Journal of Cancer Therapy
Vol.4 No.5(2013), Article ID:33769,5 pages DOI:10.4236/jct.2013.45105
The Relationship between Mouth Squamous Cell Carcinoma (MSCC) with HPV Infection and the Presence of p53 & c-myc Mutation
![]()
1Department of Dental and Mouth Disease, Faculty of Medicine, University of Sebelas Maret, Surakarta, Indonesia; 2Department of Pathobiology, Faculty of Medicine, University of Airlangga, Surabaya, Indonesia.
Email: drgadiprayitno@yahoo.com
Copyright © 2013 Adi Prayitno. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 17th, 2013; revised April 20th, 2013; accepted April 28th, 2013
Keywords: HPV; p53 and c-myc; Mutation; MSCC
ABSTRACT
Introduction: Now the molecular epidemiology is a new experience. It’s was noted that ninety percent of mouth cancers are squamous cell carcinomas and recorded 0.96% year of all cancers in Indonesia. Human papilloma virus (HPV) was implicated in pathogenesis of cancer. As a remark, that mutations of p53 and c-myc are found 50% in cancer. Objective: Aims of this research were to know the relationship between the mouth squamous cell carcinoma (MSCC) with HPV infection, the presence of p53, and c-myc genes mutation. Methods and Material: Tissue biopsy frozen sections from Benign Mouth Squamous Cell (BMSC) and MSCC patients were collected from Mouth and Dental Department of Muwardi District Hospital in Solo—Indonesia. To amplify L1-HPV gene for fixed the HPV etiology, amplified p53 and c-myc genes continued with SSCP analysis and followed with measurement using densitometer to see mutation existence. The collected data were analyzed with Chi Square Test. Results: None of the sample of patients with BMSC with positive HPV showed p53 gene mutation or c-myc gene. From eleven samples obtained from patients with MSCC who were positive HPV showed 18.2% had mutations in the p53 gene and 9.1% had mutations in c-myc gene. The chisquare test was shown to have significant differences between the MSCC with HPV infection and the presence of p53 and c-myc genes mutation. Conclusion: HPV is a risk ingredient for MSCC.
1. Introduction
Cause of cancer may divide into two groups: environmental cause and hereditary genetic cause. Cancer is primarily an environmental disease, though genetics influence the risk of some cancers. Environmental factors include: environmental pollutants, diet and obesity, tobacco, infection, radiation, and lack of physical activity. These environmental factors cause or enhance abnormalities in the genetic of cells [1,2]. Some researchers found that human papilloma virus (HPV) was implicated in Mouth Squamous Cell Carcinoma (MSCC) pathogenesis [2-4]. Therefore, if the pathogenesis of MSCC can’t be explained, the morbidity and mortality may increase and led to decrease the human resources.
The role of HPV, expression of Early gene 6 (E6) and E7 protein virus, expression of p53 and c-myc proteins host may be explained by pathobiology examination
[5,6]. This concept gives a chance to explain the increasing MSCC.
In the disease with HPV infection, it’s known that the viral genome integrates into the host genome. Many studies were shown that an integrated part of the genome corresponding to the E6 and E7. Therefore, E6 and E7 sequences are directly involved in the cellular cycle by inhibiting the normal function of p53 and pRb. Protein 53 kDa (p53) may provoke arrest cell division and guarantee for the repair of DNA. If the damage cannot be repaired, p53 may induce apoptosis and prevent the spread of DNA damage in the next generation of cells. E7 protein interacts with pRb protein that is an important ingredient for the control of cellular cycle. This interaction causes the release of the E2F transcription factor that is now free to act and may stimulate cellular division via c-myc proten. This means that certain types of HPV may cause malignant lesions even without other co-factors actions [7,8].
2. Methods
Fourty frozen section BMSC tissue and five-teen frozen section MSCC tissue patients collected from Mouth and Dental Clinic of Muwardi District Hospital in Solo from January 2007 to January 2008.
Parrafin blocks were made from cutting I that was subsequently stains with Haematoxyline Eosine (HE) to ascertain the kind of neoplasm.
Cutting II was subjected to Deoxyribonucleic Acid (DNA) isolation was made by Henk Schmits method with some modifications. Cut up to 25 mgr of tissue into small pieces, place in 1.5 ml a micro-tube volume, and add 200 µl of DNA extraction buffer. Add 20 µl of Proteinase K stock solution, mix by vortexing, and incubate at 55˚C overnight [9].
The DNA isolation results were subjected to Polymerase Chains Reaction (PCR) to amplify L1-HPV for fixed the HPV stressor. Diagnose related HPV infections are made by Henk Schmits and/or Nigel McMillan and Nina Fowler PCR-method with some modifications. 25 µl micro-tube Ready To Go PCR Bead (Amersham Pharmacia Biotech) mixed with 2 µl HPV consensus primers (MY09: 5’GC(A/C)CAGGG(A/T)CATAA(C/T)AATGC3’ and MY11: 5’CGTCC(A/C)A(A/G)(A/G)GGA(A/T)ACTATC3’) (CYBERGENE AB) and 2 µl DNA template. PCR protocol for both amplifications are 94˚C for 50 seconds, 59˚C for 50 seconds, 72˚C for 50 seconds and 4˚C soak. The Amplification of HPV-L1 gene produced 450 bp long. Other DNA isolation results were subjected to Polymerase Chains Reaction (PCR) to amplify p53 and c-myc gene and continued Single Strand Conformational Polymorphism (SSCP) analysis and followed with measurement using densitometer to see mutation existence. To amplify of p53 and c-myc made by Henk Schmits and/or Nigel McMillan and Nina Fowler PCR-method with some modifications [9,10]. 25 µl micro-tube Ready To Go PCR Bead (Amersham Pharmacia Biotech) mixed with 2 µl p53 primer (forward primer 5’TATCCTGAGTAGTGGTAATC3’ and backward primer 3’AAGTGAATCTGAGGCATAAC’) (Cybergene AB) and 2 µl sample DNA (template). The other hand 25 µl micro-tube Ready To Go PCR Bead (Amersham Pharmacia Biotech) mixed with 2 µl c-myc primer (forward primer 5’CCGTCCCTGGCTCCCCTCCTG3’ and backward primer 3’GTATCGTCGCCCGCCCGTTGAAAC5’) (Cybergene AB) and 2 µl sample DNA (template). PCR protocol for both amplifications are 94˚C for 50 seconds, 59˚C for 50 seconds, 72˚C for 50 seconds and 4˚C soak. The Amplification of p53 gene produced 216 bp and cmyc gene produced 347 bp. Single Strand Conformational Polymorphism (SSCP) analysis followed with measurement using densitometer (Shimadzu CS 930, Dual Wave Length Cromato Scanner) to see mutation existence [11].
3. Results
We analyzed 55 samples of tissue, being 40 obtained from patients with lesions classified with BMSC and 15 with lesions classified as MSCC. Of this total, 20 were positive for amplification of a 450 bp segment L1 viral gene (Figure 1), revealing an overall prevalence rate of 36.4%. The results of this research show that nine of 40 samples obtained from patients with BMSC were positive for HPV, revealing a prevalence rate of 22.5% and eleven of 15 samples from patients with MSCC were positive for HPV, representing a prevalence rate of 73.3% (Table 1).
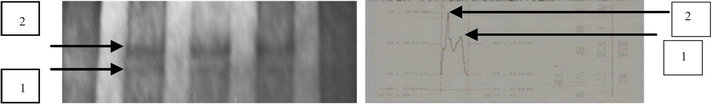
The HPV-positive samples were subjected to PCR amplification in separate reactions of a segment of 216 bp gene of the p53 and of a 347 bp of c-myc gene. The products of the PCRs were analyzed by Single Strand Conformational Polymorphism (SSCP) analysis of p53 gene (Figure 2(a)) and c-myc gene (Figure 2(c)) to detect the presence or absence of mutation and then by densintometry to confirm the existence of mutation in the p53 gene (Figure 2(b)) and c-myc gene (Figure 2(d)).
It was observed that no samples from BMSC patients with HPV positive either for p53 and c-myc genes mutation which when analyzed by SSCP and densintometry. Another samples group obtained from patients with MSCC observed that 2 of 11 samples (18.2%) were found mutation in the p53 gene and 1 of 11 samples (9.1%) were found mutations in the c-myc gene. Similar results were observed by reading the results of densitometry as confirmatory tests in Table 2.
4. Discussion
Based on shape analysis, oral health problems most prominent among low socio-economic communities is
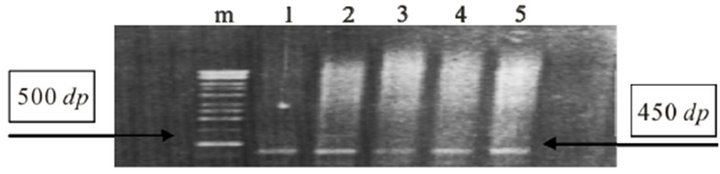
Figure 1. Polymerase Chains Reaction (PCR) L1-HPV gene, amplified 450 bp long (right arrow) in MSCC. Marker 500 bp long (left arrow).
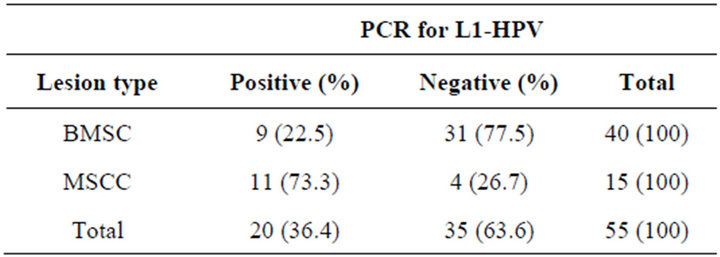
Table 1. Prevalence of HPV detected by PCR in samples from patients with BMSC and with MSCC.
 (a)
(a) (b)
(b)
Figure 2. Single Strand Conformational Polymorphism (SSCP) Analysis of p53 (a) and c-myc (c) genes. In the picture there were two bands (heterogenous) equal in all samples MSCC with HPV infection, which means there are no mutations in the gene. Whereas when you see the two bands are not equal then it is possible that a mutation in the genes. Measurement using densitometry to see mutation existence from Single Strand Conformational Polymorphism (SSCP) analysis of p53 (b) and c-myc (d) (have 2 pick-arrow). Two peaks mean that the gene is concentrated in the area. If it appears there was a significant shift in the peak, it is possible in these gene mutations.
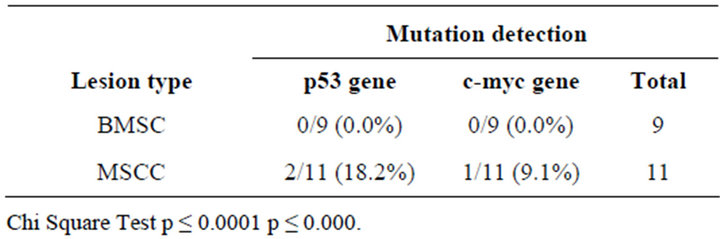
Table 2. Occurrence of mutation in the p53 gene and c-myc in a sample of patients with HPV infection.
e.g. the oral cancer. The prevalence of oral cancer is also increased. Incidence rates for oral and pharyngeal cancer is estimated at 25 cases per 100,000 in developing countries [12]. So the main risk factor is the abuse of tobacco, smokers represent 90% of oral cancer patients. Another risk in research is a viral infection such as HSV and HPV. [3,13-15].
The data about epidemiology of HPV positive in MSCC was lot variation. Many research founded that HPV was detected in 15.1% of tumors [16]. HPV was also detected in 64% of tonsil tumors, 52% oropharyngeal tumors, and 5% oral cavity tumors [17]. Other publication reported that epidemiology of associated HPV and MSCC were from the overall samples was 34.5%. With regard to the detection method, PCR-based studies reported a higher prevalence rate than ISH-based rates (34.8% versus 32.9%), especially in the MSCC subgroup that MSCC PCR based account 39.9%) [18].
Protein 53 is a gene that codes for a protein that regulates the cell cycle and hence functions as tumor suppression. Protein 53 has been described as the guardian of the genome, referring to its role in conserving stability by preventing genome mutation. The name is due to its molecular mass, it is in the 53 kilodalton fraction of cell proteins. It was plays an important role in cell cycle control and apoptosis. Defective p53 could allow abnormal cells to proliferate, resulting in cancer. As many, 50% of all human tumors contain p53 mutants. [19] And c-myc codes for a protein that binds to the DNA of other genes and is therefore a transcription factor. The healthy version of the gene that it is derived from is called a protooncogene. myc is believed to regulate expression of 15% of all genes [20]. myc is also regulate global chromatin structure by regulating histone acetylation both in generich regions and at sites far from any known gene [21]. Mutation of p53 was found in 7.8% in cervical and 52.5% in vulvae tumors [22]. In HPV-positive Squamous Cell Carcinoma Head and Neck, p53 interacts with the E6 protein, which leads to increased ubiquitin-dependent proteolysis of p53. The Mutation of Rb gene was rare in MSCC. The protein of Rb is a key regulator of cell cycle progression enabling it to form a complex with the transcription factor E2F, thereby inhibiting E2F-mediated transcription of the genes that regulate DNA synthesis. Upon mitogen stimulation, Rb becomes phosphorylated and the Rb-E2F complex dissociates, freeing E2F to activate transcription of c-myc and cell proliferation is going on [8].
Failure of apoptosis regulation is the key principle for the success of carcinogenesis, i.e. inhibition of apoptosis events [23,24]. Increased cell proliferation is also a key to the success of carcinogenesis [25].
5. Conclusion
HPV is a risk factor for OSCC, and not always that the incidence of cancer is caused by mutations in genes, eg protein-protein misfolding events that resulted in the passage of the triggers of apoptosis and inhibition of proliferation of cancer cells.
6. Acknowledgements
We thank to acknowledge Inter University Center (IUC) for Biotechnology of Gadjah Mada University Yogyakarta, Airlangga University Surabaya and Muwardi Distric Hospital Surakarta for his laboratory facilities. And Prof. José Veríssimo Fernandes from Federal University of Rio Grande do Norte, Natal—Brazil for his suggestion. We acknowledge to with J. B. Dalono, Retno Puji Rahayu, Ni Nyoman Tri Puspaningsih, Kuntoro, J. M. Harjanto.
REFERENCES
- R. Doll and R. Peto, “The Causes of Cancer: Quantitative Estimates of Avoidable Risks of Cancer in the United States Today,” Journal of the National Cancer Institute, Vol. 66, No. 6, 1981, pp. 1191-308.
- K. W. Kinzler and B. Vogelstein, “Introduction. The Genetic Basis of Human Cancer,” McGraw-Hill, Medical Publishing Division, New York, 2002, p. 5.
- National Cancer Institute, US National Institute of Health, “Evidence of Benefit,” 2009. http://www.cancer.gov/cancertopics/pdq/prevention/oral/HealthProfessional/page4
- R. I. Morimoto, “Regulation of the Heat Shock Transcriptional Response: Cross Talk between a Family of Heat Shock Factors, Molecular Chaperones, and Negative Regulators,” Genes & Development, Vol. 12, No. 24, 1998, pp. 3788-3796. doi:10.1101/gad.12.24.3788
- C. R. Young and C. J. Welsh, “Stress, Health, and Disease,” Cell Science, Vol. 2, No. 2, 2005, pp. 132-159.
- E. H. Vogel, M. E. Castro, P. A. Solar and F. A. Soto, “Enhancement of Pavlovian Conditioned Immunosuppression in Rats,” Acta Neurobiologiae Experimentalis, Vol. 67, No. 1, 2007, pp. 71-81.
- M. A. González Intxaurraga, R. Stankovic, R. Sorli and G. Trevisan, “HPV and Carcinogenesis,” Acta Dermatovenerologica, Vol. 11, No. 3, 2002, pp. 1-8.
- S, Choi and J. N. Myers, “Molecular Pathogenesis of Oral Squamous Cell Carcinoma: Implications for Therapy,” Journal of Dental Research, Vol. 87, No. 1, 2008, pp. 14- 32. doi:10.1177/154405910808700104
- H. L. Schmits, “Species Diagnostics Protocols: PCR and Other Nucleic Acid Methods,” In: J. P. Clapp, Ed., Methods in Molecular Biology, Humana Press, Totowa, 1996, 416 pp.
- M. M. Nigel and F. Nina, “Typing of HPV Isolates by PCR: Molecular Approach toward Vaccine Development for Viral Infection,” Icro-Unesco Training Course, Inter University Center for Biotecnology, Gajah Mada University, Yogyakarta, 1998, pp. 18-28.
- T. Hessel, S. P. Dhital, R. Plank and D. Dean, “Immune Response to Chlamydial 60-Kilodalton Heat Shock Protein in Tears from Nepali Trachoma Patients,” Infection and Immunity, Vol. 69, No. 8, 2001, pp. 4996-5000. doi:10.1128/IAI.69.8.4996-5000.2001
- P. E. Petersen, “Oral Cancer Prevention and Control— The Approach of the World Health Organization,” 2008. www.elsevier.com/locate/oraloncology
- N. W. Johnson, S. Warnakulasuriya, E. Gupta, P. C. Dimba, M. Chindia, E. C. Otoh, R. Sankaranarayanan, J. Califano and L. Kowalski, “Global Oral Health Inequalities in Incidence and Outcomes for Oral Cancer: Causes and Solutions,” Advances in Dental Research, Vol. 23, No. 2, 2011, pp. 237-246. doi:10.1177/0022034511402082
- Cancer Research UK, “Oral Cancer—Risk Factors,” 2009. http://info.cancerresearchuk.org/cancerstats/types/oral/riskfactors/index.htm
- R. Mehrotra and S. Yadav, “Oral Squamous Cell Carcinoma: Etiology, Pathogenesis and Prognostic Value of Genomic Alterations,” Indian Journal of Cancer, Vol. 43, No. 2, 2006, pp. 60-66. doi:10.4103/0019-509X.25886
- S. R. Schwartz, B. Yueh, J. K. Mc Dougall, J. R. Daling and S. M. Sswartz, “Human Papillomavirus Infection and Survival in Oral Squamous Cell Cancer: A PopulationBased Study,” Otolaryngology—Head and Neck Surgery, Vol. 125, No. 1, 2001, pp. 1-9. doi:10.1067/mhn.2001.116979
- R. Elin, P. Edward, H. Masayuki, P. Marshall, L. Mei and T. K. Karl, “Human Papillomavirus Type 16 and Squamous Cell Carcinoma of the Head and Neck,” Clinical Cancer Research, Vol. 8, No. 10, 2002, pp. 3187-3192.
- N. Termine, V. Panzarella, S. falaschini, A. Russo, D. Matranga, L. Lo Muzio and G. Campisi, “HPV in Oral Squamous Cell Carcinoma vs Head And Neck Squamous Cell Carcinoma Biopsies: A Meta-Analysis (1988-2007),” Oxford Journals Medicine Annals of Oncology, Vol. 19, No. 10, 2008, pp. 1681-1690. doi:10.1093/annonc/mdn372
- A. O. Robert, G. N. Nikolaos and J. S. John, “Screening the Non-High Risk Oral Cancer Patient—Part 1,” Oral Cancer Foundation, University of Maryland, Baltimore Dental School, Baltimore, OCF Inc. Copyright 2001-2009. http://oralcancerfoundation.org/dental/why_screening_works.htm
- A. P. Read and T. Strachan, “Chapter 18: Cancer Genetics,” In: T. Strachan and A. P. Read, Eds., Human Molecular Genetics, Wiley, New York, 1999.
- R. Cotterman, V. X. Jin, S. R. Krig, J. M. Lemen, A. Wey, P. J. Farnham and P. S. Knoepfler, “N-myc Regulates a Widespread Euchromatic Program in the Human Genome Partially Independent of Its Role as a Classical Transcription Factor,” Cancer Research, Vol. 68, No. 23, pp. 2008, 9654-9662.
- K. Milde-Langosch, K. Albrecht, S. Joram, H. Schlechte, M. Giessing and T. Löning, “Presence and Persistence of HPV Infection and p53 Mutation in Cancer of the Cervix Uteri and the Vulva,” International Journal of Cancer, Vol. 63, No. 5, 1995, pp. 639-645. doi:10.1002/ijc.2910630507
- M. P. Mayer and B. Bukau, “Hsp70 Chaperones: Cellular Functions and Molecular Mechanism,” Cellular and Molecular Life Sciences, 2005, Vol. 62, No. 6, 2005, pp. 670-684. doi:10.1016/S0022-2143(97)90130-X
- R. S. Fife, B. T. Rougraff, C. Proctor and G. W. Sledge Jr., “Inhibition of Proliferation and Induction of Apoptosis by Doxycycline in Cultured Human Osteosarcoma Cells,” Journal of Laboratory and Clinical Medicine, Vol. 130, No. 5, 1997, pp. 530-534.
- P. F. Andrew, O. Rolf and H. Steven, “The Epigenetic Progenitor Origin of Human Cancer,” Nature Reviews Genetics, Vol. 7, No. 1, 2006, pp. 21-33.