al., “The in Vitro and in Vivo Antitumor Activity of Vitamin C: K3 Combinations against Prostate Cancer,” In: J. L. Lucas, Ed., Prostate Cancer, Nova Science Publishers, Inc., New York, 2005, pp. 189-236.
- B. Tareen, J. L. Summers, J. M. Jamison, et al., “A 12 Week, Open Label, Phase I/IIa Study Using Apatone® for the Treatment of Prostate Cancer Patients Who Have Failed Standard Therapy,” International Journal of Medical Science, Vol. 5, No. 2, 2008, pp. 62-67. doi:10.7150/ijms.5.62
- J. Gilloteaux, J. M. Jamison, D. Arnold, et al., “Autoschizis: A New Cell Death Found in Tumour Cells Induced by an Oxidative Stress Mechanism,” In: A. Mé- ndez-Vilas and J. Díaz, Eds., Microscopy: Science, Technology, Applications and Education, Formatex Research Center, Badajoz, 2010, pp. 1-12.
- J. Gilloteaux, J. M. Jamison, D. R. Neal, et al., “Xenotransplanted Human Prostate Carcinoma (DU145) Cells Develop into Carcinomas and Cribriform Carcinomas: Ultrastructural Aspects,” Ultrastructural Pathology, Vol. 36, No. 5, 2012, pp. 294-311. doi:10.3109/01913123.2012.708472
- J. Gilloteaux, J. M. Jamison, D. R. Neal, et al., “Human Prostate DU145 Carcinoma Cells Implanted in Nude Mice Remove the Peritoneal Mesothelium to Invade and Grow as Carcinomas,” The Anatomical Record, Vol. 296, No. 1, 2013, pp. 40-55. doi:10.1002/ar.22607
- E. Lasalvia-Prisco, S. Cucchi, J. Vázquez, et al., “Serum Markers Variation Consistent with Autoschizis Induced by Ascorbic Acid-Menadione in Patients with Prostate Cancer,” Medical Oncology, Vol. 20, No. 1, 2003, pp. 45-52. doi:10.1385/MO:20:1:45
- D. B. Agus, J. C. Vera and D. W. Golde, “Stromal Cell Oxidation: A Mechanism by Which Tumors Obtain Vitamin C,” Cancer Research, Vol. 59, No. 18, 1999, pp. 4555-4558.
- B. Meier, A. R. Cross, J. T. Hancock, et al., “Identification of a Superoxide Generating NADPH Oxidase System in Human Fibroblasts,” Biochemical Journal, Vol. 275, No. 1, 1991, pp. 241-245.
- J. C. Vera, C. I. Rivas, J. Fischbarg, et al., “Mammalian Facilitative Hexose Transporters Mediate the Transport of Dehydroascorbic Acid,” Nature, Vol. 364, No. 6432, 1993, pp. 79-82. doi:10.1038/364079a0
- J. C. Vera, C. I. Rivas, F. V. Velasquez, et al., “Resolution of the Facilitated Transport of Dehydroascorbic Acid from Its Intracellular Accumulation as Ascorbic Acid,” The Journal of Biological Chemistry, Vol. 270, No. 40, 1995, pp. 23706-23712.
- M. M. Bradford, “A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding,” Analytical Biochemistry, Vol. 72, No. 1-2, 1976, pp. 248-254. doi:10.1016/0003-2697(76)90527-3
- J. A. Buege and S. D. Aust, “Microsomal Lipid Peroxidation,” Methods in Enzymology, Vol. 52, 1978, pp. 302-310. doi:10.1016/S0076-6879(78)52032-6
- K. Nitahara, A. Kittel, S. D. Liang, et al., “A1-ReceptorMediated Effect of Adenosine on the Release of Acetylcholine from the Myenteric Plexus: Role and Localization of Ecto-ATPase and 5’-Nucleotidase,” Neuroscience, Vol. 67, No. 1, 1995, pp. 159-168. doi:10.1016/0306-4522(94)00585-S
- J. F. Nagelkerke, P. Dogterom, H. J. De Bont, et al., “Prolonged High Intracellular Free Calcium Concentrations Induced by ATP Are Not Immediately Cytotoxic in Isolated Rat Hepatocytes,” Biochemical Journal, Vol. 263, No. 2, 1989, pp. 347-353.
- M. J. González, J. R. Miranda-Massari, E. M. Mora, et al., “Orthomolecular Oncology Review: Ascorbic Acid and Cancer 25 Years Later,” Integrative Cancer Therapies, Vol. 4, No. 1, 2005, pp. 32-44. doi:10.1177/1534735404273861
- J. B. Liehr, “Vitamin C Reduces the Incidence and Severity of Renal Tumors Induced by Estradiol and Diethylstilbesterol,” The American Journal of Clinical Nutrition, Vol. 54, No. 6, 1991, pp. 1256S-1260S.
- Y. Zaizen, A. Nakagawara and K. Ikeda, “Patterns of Destruction of Mouse Neuroblastoma Cells by Extracellular Hydrogen Peroxide Formed by 6-Hydroxydopamine and Ascorbate,” Journal of Cancer Research and Clinical Oncology, Vol. 111, No. 2, 1986, pp. 93-97. doi:10.1007/BF00400743
- J. J. Casciari, N. H. Riordan, T. L. Schmidt, et al., “Cytotoxicity of Ascorbate, Lipoic Acid, and Other Antioxidants in Hollow Fibre in Vitro Tumours,” British Journal of Cancer, Vol. 84, No. 11, 2001, pp. 1544-1550. doi:10.1054/bjoc.2001.1814
- G. J. Koch and J. E. Biaglow, “Toxicity, Radiation Sensitivity Modification and Biological Effects of Dehydroascorbate and Ascorbate in Mammalian Cells,” Journal of Cellular Physiology, Vol. 94, No. 3, 1978, pp. 299- 306. doi:10.1002/jcp.1040940307
- L. M. Nutter, A. L. Cheng, H. I. Hung, et al., “Menadione: Spectrum of Anticancer Therapy and Effects on Nucleotide Metabolism in Human Neoplastic Cell Lines,” Biochemical Pharmacology, Vol. 41, No. 9, 1991, pp. 1283-1292. doi:10.1016/0006-2952(91)90099-Q
- W. C. Su, T. P. Sun and F. Y. Wu, “The in Vitro and in Vivo Cytotoxicity of Menadione (Vitamin K3) against Rat Transplantable Hepatoma Induced by 3’-Methyl-4-dimethyl-aminoazobenzene,” Gaoxiong Yi Xue Ke Xue Za Zhi, Vol. 7, No. 9, 1991, pp. 454-459.
- T. W. Gant, D. N. Rao, R. Mason, et al., “Redox Cycling and Sulphydryl Arylation; Their Relative Importance in the Mechanism of Quinone Cytotoxicity to Isolated Hepatocytes,” Chemico-Biological Interactions, Vol. 65, No. 2, 1988, pp. 157-173. doi:10.1016/0009-2797(88)90052-X
- F. Mirabelli, A. Salis, M. Vairetti, et al., “Cytoskeletal Alterations in Human Platelets Exposed to Oxidative Stress Are Mediated by Oxidative and Ca2+-Dependent Mechanisms,” Archives of Biochemistry and Biophysics, Vol. 270, No. 2, 1989, pp. 478-488. doi:10.1016/0003-9861(89)90529-8
- C. R. Stubberfield and C. R. Cohen, “Interconversion of NAD(H) to NADP(H): A Cellular Response to QuinoneInduced Oxidative Stress in Isolated Hepatocytes,” Biochemical Pharmacology, Vol. 38, No. 16, 1989, pp. 2631-2637. doi:10.1016/0006-2952(89)90548-0
- V. Noto, H. S. Taper, Y. H. Jiang, et al., “Effects of Sodium Ascorbate (Vitamin C) and 2-Methyl-1,4-naphthoquinone (Vitamin K3) Treatment on Human Tumor Cell Growth in Vitro, I: Synergism of Combined Vitamin C and K3 Action,” Cancer, Vol. 63, No. 5, 1989, pp. 901- 906. doi:10.1002/1097-0142(19890301)63:5<901::AID-CNCR2820630518>3.0.CO;2-G
- W. De Loecker, J. Janssens, J. Bonte, et al., “Effects of Sodium Ascorbate (Vitamin C) and 2-Methyl-1,4-naphthoquinone (Vitamin K3) Treatment on Human Tumor Cell Growth in Vitro, II: Synergism with Combined Chemotherapy Action,” Anticancer Research, Vol. 13, No. 1, 1993, pp. 103-106.
- R. Jarabak and J. Jarabak, “Effect of Ascorbate on the DT-Diaphorase-Mediated Redox Cycling of 2-Methyl- 1,4-naphthoquinone,” Archives of Biochemistry and Biophysics, Vol. 318, No. 2, 1995, pp. 418-423. doi:10.1006/abbi.1995.1249
- M. Comporti, “Three Modes of Free Radical-Induced Cell Injury,” Chemico-Biological Interactions, Vol. 72, No. 1-2, 1989, pp. 1-56.
- D. Di Monte, G. Bellomo, H. Thor, et al., “MenadioneInduced Cytotoxicity Is Associated with Protein Thiol Oxidation and Alteration in Intracellular Ca2+ Homeostasis,” Archives of Biochemistry and Biophysics, Vol. 235, No. 2, 1984, pp. 343-350. doi:10.1016/0003-9861(84)90207-8
- J. M. Cárcamo, A. Pedraza, O. Bórquez-Ojeda, et al., “Vitamin C Suppresses TNF Alpha-Induced NF Kappa B Activation by Inhibiting I Kappa B Alpha Phosphorylation,” Biochemistry, Vol. 41, No. 43, 2002, pp. 12995-13002. doi:10.1021/bi0263210
- V. S. Akatov, Y. V. Evtodienko, V. V. Leshchenko, et al., “Combined Vitamins B12b and C Induce the Glutathione Depletion and the Death of Epidermoid Human Larynx Carcinoma Cells HEp-2,” Bioscience, Vol. 20, No. 5, 2000, pp. 411-417. doi:10.1023/A:1010386102562
- S. Biswas, X. Zhao, A. P. Mone, et al., “Arsenic Trioxide and Ascorbic Acid Demonstrate Promising Activity against Primary Human CLL Cells in Vitro,” Leukemia Research, Vol. 34, No. 7, 2010, pp. 925-931.
- H. Zhang, “Anticancer Activities of Resveratrol Alone and in Combination with Ascorbic Acid,” Master’s Thesis, Kent State University, Kent, 2010.
- S. Prakash, J. Sunitha and M. Hans, “Role of Coenzyme Q10 as an Antioxidant and Bioenergizer in Periodontal Diseases,” Indian Journal of Pharmacology, Vol. 42, No. 6, 2010, pp. 334-337. doi:10.4103/0253-7613.71884
- A. W. Linnane, M. Kios and L. Vitetta, et al., “Coenzyme Q(10)—Its Role as a Prooxidant in the Formation of Superoxide Anion/Hydrogen Peroxide and the Regulation of the Metabolome,” Mitochondrion, Vol. 7, 2007, pp. S51-S61. doi:10.1016/j.mito.2007.03.005
- V. A. Roginsky, G. Bruchelt and O. Bartuli, “Ubiquinone-0 (2,3-Dimethoxy-5-methyl-1,4-benzo-qui-none) as Effective Catalyzer of Ascorbate and Epinephrine Oxidation and Damager of Neuroblastoma Cells,” Biochemical Pharmacology, Vol. 55, No. 1, 1998, pp. 85-91. doi:10.1016/S0006-2952(97)00434-6
- C. Spielholz, D. W. Golde, A. N. Houghton, et al. “Increased Facilitated Transport of Dehydroascorbic Acid without Changes in Sodium-Dependent Ascorbate Transport in Human Melanoma Cells,” Cancer Research, Vol. 57, No. 12, 1997, pp. 2529-2537.
- K. M. McGuire, “Characterization of Apatone and Tolecine Induced Cell Death Mechanisms in Bladder and Ovarian Cancer,” Ph.D. Dissertation, Kent State University, Kent, 2012.
- L. Dyrskjot, M. Kruhoffer, T. Thykjaer, et al., “Gene Expression in the Urinary Bladder: A Common Carcinoma in Situ Gene Expression Signature Exists Disregarding Histopathological Classification,” Cancer Research, Vol. 64, No. 11, 2004, pp. 4040-4048. doi:10.1158/0008-5472.CAN-03-3620
- K. M. Owens, M. Kulawiec, M. M. Desouki, et al., “Impaired OXPHOS Complex III in Breast Cancer,” PLoS One, Vol. 6, No. 8, 2011, Article ID: e23846. doi:10.1371/journal.pone.0023846
- S. Eleff, N. G. Kennaway, N. R. M. Buist, et al., “31P NMR Study of Improvement in Oxidative Phosphorylation by Vitamins K3 and C in a Patient with a Defect in Electron Transport at Complex III in Skeletal Muscle,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 81, No. 11, 1984, pp. 3529-3533. doi:10.1073/pnas.81.11.3529