Advances in Bioscience and Biotechnology
Vol.2 No.6(2011), Article ID:16498,7 pages DOI:10.4236/abb.2011.26058
High level of soluble expression and purification of catalytically active native UDP-galactose 4-epimerase of Aeromonas hydrophila in E. coli
![]()
Gene Regulation Laboratory, School of Biotechnology, Jawaharlal Nehru University, New Delhi, India.
Email: *adixit7@gmail.com, adix2100@mail.jnu.ac.in
Received 12 September 2011; revised 22 October 2011; accepted 5 November 2011.
Keywords: Tag-Less GalE; Aeromonas hydrophila; Recombinant Expression; Kcat
ABSTRACT
The ubiquitous Aeromonas hydrophila is responsible for several pathological conditions in fish and human. Like most gram negative bacteria, its virulence relies on outer membrane lipopolysachharide (LPS). The Leloir pathway enzyme UDP-galactose 4-epimerase (GalE), plays an important role in the LPS biosynthesis, and therefore is a potential drug target. We have earlier carried out extensive biochemical and biophysical studies with histidine-tagged recombinant GalE. However, for effective drug design it is desirable to understand the structure-function relation of a protein in its native form without any additional sequences or tags. In the present study, we report the high level expression, purification and characterization of recombinant GalE (rGalE) of Aeromonas hydrophila in its native form in E coli. The rGalE expressed as a soluble protein was purified to near homogeneity. From 1 L of shake flask culture ~15 mg of purified rGalE was obtained. The purified protein was biologically active with Km and Kcat values of 0.7 mM and 28.8 s–1, respectively. The enzyme exhibited a temperature and pH optima of 37˚C and 7 - 9, respectively. Thus, the present study employed for soluble expression and purification of functionally active rGalE without any tag bypasses the need for cumbersome strategies associated with removal of tag from purified protein.
1. INTRODUCTION
Aeromonas hydrophila are ubiquitous, facultative anaerobe, gram-negative bacteria found in fresh, brackish, marine, chlorinated and non-chlorinated water supplies worldwide [1-3]. It is one of the major disease causing pathogens responsible for a variety of fish pathological conditions [4,5]. In recent years, a sharp increase in the acute diarrohoeal incidences in human by A. hydrophila has generated a great interest in Aeromonas sp. Lipopolysaccharide (LPS), the major integral component of the outer membrane possesses endotoxic properties [6,7] and is one of the important virulent factors of gramnegative bacteria including A. hydrophila. UDP-galactose 4-epimerase (GalE) is an essential enzyme of the Leloir pathway of galactose metabolism and catalyzes the interconversion of UDP-galactose and UDP-glucose [8-11]. UDP-galactose thus formed serves as galactose donor for the biosynthesis of galactosyl residues in glycoproteins and complex polysaccharides of the LPS [12,13]. The GalE mutants of a number of pathogenic bacteria have been reported to be avirulent [14-16].
Due to its involvement in LPS biosynthesis GalE has potential to be used as drug target, for which high level production of the enzyme in its native form is necessary. Use of affinity tags is employed for ease of protein purification, as it is reported to improve protein yield [17], prevent proteolysis [18] and enhance protein solubility [19]. However, there are several reports pointing out that the affinity tags can affect the properties of recombinant proteins by altering the protein conformation, undesired flexibility in structure studies [20], crystallization difficulties [21] and inhibition of enzyme activity [22,23]. There are limitations of histidine tag like; proteins are retained on Ni2+-NTA columns according to number of accessible histidines, the Ni2+-NTA resin can also act as a weak ionic exchange column and some recombinant protein does not interact in its native form with Ni2+- NTA resin [24]. For these reasons, it is desirable to remove affinity tags after purification prior to structure function studies. Although there are several methods to remove affinity tag after protein purification, these have their own limitations in terms of partial removal of tag, effect of cleavage on protein solubility and impairing the target protein by side reactions [25]. Also for an enzyme to be used as a drug target, it is important to have the knowledge of its crystal structure in its native form. In order to overcome these problems, it is essential to express the protein in its native form without any tag.
In the present report, we describe the over-expression and purification strategy for preparation of large quantities of soluble GalE of A. hydrophila without any tag and its biochemical and functional characteristics. The rGalE got expressed in soluble form with late log phase induction and was purified to near homogeneity by Diethylaminoethane(DEAE)-sepharose anion exchange chromatography followed by gel filtration.
2. MATERIALS AND METHODS
2.1. Materials
E. coli BL21 (DE3) (Novagen, USA) were used for expression studies. All the chemicals required for DNA manipulation (restriction enzymes and other chemicals) were from New England Biolabs (Beverly, MA, USA) and Promega Corporation (Madison, WI, USA). QIA quick spin columns, plasmid purification kit and Ni2+- NTA agarose for protein purification were purchased from Qiagen (Germany). DEAE-sepharose anion exchange resin and Pre-packed Sephacryl S-200HR HiPrep™ column (HR 16/60) were from GE Healthcare Bio Sciences AB, Sweden. Oligonucleotides were synthesized by Microsynth (Balgach, Switzerland). All other chemicals were of analytical grade and were purchased from Sigma Chemicals, USA, unless otherwise stated.
2.2. Cloning and Expression of rGalE
For producing rGalE without any tag, the galE gene cloned in pRSET-A was used [26,27] and the recombinant was designated as pRSETA-galE.
For expression analysis of rGalE without tag, E. coli BL21 (DE3) cells harboring pRSETA-galE were induced with 0.4 mM IPTG, optimized earlier for the expression of histidine-tagged rGalE (6xhis-rGalE), at different stages of growth curve at different temperatures and the culture was further grown for 4 h and/or 12 h post-induction. The cell lysates were subjected to 12% SDS-PAGE. The localization analysis of expressed rGalE in the induced cells was performed essentially as described previously [28].
2.3. Purification of Recombinant GalE
All purification procedures were performed at 4˚C unless otherwise stated. For purification of rGalE, soluble fraction of the induced culture was loaded onto a DEAESepharose column pre-equilibrated with 10 mM TrisHCl, pH 8. The column was then washed with 6 column volumes of 10 mM Tris-HCl pH 8 to remove unbound proteins. The bound recombinant rGalE was eluted with 0 - 200 mM NaCl linear gradient. Different fractions were analyzed on SDS-PAGE. The rGalE enriched fractions were concentrated by Amicon Ultra centrifugal filter device cutoff 10 kDa (Millipore, USA). The concentrated product was then purified by gel filtration chromatography using Sephacryl S-200HR HiPrep™ column (HR 16/60) pre equilibrated with 10 mM Tris-HCl, pH 8. The fractions were collected at 0.5 ml/min flow rate. Fractions eluting at 95 mM NaCl concentration of the gradient contained maximum amount of the rGalE. The purified recombinant rGalE stored at –20˚C till further use.
2.4. Determination of Protein Concentration and Enzyme Activity
The protein concentration was determined by the method of Lowry et al., [29] using bovine serum albumin (BSA) as the standard. The activity of rGalE was determined spectrophotometrically essentially as described earlier [27]. Appropriate dilutions of the purified enzyme were made in 10 mM Tris-HCl, pH 8.0 prior to activity determination, in order to obtain a linear initial velocity. For catalytic reaction the purified protein or cell lysate was added to 10 mM Tris-HCl, pH 8.0 and the reaction was initiated by 2 mM UDP-galactose in a 100 µl final reaction volume. The produced glucose was estimated using glucose estimation kit (Merckotest ®, Merck Ltd, India) based on GOD-POD assay as per the manufacturer’s instructions using spectrophotometer Lambda25 (PerkinElmer, USA). One unit of rGalE is defined as the amount of enzyme that catalyzes the conversion of 1 μmol of UDP-galactose to UDP-glucose per minute under the above assay conditions.
2.5. Biochemical Characterization of the A. Hydrophila rGalE
The optimum temperature for the rGalE without tag was determined by assaying the enzyme activity at different temperatures (4˚C - 55˚C) in 10 mM Tris-HCl buffer, pH 8. For optimum pH, rGalE activity was measured in 10 mM buffer of varying pH range from 4 - 11 (sodium acetate pH 4 and 5, sodium phosphate pH 6 and 7, Tris-HCl pH 8 and 9 and sodium carbonate buffer, pH 10 and 11).
Kinetic properties of the purified rGalE were determined as reported earlier [27]. The Michaelis-Menten constant was determined for the reaction using different concentrations of the UDP-galactose (0.16 mM - 2.0 mM) at 37˚C in 10 mM Tris-HCl (pH 8). The Km, Vmax and kcat (Vmax/molar concentration of the enzyme) were determined using Lineweaver-Burk plot. For kcat calculation, the molecular weight of the enzyme was taken as 37 kDa.
3. RESULTS
3.1. Analysis of Expression of rGalE
Initial attempts to express rGalE (without any tag) were made at 37˚C by inducing the cells at A600 of 0.6, an equal expression of the rGalE was seen both in soluble and insoluble fractions (Figure 1(a)). Induction at late log phase also did not result in any shift of expression from insoluble to soluble (Figure 1(b)). Also, the induction of culture at late log phase at 25˚C and with 4 h induction period also did not affect the expression (Figure 1(c)). However, when the rGalE expression was carried out at 25˚C by inducing the cells at late log phase and the induction period was increased to 12 h, a greater proportion of the rGalE could be seen as a soluble protein (Figure 1(d)). Thus, we could achieve soluble expression of rGalE. Therefore, for purification purposes, the cells were induced at late log phase of the cells grown at 25˚C with an induction period of 12 h.
3.2. Purification of rGalE
The rGalE was purified by DEAE-sepharose anion exchange chromatography from soluble fraction, which resulted in the removal of most of the contaminants of lower molecular weight (Figure 2(a), lanes 6 and 7). The other contaminants were further removed by gel filtration chromatography and the rGalE was found to be free of contaminating proteins (Figure 2(b)). Increase in the specific activity of the rGalE was obtained with purification (Table 1). The purified protein thus obtained was used for further characterization.
3.3. Catalytic Properties of the rGalE
Activity of the purified rGalE showed a linear increase with increasing protein concentrations as was observed earlier with 6xhis-rGalE [27]. Activity detected at different pH revealed that the rGalE was active in a wide pH range and exhibited a bell shaped curve over pH 5 to 11 (Figure 3(a)) with an optimal activity between pH 7 - 9. Temperature optima studies by assaying the enzyme activity at different temperatures (4˚C - 55˚C) showed that the enzyme retained activity within the range of 16˚C to 45˚C and the temperature optimum for the rGalE was determined to be 37˚C (Figure 3(b)). The rGalE exhibited significantly lower activity when assayed at temperatures below 16˚C or above 45˚C. Similar to 6xhisrGalE, the rGalE retained its full activity up to 45˚C, and was completely inactivated when incubated at 55˚C (Figure 3(b)). Thus, the temperature optimum of rGalE was similar to that of the 6xhis-rGalE [27].
The kinetics of the rGalE catalyzed reaction was consistent with the Michaelis–Menten equation and the Km and Vmax was calculated using Lineweaver–Burk plot (Figure 4). The Km of the rGalE was calculated to be 0.7 mM for UDP-Galactose with a Vmax and Kcat of 4.5 nmol/min and 28.8 s–1, respectively. The recombinant protein was quite active as 917 units of native rGalE with specific activity 66.5 U/mg was purified from 1 liter of culture at shake flask level.
4. DISCUSSION
The native form of an enzyme isolated from natural source is ideal to study its properties. However, most enzymes are present in low abundance at their natural source; therefore purifying large quantities for characterization would not be feasible. We have previously utilized recombinant DNA technology to clone and overexpress the UDP-galactose 4-epimerase (GalE) with 6x-
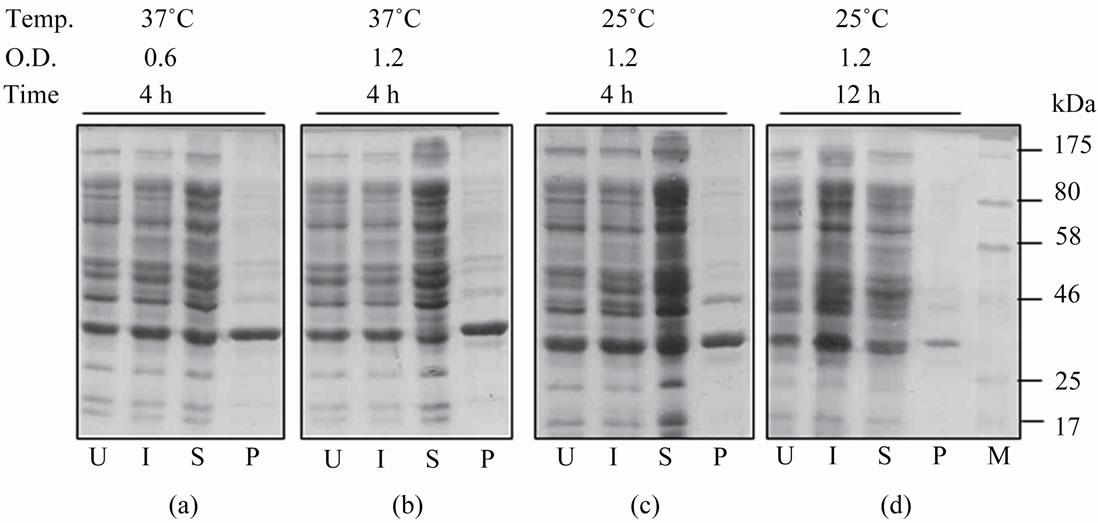
Figure 1. Expression and localization of the rGalE from pRSETA. galE in E. coli BL21 (DE3) cells. ((a)-(d)) The equal proportion of proteins of cells induced with 0.4 mM IPTG at different temperature and O.D after different time of post induction (mentioned at the top) were analyzed on SDS PAGE. U and I indicate whole cell lysates of uninduced and induced cells, respectively. S and P show soluble and pellet (insoluble) fractions of induced cells, respectively. M indicates Protein molecular weight marker.
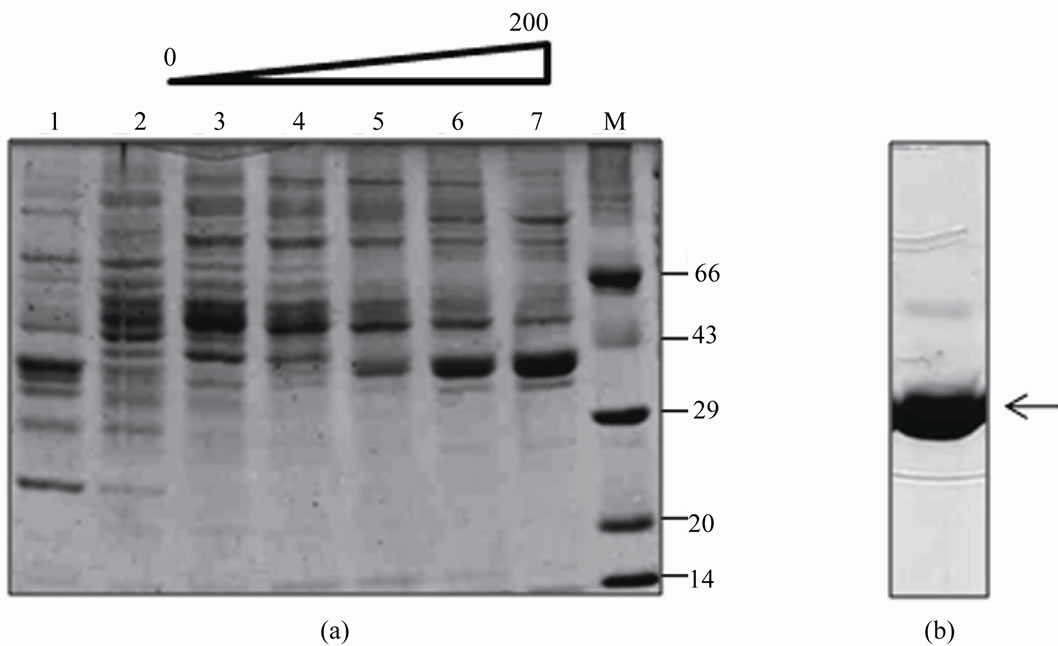
Figure 2. Purification of rGalE from E. coli BL21(DE3) cells harboring pRSETA-galE. The purification of rGalE from soluble fraction of induced cells were carried out by DEAE-sepharose anion exchange chromatography (a) followed by gel filtration chromatography (b). The soluble fraction was loaded onto a DEAE-sepharose column pre-equilibrated with 10 mM Tris-HCl pH 8. After extensive washes, the bound proteins were eluted with 0 - 200 mM NaCl gradient. The different fractions collected were analyzed on SDS-PAGE. Lanes 1 and 2 show the flow through fractions. Lanes 3 - 7 show the eluted fractions with 0 - 200 mM NaCl gradient (a). The fractions 6 and 7 were pooled, concentrated and loaded onto sephacryl S-200HR HiPrep™ gel filtration column and biochemically active peak fraction was analyzed on SDS-PAGE (b).
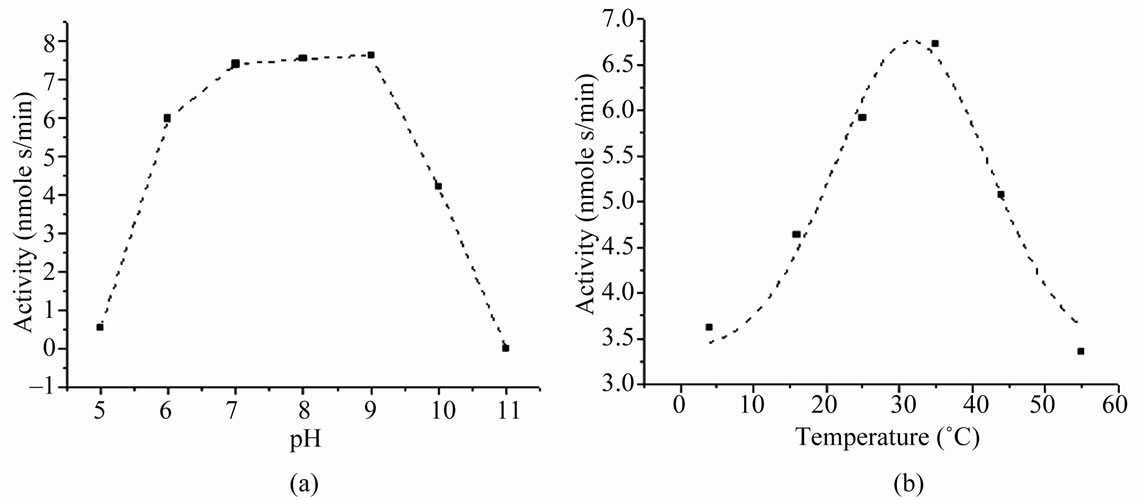
Figure 3. Effect of pH and temperature on the activity of rGalE. (a) Determination of optimum pH for the biochemical activity of rGalE. The activity of rGalE was assayed at different pH range of 5 - 11 in buffers using 100 ng of purified protein in the reaction mix at 37˚C. The bell shaped curve shows the optimum activity in the range of pH 7 - 9. (b) Determination of optimum temperature for the activity of rGalE. The activity of rGalE was assayed at different temperature conditions varying from 4˚C to 55˚C using 100 ng of purified protein in 10 mM Tris-HCl buffer, pH 8. The bell shaped activity curve shows the optimum activity at 37˚C.
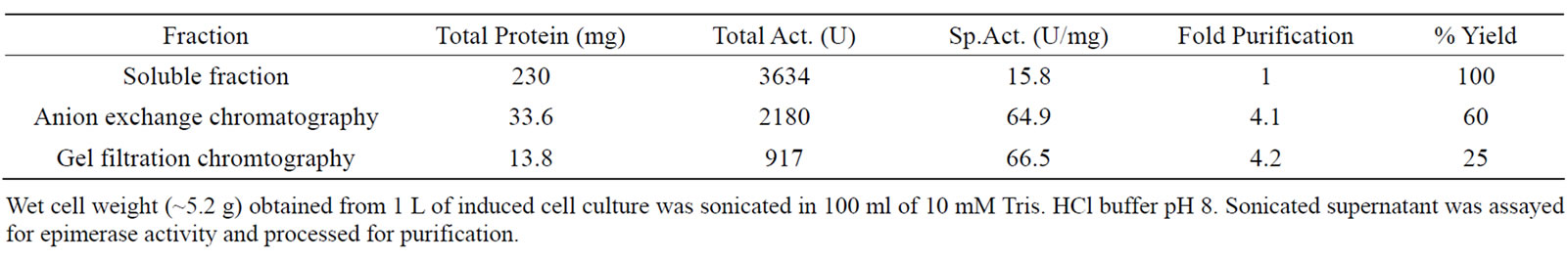
Table 1. Purification of rGalE of A. hydrophila from E. coli BL21(DE3).
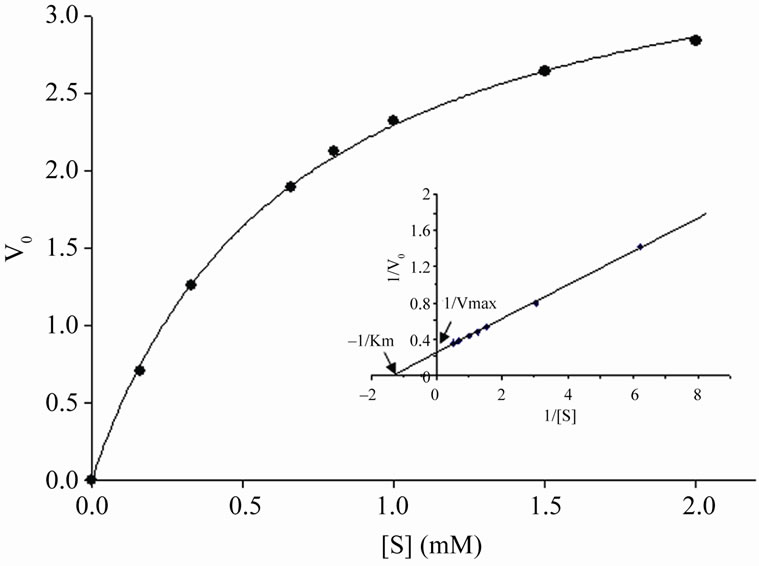
Figure 4. Enzyme kinetic analysis of rGalE. Determination of Km and Vmax of rGalE was accessed using UDP-galactose as a substrate. The enzyme activity was measured at different substrate concentrations (0.2 mM - 2.0 mM) under standard reaction conditions for 5 min. Michaelis-Menton plot (V0 verses [S]) and Lineweaver-Burk plot (inset) of the rGalE is shown. V0 indicates the initial velocity of the reaction and [S] is substrate concentration.
histidine tag and extensively characterized the biochemical and biophysical properties [27]. However, expression of the above in the selected vector (pET28a) with histidine tag resulted in addition of total 20 amino acid residues (including the 6xhistidine tag), contributed by the vector. Addition of such large stretch of amino acids are known to affect the structure and function of proteins [20-23]. Although, we have studied GalE extensively with histidine tag, there is always a need to have protein without any affinity tag for structural and drug target studies [30]. The removal of affinity tags from the protein necessitating the use of expensive proteolytic enzymes with additional purification steps. There are intricacies in affinity tag removal by cleavage ranging from non-specific cleavages generating truncated form of the protein, addition of extra amino acids and partial removal of the tags [31]. Therefore, it is always better to overexpress protein without any affinity tag in soluble form.
To compare the biochemical properties of GalE with histidine tag (6xhis-rGalE) and without any affinity tag i.e. in its native form (rGalE), the GalE gene was cloned in pET28a and pRSET-A expression vector, respectively. Upon induction with IPTG, the 6xhis-rGalE expressed only in induced whole cell lysate of E. coli BL21, showing stringent control over the expression of protein [27]. In the present study, the expression of rGalE was also visible in uninduced whole cell lysates, indicating leaky expression of recombinant protein. To date, there is no report of toxicity caused by GalE. Therefore, being native to the host cell, leaky expression would not hamper the cellular metabolism and finally can result production of recombinant protein. The induction of protein expression at lower IPTG (0.4 mM) concentration than normally used (1 mM) for other recombinant proteins would be cost effective for production of large amount of recombinant proteins. The induction of culture at 37˚C resulted in expression of rGalE both in the soluble and insoluble fractions, at almost equal level. Strategies to increase expression of soluble proteins include lowering the temperature and IPTG concentration, leading to decrease in cellular metabolism, cell numbers, therefore expression of recombinant proteins, which can be compensated by inducing the cells at late log phase [32]. Thus, the expression of rGalE was tried at lower temperature with induction at late log phase. The slowdown of metabolism and protein production at low temperature was compensated by increase in time of post induction of culture which resulted in high level expression of soluble rGalE (with induction at late log phase for 12 h at 25˚C).
The biochemical properties of rGalE were compared with 6xhis-rGalE to analyze the functional integrity of recombinant protein. Although, the specific activity of purified rGalE was calculated to be 66.5 U/mg, which is slightly lower than that of 6xhis-rGalE (128.5 U/mg). However, it was within the range of 0.012 - 210 U/mg, reported for UDP-galactose 4-epimerase from a variety of sources [33], which can be accounted for the number of steps involved in purifications. The rGalE exhibited slightly broader pH optima when compared to that reported earlier for 6xhis-rGalE [27]. The broader pH optima for rGalE are in line with the observed pH optima for native GalE reported from other organisms [34,35]. Secondary structure prediction using Network Protein Sequence Analysis tool [36] did not show any significant difference in the secondary structures of the tagged or un-tagged rGalE. However, the enzyme kinetics of rGalE was diminutively weak when compared to that of 6xhisrGalE, with slightly higher Km and lower Vmax. However, the higher Kcat of the rGalE (28.8 s–1) in comparison to that of 6xhis-rGalE (12.9 s–1) shows higher catalytic activity and thus can in part, compensate for lower yields (~15 mg/L) when compared with that of 6xhis-rGalE (90 mg/L) [27].
Thus, the present study describes the optimized protocol for purification of highly pure, enzymatically active rGalE of Aeromonas hydrophila in large quantity for further structural, functional and drug target studies.
5. ACKNOWLEDGEMENTS
This work is supported by a research grant from the Indian Council of Agricultural Research, New Delhi, India to AD. The University Grant Commission, and the Council of Scientific and Industrial Research, New Delhi are acknowledged for providing research fellowships to KG and SA, respectively.
REFERENCES
- Janda, J.M. (1991) Recent advances in the study of taxonomy, pathogenicity, and infectious syndromes associated with the genus Aeromonas. Clinical Microbiology Reviews, 4, 397-410.
- Kaper, J.B., Lockman, H., Colwell, R.R. and Joseph, S.W. (1980) Aeromonas hydrophila: Ecology and toxigenicity of isolates form an estuary. Journal of Applied Microbiology, 50, 359-377. doi:10.1111/j.1365-2672.1981.tb00900.x
- Van der Kooj, D. (1988) Properties of Aeromonads and their occurrence and hygienic significance in drinking water. Zentralblatt Bakteriology Hygien B, 187, 1-17.
- Hazen, T.C., Flierman, C.B., Hirsch, R.P. and Esch, G.W. (1978) Prevalence and distribution of Aeromonas hydrophila in the United States. Applied and Environmental Microbiology, 36, 731-738.
- Janda, J.M. and Duffy, P.S. (1998) Mesophilic Aeromonads in human disease: Current taxonomy, laboratory identification, and infectious disease spectrum. Reviews of Infectious Diseases, 10, 980-997. doi:10.1093/clinids/10.5.980
- Pierson, D.E. and Carlson, S. (1996) Identification of the gale gene and a gale homolog and characterization of their roles in the biosynthesis of lipopolysaccharide in a serotype O: 8 strain of Yersinia enterocolitica. Journal of Bacteriology, 178, 5916-5924.
- Fry, B.N., Feng, S., Chen, Y.Y., Newell, D.G., Coloe, P.J. and Korolik V. (2000) The GalE gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and virulence. Analytical Biochemistry, 283, 64-70.
- Maxwell, E.S. (1957) The enzymic interconversion of uridine diphosphogalactose and uridine diphosphoglucose. Journal of Biological Chemistry, 229, 139-151.
- Vorgias, C.E., Lemaire, H.G. and Wilson, K.S. (1991) Over expression and purification of the galactose Operon enzymes from Escherichia coli. Protein Expression and Purifaction, 2, 330-338. doi:10.1016/1046-5928(91)90091-V
- Wilson, B.D. and Hogness, D.S. (1966) Galactokinase and Uridine diphosphogalactose 4-epimerase from Escherichia coli. Methods in Enzymology, VIII, Academic Press, New York, 229-240.
- Wilson, B.D. and Hogness, D.S. (1964) The enzymes of the galactose Operon in Escherichia coli I. Purification and characterization of uridine diphosphogalactose 4- epimerase. Journal of Biological Chemistry, 239, 2469- 2481.
- Houng, H.H., Kopecko, D.J. and Baron, L.S. (1990) Molecular cloning and physical and functional characterization of the Salmonella typhimurium and Salmonella typhi galactose utilization operons. Journal of Bacteriology, 172, 4392-4398.
- Potter, M.D. and Lo, R.Y. (1996) Cloning and characterization of the galE locus of Pasteurella haemolytica A1. Infection and Immunity, 64, 855-860.
- Stevenson, G. and Manning, P.A. (1985) Galactose epimeraseless (GalE) mutant G30 of Salmonella typhimurium is a good potential live oral vaccine carrier for fimbrial antigens. FEMS Microbiology Letters, 28, 317-321. doi:10.1111/j.1574-6968.1985.tb00813.x
- Fernández de Henestrosa, A.R., Badiola, I., Saco, M., Perez de Rozas, A.M., Campoy, S. and Barbé, J. (1997) Importance of the galE gene on the virulence of Pasteurella multocida. FEMS Microbiology Letters, 154, 311- 316. doi:10.1016/S0378-1097(97)00347-9
- Petrovska, L., Hewinson, R.G., Dougan, G., Maskell, D.J. and Woodward, M.J. (1999) Brucella melitensis 16M: Characterisation of the galE gene and mouse immunisation studies with a galE deficient mutant. Veterinary Microbiology, 65, 21-36. doi:10.1016/S0378-1135(98)00281-8
- Sun, Q.M., Chen, L.L., Cao, L., Fang, L., Chen, C. and Hua, Z.C. (2005) An improved strategy for high-level production of human vasostatin120-180. Biotechnology Progress, 21, 1048-1052. doi:10.1021/bp049583x
- Tang, W., Sun, Z.Y., Pannell, R., Gurewich, V. and Liu, J.N. (1997) An efficient system for production of recombinant urokinase-type plasminogen activator. Protein Expression and Purification, 11, 279-283. doi:10.1006/prep.1997.0800
- Busso, D., Kim, R. and Kim, S.H. (2003) Expression of soluble recombinant proteins in a cell-free system using a 96-well format. Journal of Biochemical and Biophysical Methods, 55, 233-240. doi:10.1016/S0165-022X(03)00049-6
- Amor-Mahjoub, M., Suppini, J.P., Gomez-Vrielyunck, N. and Ladjimi, M. (2006) The effect of the hexahistidinetag in the oligomerization of HSC70 constructs. Journal of Chromatography B, Analytical Technologies in the Biomedical and Life Sciences, 844, 328-334. doi:10.1016/j.jchromb.2006.07.031
- Bucher, M.H., Evdokimov, A.G. and Waugh, D.S. (2002) Differential effects of short affinity tags on the crystallization of Pyrococcus furiosus maltodextrin-binding protein. Acta Crystallographica D, 58, 392-397. doi:10.1107/S0907444901021187
- Cadel, S., Gouzy-Darmon, C., Petres, S., Piesse, C., Pham, V.L., Beinfeld, M.C., Cohen, P. and Foulon, T. (2004) Expression and purification of rat recombinant aminopeptidase B secreted from baculovirus-infected insect cells. Protein Expression and Purification, 36, 19-30. doi:10.1016/j.pep.2004.03.013
- Freydank, A.C., Brandt, W. and Dräger, B. (2008) Protein structure modeling indicates hexahistidine-tag interference with enzyme activity. Proteins, 72, 173-183. doi:10.1002/prot.21905
- Schmitt, J., Hess, H. and Stunnenberg, H.G. (1993) Affinity purification of histidine-tagged proteins. Molecular Biology Reports, 18, 223-230. doi:10.1007/BF01674434
- Jenny, R.J., Mann, K.G. and Lundblad, R.L. (2003) A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expression and Purification, 31, 1-11. doi:10.1016/S1046-5928(03)00168-2
- Agarwal, S., Gopal, K., Chhabra, G. and Dixit, A. (2009) Molecular cloning, sequence analysis and homology modeling of galE encoding UDP-galactose 4-epimerase of Aeromonas hydrophila. Bioinformation, 4, 216-222.
- Agarwal, S., Gopal, K., Upadhyaya, T. and Dixit, A. (2007) Biochemical and functional characterization of UDP-galactose 4-epimerase from Aeromonas hydrophila. Biochimica et Biophysica Acta, 1774, 828-837.
- Mathur, D. and Garg, L.C. (2007) Functional phosphoglucose isomerase from Mycobacterium tuberculosis H37 Rv: Rapid purification with high yield and purity. Protein Expression and Purification, 52, 373-378. doi:10.1016/j.pep.2006.10.006
- Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall, R.J. (1951) Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry, 193, 265-275.
- Smyth, D.R., Mrozkiewicz, M.K., McGrath, W.J., Listwan, P. and Kobe, B. (2003) Crystal structures of fusion proteins with large-affinity tags. Protein Science, 12, 1313-1322. doi:10.1110/ps.0243403
- Kenig, M., Peternel, S., Gaberc-Porekar, V. and Menart, V. (2006) Influence of the protein oligomericity on final yield after affinity tag removal in purification of recombinant proteins. Journal of Chromatography A, 1101, 293-306. doi:10.1016/j.chroma.2005.09.089
- Galloway, C.A., Sowden, M.P. and Smith, H.C. (2003) Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. BioTechniques, 34, 524-530.
- Roper, J.R. and Ferguson M.A.J. (2003) Cloning and characterization of the UDP-glucose 4-epimerase of Trypanosoma cruzi. Molecular and Biochemical Parasitology, 132, 47-53. doi:10.1016/j.molbiopara.2003.07.002
- Dormann, P. and Benning, C. (1996) Functional expression of uridine 5’-diphosphoglucose 4-epimerase (EC 5.1.3.2) from Arabidopsis thaliana in Saccharomyces cerevisiae and Escherichia coli. Archives of Biochemistry and Biophysics, 327, 27-34. doi:10.1006/abbi.1996.0088
- Berger, E., Arabshahi, A., Wei, Y., Schilling, J.F. and Frey, P.A. (2001) Acid-base catalysis by UDP-galactose 4-epimerase: Correlations of kinetically measured acid dissociation constants with thermodynamic values for tyrosine 149. Biochemistry, 40, 6699-6705. doi:10.1021/bi0104571
- Combet, C., Blanchet, C., Geourjon, C. and Deléage, G. (2000) NPS@: Network Protein Sequence Analysis. Trends in Biochemical Sciences, 25, 147-150. doi:10.1016/S0968-0004(99)01540-6