Journal of Biophysical Chemistry
Vol.1 No.3(2010), Article ID:3194,12 pages DOI:10.4236/jbpc.2010.13017
A spectroscopic study of the interaction ofantioxidant naringin with bovine serum albumin
Department of Chemistry Indian Institute of Technology, Kharagpur, India; *Corresponding Author: swagata@chem.iitkgp.ernet.in
Received 9 September 2010; revised 12 October 2010; accepted 19 October 2010.
Keywords: Naringin; Bovine Serum Albumin;Fluorescence; Binding; Warfarin; Docking
ABSTRACT
The interaction of naringin with bovine serum albumin has been performed using fluorescence, circular dichroism and fourier transform infrared spectroscopy in 20 mM phosphate buffer of pH 7.0 as well as molecular docking studies. The changes in enthalpy (ΔH°) and entropy (ΔS°) of the interaction were found to be +18.73 kJ/mol and +143.64 J mol-1 K-1 respectively, indicating that the interaction of naringin with bovine serum albumin occurred mainly through hydrophobic interactions. Negative values of free energy change (ΔG°) at different temperatures point toward the spontaneity of the interaction. Circular dichroism studies reveal that the helical content of bovine serum albumin decreased after interaction with naringin. According to the Förster non-radiative energy transfer theory the distance between Trp 213 residue and naringin was found to be 3.25 nm. Displacement studies suggest that naringin binds to site 1 (subdomain IIA) of bovine serum albumin (BSA) which was also substantiated by molecular docking studies.
1. INTRODUCTION
Plant sources are abundant in the common polyphenolic compounds known as flavonoids that are known to possess various types of pharmacological effects including antioxidant, anti-microbial, anti-inflammatory, anti-allergic and anti-cancer activity [1-7]. The importance of the antioxidant activity of these compounds lies in their ability to scavenge free radicals [8] that are responsible for DNA damage. Naringin (Figure 1), a major bioflavonoid in grapefruit has been shown to reduce radiation-induced damage to DNA [9]. Naringin also acts as the inhibitor of VEGF release, which causes angiogenesis [10] and has been proven to be effective against ethanol injury in rats [11]. The chemopreventive role of naringin against protoxicants, its action as anti-atherogenic agents in rabbits and its ability to significantly reduce the level of total cholesterol have also been established [12-14]. Apart from being able to provide protection against the autophagy-inhibitory effect of okadaic acid, it also possesses anti-apoptotic properties [15,16]. The oral bioavailability of quinine in rats has been shown to increase after pretreatment with naringin [17].
Serum albumins are the most abundant carrier proteins of blood plasma that promote the transportation and disposition of exogenous and endogenous materials in blood. They are able to bind with different biologically active compounds (drugs, fatty acids, steroids, dyes etc) in the body [18-20]. The structure of BSA is homologous to HSA (human serum albumin) and consists of three linearly arranged domains (I-III) that are composed of two subdomains (A and B) [21-23]. There are two tryptophans (Trp 134 and Trp 213) in BSA, of which Trp 134 is located on the surface of the molecule and Trp 213 resides in the hydrophobic pocket similar to Trp 214 in HSA [24,25]. Like other serum albumins bovine serum albumin (BSA) possesses a wide range of physiological functions associated with the binding, transport and distribution of biologically active compounds. The binding affinity to serum albumins also influences the effectiveness of the drugs at the active site. Thus the study of the interactions of small drug molecules with serum albumins provides a good insight into an understanding of the recognition pattern under physiological conditions [26]. In this regard, BSA is extensively used because of its intrinsic structural similarity with HSA [27-30].
Analyses of the interactions of small ligands with biomacromolecules include the use of optical spectroscopy as an important tool [31-35]. Studies of the interaction of antioxidant flavonoids with serum albumins are of pharmaceutical importance. The binding parameters derived from the spectroscopic methods
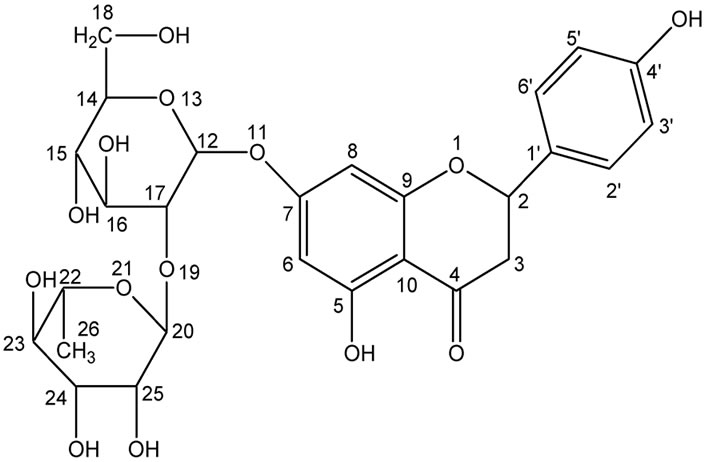
Figure 1. Structure of naringin.
play a major role in pharmacokinetics [36,37]. To this effect, the interactions of hesperidin and naringin with the serum albumins have been preliminary reported upon by fluorescence and absorption spectroscopy as this report was in preparation [38,39]. Our studies have further investigated the interactions between naringin and BSA by fluorescence, circular dichroism and Fourier Transform infrared spectroscopy. Molecular displacement studies to identify the site of binding have also been conducted along with synchronous fluorescence experiments. Thermodynamic parameters determined from measurements at four different temperatures, indicated that hydrophobic interactions play an important role in the association. These observations were further substantiated by the FTIR studies providing additional insight into the mode of binding. Finally, protein ligand docking studies have been conducted to identify the residues likely to be involved in the binding.
2. MATERIALS AND METHODS
2.1. Materials
BSA and the other analytical grade reagents were purchased from SRL (India). Naringin, warfarin, ibuprofen and 8-anilino-1-naphthalene sulfonic acid (ANS) were obtained from Sigma (USA). Stock solutions of naringin, warfarin and ibuprofen were prepared in doubled distilled water. BSA was dissolved in 20 mM phosphate buffer of pH 7.0. The concentrations were measured spectrophotometrically (Shimadzu UVPC 2450) using their molar extinction coefficient values: ε280 (BSA) = 43800 M-1 cm-1 [40], ε308 (warfarin) = 12500 M-1 cm-1 [41], ε280 (naringin) = 17400 M-1 cm-1 [42], ε222 (ibuprofen) = 12700 M-1 cm-1 [43]. All the experiments were carried out in 20 mM phosphate buffer of pH 7.0.
2.2. Fluorescence Spectroscopy
2.2.1. Fluorescence Quenching Studies
All the fluorometric experiments were carried out in a Horiba Jobin Yvon spectrofluorometer (Fluoromax 4) using a 1 cm quartz cell. A 3.0 mL solution of 2 μM BSA was titrated with successive addition of naringin (0 to 36 μM) at four different temperatures (293, 299, 305 and 311 K). The excitation wavelength was 295 nm and the emission spectra were recorded from 305 to 500 nm. The intensities of emission maxima were used for the calculation of binding constants, occupation of binding sites and thermodynamic parameters.
The Stern-Volmer equation can be used to describe the fluorescence quenching process [44].
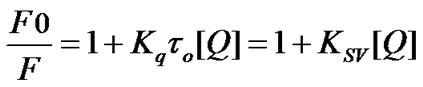 (1)
(1)
where τ0 (5 ns [45]) is the lifetime of the fluorophore in absence of quencher and kq is the bimolecular quenching constant. KSV is the Stern-Volmer quenching constant and [Q] is the quencher concentration. KSV were obtained by plotting F0/F versus [Q] and if KSV is much larger than 2×1010 M-1 s-1, it can be concluded that the quenching is static in nature. The modified Stern-Volmer equation was also taken to analyze the fluorescence quenching method using the equation given below:
 (2)
(2)
where ΔF = F0 – F; F0 and F are the relative fluorescence intensities in absence and presence of the quencher, respectively, fa the fraction of fluorophore accessible to the quencher, [Q] the concentration of the quencher and KA is the Modified Stern-Volmer quenching constant. The plot of F0 /ΔF versus 1/[Q] yields 1/fa as the intercept, and 1/KA fa as the slope. From the intercept and slope the values for fa and KA can be obtained.
The number of binding sites and the binding constant of the interaction of naringin with BSA can be obtained from the following equation [46].
 (3)
(3)
where ΔF = F0 – F, F0 and F are the fluorescence intensities of protein in presence and presence of quencher, n is the number of binding sites and Kb the equilibrium binding constant.
The mode of interaction of naringin with BSA was ascertained from the change in enthalpy (ΔH°) and entropy (ΔS°) [47]. This can be done by using the van’t Hoff equation Eq.4:
 (4)
(4)
where Kb is the equilibrium binding constant at the corresponding temperature and R the universal gas constant. The free energy change (ΔG°) associated with the interaction of naringin with BSA can be calculated from the values of ΔH° and ΔS° using the following equation:
 (5)
(5)
2.2.2. Synchronous Fluorescence Quenching of BSA by Naringin
Synchronous fluorescence spectra of BSA (2 μΜ) were recorded with increasing concentrations of naringin (0 to 14 μΜ), by setting Δλ = 60 nm and Δλ = 15 nm for tryptophan and tyrosine residues respectively [48].
2.2.3. 8-Anilino-1-Naphthalene Sulfonic acid(ANS) Displacement Study
2 μΜ BSA was saturated with 6 μΜ ANS [49] in 20 mM phosphate buffer of pH 7.0 and incubated for 1 hour. The mixture was then titrated successively with addition of naringin (21 μΜ). The concentration of ANS was determined spectrophotometrically using its molar absorption coefficient of 4950 M-1 cm-1 at 350 nm [50]. The excitation wavelength for ANS-bound BSA was set at 375 nm and emission observed at 474 nm.
2.2.4. Warfarin and Lbuprofen DisplacementStudies
For the displacement study of warfarin bound to BSA, a 3 mL solution was prepared containing 2 μM of warfarin and BSA and kept for 1 hour. Titrations were performed with increasing concentrations of naringin (0 to 40 μM) on excitation at 308 nm. Another set containing 1 μM ibuprofen and 2 μM BSA was prepared to check the displacement of ibuprofen bound BSA. After a 1 hour incubation period titration was done by successive addition of naringin (0 to 21 μM) using an excitation wavelength of 265 nm.
2.3. Fluorescence Resonance Energy Transfer
The rate of energy transfer depends on the extent of overlap between the emission spectrum of the donor (protein) and the absorption spectrum of the acceptor (ligand) and the relative orientations of the transition dipoles. In accordance to Förster’s theory, the efficiency of energy transfer, E, is estimated using the following Eq. (1) [51].
 (6)
(6)
where F0 and F are the fluorescence intensities of the BSA in the absence and presence of naringin. The distance from the ligand to the fluorophore of the protein is r and R0 is the Förster distance at which 50% of the excitation energy is transferred to the acceptor. The value of R0 can be obtained from Eq.2.
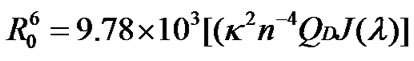 (7)
(7)
where QD is the quantum yield of the donor in absence of acceptor, n the refractive index of the medium, J(λ) is the overlap integral of the emission spectra of donor and absorption spectra of the acceptor and κ2 is the relative orientations in space of the transition dipoles of the donor and acceptor. The overlap integral J(λ) is calculated from the following equation:
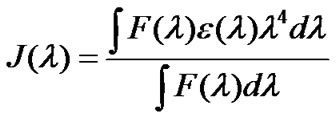 (8)
(8)
where F(λ) is the corrected fluorescence intensity of the donor in the range of λ to λ + Δλ with the total intensity normalized to one with the quantity F(λ) dimensionless, ε(λ) is the molar extinction coefficient of the acceptor at λ nm.
2.4. Circular Dichroism Measurements
All CD experiments were carried out on a Jasco-810 automatic spectrophotometer using a 0.1 cm cell length at room temperature. The concentration of BSA used for the CD study was 9 μM in 20 mM phosphate buffer of pH 7.0. Five sets of solutions were prepared containing BSA and naringin in the following proportions 1:0, 1:2, 1:4, 1:6 and 1:8 respectively. Spectra were recorded in the range of 190 to 240 nm with a scan rate of 50 nm/min and the response time was fixed at 4 s during the measurements. The mean residual ellipticity (MRE) in deg cm2 dmol-1 can be estimated using Eq.4 [52].
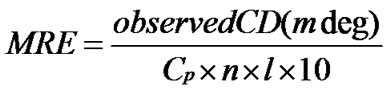 (9)
(9)
where n is the number of residues in the protein, Cp is the molar concentration of the protein and l is the path length. From the MRE values at 208 nm we can calculate the α-helical content of free and complexed BSA using the following Eq.5 [52,53].
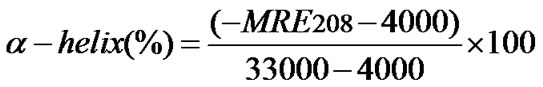 (10)
(10)
Where MRE208 is to the observed MRE values, 33000 and 4000 are the MRE values of a pure α-helix and of the β-form and random coil conformation at 208 nm respectively.
2.5. FT-IR Spectroscopic Measurements
FT-IR measurements were carried out on Nexus-870 FT-IR spectrometer equipped with a DTGS detector, a KBr beam splitter, Zn-Se attenuated total reflection (ATR) accessory at room temperature. A 256-scan interferogram with 4 cm-1 resolution were used to collect the spectra. Eighteen milligrams of BSA was dissolved in 20 mM phosphate buffer of pH 7.0 giving a concentration of 0.27 mM. Three additional sets of samples were prepared keeping the ratio of BSA and naringin as 1:1, 1:2

Table 1. KSV and Kq values for the binding interaction of naringin with BSA in 20 mM phosphate buffer of pH 7.0.
and 1:3. The background was corrected before scanning the samples and the buffer spectrum collected. Blank spectra were taken to subtract them from the BSA-naringin combined spectra. The secondary structural analyses were performed according to Byler and Susi [54].
2.6. Molecular Docking Study
The structure of BSA is unavailable in the Protein Data Bank [55], so a homology model was used for the docking with naringin. The BSA sequence [Swissport sequence ALBU_BOVIN (P02769)] has 75% similarity with HSA. The model structure was obtained from SAM_T06 server [56-60]. The energy minimized structure of naringin was obtained by Sybyl 6.92 (Tripos Inc., St. Louis, USA) using Amber 7.0 force field and Gasteiger-Huckel charges. FlexX in the Sybyl suite carried out the docking of naringin with BSA. PyMol was used to visualize the docked conformation [61]. The accessible surface area (ASA) of free and complexed BSA was calculated with the help of NACCESS [62]. The change in ASA on complex formation for a specific residue n was calculated using ΔASAn = ASAn (free BSA) – ASAn (naringin-BSA). If any residue lost more than 10 Å2 on complex formation it was assumed that it participated in the association.
3. RESULTS AND DISCUSSION
3.1. Fluorescence Spectroscopy
3.1.1. Fluorescence Quenching Mechanismand Fractional Accessibility of theQuencher
The fluorescence emission spectra of BSA in absence and presence of naringin is given in Figure 2. BSA exhibits a strong emission at 347 nm on excitation at 295 nm with naringin being non-fluorescent under the experimental conditions. The fluorescence intensity of BSA decreases with successive addition of naringin in 20 mM phosphate buffer of pH 7 indicating that naringin can quench the intrinsic fluorescence of BSA. Generally, fluorescence quenching occurs through both dynamic (collisional) and static means. In the latter there is complex formation between the protein and the quencher. Most commonly the processes are distinguished by their dependence on temperature. The maximum value possible for the collision quenching constant of the biomacromolecule is 2 × 1010 M-1 s-1 in aqueous medium. The interaction of naringin with BSA involves static quenching, as the KSV values at the four different temperatures studied are greater than this value (Table 1). The SternVolmer plots at four different temperatures are shown in supplementary, Figure S1. The difference spectra from a UV-Vis study were also used to analyze the quenching mechanism of the interaction. For dynamic quenching, it is necessary that the UV-Vis spectrum of BSA can be superimposed with the difference spectrum of BSANaringin minus naringin [63]. However, in the present study this was not observed which ruled out any dynamic quenching process (Supplementary Figure S2).
From the intercept and slope the values for fa and KA can be obtained (Supplementary Figure S3a). The values of fa of Trp 213 were calculated at four different temperatures using Eq.7. The average value of fa was 0.81 ± 0.09 indicating that naringin could easily bind to the hydrophobic pocket of site 1 of subdomain IIA and that ~80% of the total fluorescence of Trp 213 is accessible to the quencher.
3.1.2. Binding Constant and Mode of Binding from Thermodynamic Parameters
The values of Kb have been calculated at each temperature using Eq.8 with the corresponding plots in supplementary Figure S3b. The binding constants are found to be in the order of 104 M-1 as reported earlier [39]. It has been observed that there is an increase in the binding constant with rise in temperature. The binding constant and the number of binding sites are listed in the Table 2.
The average value of ΔG° obtained is -24.65 ± 1.11 kJ/mol. The values of change in enthalpy (ΔH°) and entropy (ΔS°) for the interaction were found to be + 18.73 kJ/mol and + 143.64 J mol-1 K-1 respectively (Supplementary, Figure S4). The mode of interaction of small ligands with the biomacromolecules based on the signs of the thermodynamic parameters obtained has been characterized by Ross and Subramanian [47]. The positive sign for both ΔH° and ΔS° indicates a predominantly hydrophobic interaction between naringin and BSA. This is also supported by the spectral blue shift observed in the emission spectra of BSA in presence of naringin (Figure 2). The values indicate that the major contribution to ΔG° arises from the change in entropy rather than the change in enthalpy as shown in the bar diagram (Figure 3). Hence we can conclude that the binding process is entropy driven process.
3.1.3. Synchronous Fluorescence Analysis
Synchronous fluorescence studies were conducted to provide information about the molecular environment of
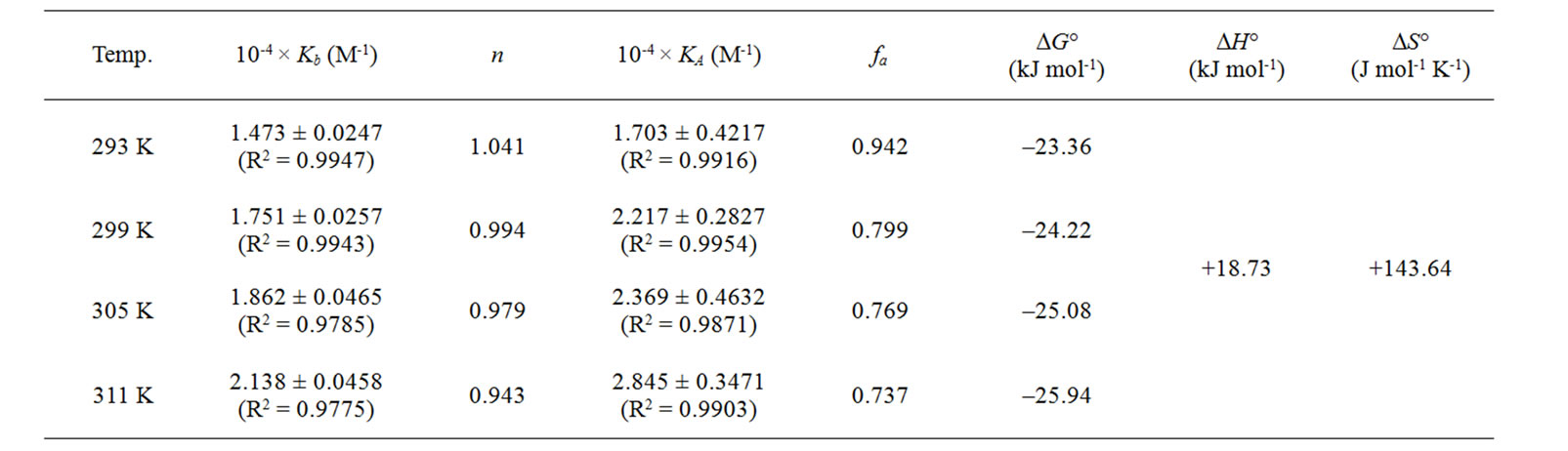
Table 2. Binding and thermodynamic parameters for the complexation of naringin with BSA at different temperatures.
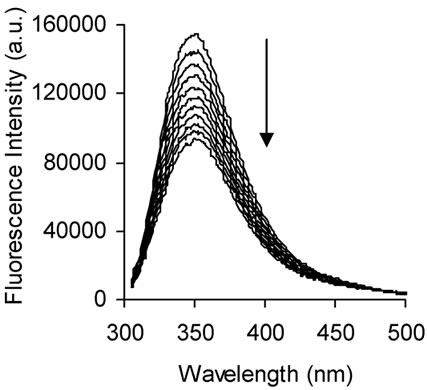
Figure 2. Fluorescence emission spectra of BSA (2 μΜ) in absence (top line) and presence (bottom lines) of naringin (0 to 36 μΜ) at 299 K in 20 mM phosphate buffer of pH 7.0; λex = 295 nm. Arrows indicate the progress of titration.

Figure 3. The bar diagram for interaction of naringin (0 to 36 μΜ) with BSA (2 μΜ) in 20 mM phosphate buffer of pH 7.0. λex = 295 nm.
the chromophores present in the protein [64]. The synchronous fluorescence intensity of tryptophan residues decreased after addition of naringin with an associated blue shift from 347 to 345 nm (Supplementary Figure S5a). In accordance with the thermodynamic parameters, the blue shift in the synchronous emission spectra clearly indicates that naringin binds to the hydrophobic cavity of BSA where Trp 213 is located. On the other hand, the fluorescence intensity of tyrosine decreases regularly with increasing concentration of naringin, but no significant change in λem was observed (Supplementary Figure S5b). This observation suggests that the microenvironment around tyrosine remains unaffected after the addition of naringin.
3.1.4. Analysis of site Marker Displacement Studies
It is known that ANS, a fluorescent hydrophobic probe binds to the non-polar region of proteins, which corresponds to site 1 comprising subdomains IIA and IIIA of HSA and BSA [38,65]. The displacement of ANS bound to BSA after the addition of naringin (Supplementary Figure S6) indicates that naringin does bind to site 1 of the protein [66]. This displacement also indicates the involvement of a hydrophobic interaction in the complexation process that has also been found from the thermodynamic parameters.
The decrease in fluorescence intensity observed in case of bound warfarin to BSA (Supplementary Figure S7) supports the observation that naringin binding competes for the same site in BSA (subdomain IIA and IIIA). An additional experiment with ibuprofen bound to BSA was also performed (Supplementary Figure S8) to check whether naringin binds to site 2 (subdomain IIIA). The binding constants of the interaction of naringin with BSA were calculated in presence of warfarin and ibuprofen according to Eq.8 at 299 K (Supplementary Figure S9).
The binding constant for the complex formation at this temperature is 1.75 x 104 M-1 (the mean ±sd being 1.80 ± 0.28 x 104 M-1). In case of warfarin and ibuprofen the values were found to be 1.49 × 104 and 2.29 × 104 M-1. This implies that the addition of naringin to warfarin bound protein was able to disrupt the binding as there is a decrease in the binding constant. In case of ibuprofen bound BSA the binding constant does not decrease. These results suggested that naringin binds preferentially to site 1 (subdomain IIA) where warfarin binds. This result is also supported by the binding of naringin to HSA where it is also found to bind to the same site [36]. Docking results (below) also support this observation.
3.2. Energy Transfer from Donor to Acceptor
The overlap of the absorption spectrum of naringin with the fluorescence emission spectrum of BSA is shown in Figure 4. The values of κ2, N and QD are 2/3, 1.336 and 0.15 respectively [67]. The value of J(λ) was estimated from Eq.3 with the help of Origin 6.0, from 305 to 450 nm and was found to be 6.1 × 10-15 M-1 cm3.
Using Eq.2 R0 was calculated to be 2.32 nm. The value of r from Eq.1 using E = 0.117 was 3.25 nm. The donor-to-acceptor distance r < 7 nm and 0.5R0 < r < 2 R0 [44], indicated that the energy transfer from the Trp 213 residue to naringin occurred with high propensity. This also explains the quenching of tryptophan fluorescence.
3.3. Circular Dichroism
The CD spectrum of BSA exhibits two negative bands in the ultraviolet region at 208 (π→π* transition) and 222 nm (n→π* transition) as shown in Figure 5 and this is characteristic of the α-helical structure of a protein. The interaction between naringin and BSA caused a slight decrease in band intensity at all wavelengths of the far UV CD without any significant shift in the peak position. The α-helical content of free BSA was 65%. The α-helical content of BSA decreased to 63, 62, 61 and 59% on 1:2, 1:4, 1:6 and 1:8 binding with naringin. This can be treated as a minor conformational change in protein structure in presence of the external ligand. In presence of naringin the α-helical content of HSA is also found to decrease [38].
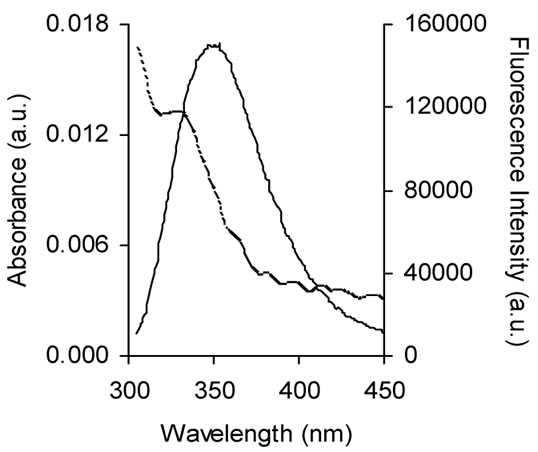
Figure 4. Spectral overlap between fluorescence emission spectrum of BSA (solid line) with the absorption spectrum of naringin (dotted line) in 20 mM phosphate buffer of pH 7.0. [naringin]/[BSA] = 1:1, λex = 295 nm; λem = 347 nm.

Figure 5. CD spectra of free BSA (dotted line) and complexed (solid lines) with naringin ([BSA]:[naringin] ratios are 1:2, 1:4, 1:6 and 1:8) in 20 mM phosphate buffer of pH 7.0.
3.4. FT-IR Analysis
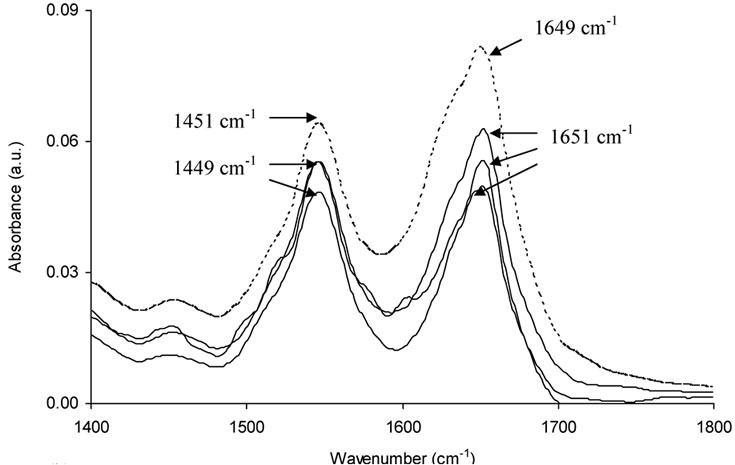
The secondary structure of proteins correspond to 1650-1654 cm-1 (amide I, due to C=O stretching) and 1548-1560 cm-1 (amide II, due to C-N stretching coupled with N-H bending) [54]. The FT-IR spectra of BSA (Figure 6A) show the effect on the secondary structure of BSA due to complexation. The amide I of free BSA has shifted from 1649 cm-1 to 1651 cm-1. The peaks near 1552 cm-1 shifted to 1549 cm-1 showing the effect on binding of naringin to BSA on the amide II region [68,69]. Free BSA shows peaks at 1456 cm-1, 1438 cm-1 and 1423 cm-1 that arise due to antisymmetric angle bending of CH3 groups and angle bending of CH2 of the side chains [67]. After complexation with naringin the peaks shift to some extent (Figure 6b) indicating the involvement of a hydrophobic interaction in the binding of naringin to BSA. The difference spectra (Supplementary, Figure S11) show positive bands at 1618, 1635, and 1648 cm-1 (amide I) and 1520, 1552 and 1568 cm-1 (amide II) for a 1:1 complex. Negative peaks arise in the difference spectra at 1:2 and 1:3 ratios indicating the presence of H-bonding of naringin with BSA [54,68]. It was also observed that the secondary structure of BSA was perturbed in presence of naringin. The α-helical content of BSA decreases from 66 to 47% as observed in the CD studies. The difference in values for the % content arise due to the calculations arising from differences in electronic transitions and vibrational transitions which are the observed responses in case of circular dichroism and infrared spectra respectively. The % α-helical content was calculated from the deconvoluted spectra of BSA and BSA-naringin complex (Supplementary, Figure S10).
3.5. Docking Results
The docked conformation of naringin with BSA is
 (a)
(a) (b)
(b)
Figure 6. (a) FT-IR spectra of BSA (dotted line) and BSA bound with naringin (solid lines) in the region 1800-1400 cm-1 and (b) in the region 1460-1400 cm-1 in 20 mM phosphate buffer of pH 7.0. The contribution of naringin was subtracted from the BSA-Naringin complexes.
given in Figure 7. From the docking study it is clear that the drug naringin binds to hydrophobic site 1 (subdomain IIA) where Trp 213 residue is located. This also supports the hydrophobic character of binding of naringin with BSA, which is corroborated by the thermodynamics of the interaction. Fluorescence studies that show a blue shift on complex formation also support the hydrophobic association. Changes in ASA of specific residues involved in the interaction are given in Table 3. There is a possible hydrogen bond interaction between 4-C=O of naringin with Arg 198 and Arg 217; 23-OH and Asp 450; 25-OH and –NH of Lys 187. This interaction decreases the hydrophilicity and increases the hydrophobicity that results in the stabilization of the BSAnaringin complex. A similar observation has also been reported in case of the interaction of naringin with HSA [36].
4. CONCLUSION
The binding of naringin with BSA under physiological conditions has been presented by fluorescence spectros

Figure 7. The docking pose of naringin with BSA.
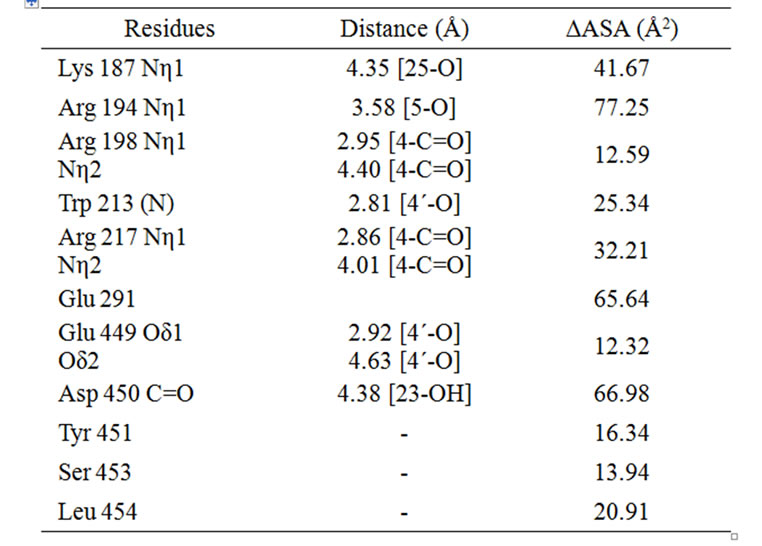
Table 3. The distances (Ǻ) between naringin and BSA and change in accessible surface area (ΔASA) of residues of BSA after interaction with naringin.
copy in combination with CD, FTIR and molecular docking studies. We observe that naringin binds to the hydrophobic site 1 (subdomain IIA) of BSA as confirmed from both experimental and docking results. The binding constants were obtained in order of 104. The distance between Trp 213 and naringin was 3.25 nm, which predicts that there is a possibility of energy transfer from donor to acceptor. The negative value of ΔG° implies the spontaneity of the complexation. According to the docking studies naringin is within hydrogen bonding distance of Trp 213. The study of the interactions of naturally occurring polyphenols with serum albumins is essential for the understanding of how the molecules may be modified to target specific diseases, since the polyphenols are widely known to have a multitude of pharmacological activities.
5. ACKNOWLEDGEMENTS
SDG is grateful to Department of Science and Technology, Government of India (DST, Project no. SR/SO/BB-54/2007). We are also thankful to Central Research Facility, IIT Kharagpur for providing experimental facilities. ASR and DRT thank CSIR, New Delhi for Junior Research fellowship.
REFERENCES
- Spencer, J.P.E. (2008) Flavonoids: Modulators of brain function? Brit. J. Nutr. Vol. 99, May, pp. ES60- ES77.
- S. S. Han and I. J. You, “Studies on antimicrobial activities and safety of natural naringin,” Korean J. Mycol. Vol. 16, March 1988, pp. 33-40.
- T. P. T. Cushnie and A.J. Lamb, “Antimicrobial Activity of flavanoids,” Int. J. Antimicrob. Agents, Vol. 26, August 2005, pp. 343-356.
- Y. Yamamoto and R. B. Gaynor, “Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer,” J. Clin. Invest., Vol. 107, February 2001, pp. 135-142.
- G. G. Duthic, S. J. Duthic and J. A. M. Kyle, “Plant polyphenols in cancer and heart disease: implications as nutritional antioxidants,” Nutr. Res. Rev., Vol. 13, June 2000, pp. 79-106.
- R. Francis, T. K. Shetty and R. K. Bhattacharya, “Modulating effect of plant flavonoids on the mutagenicity of N-methyl-N'-nitro-N-nitrosoguanidine,” Carcinogenesis, Vol. 10, October 1989, pp. 1953-1955.
- W. L. Bear and R. W. Teel, “Effect of citrus phytochemicals on liver and lung cytochrome P450 activity and on the in vitro metabolism of the tobaccospecific nitrosamine NKP,” Anticancer Res., Vol. 20, September 2000, pp. 3323-3329.
- S. V. Jovanovic, S. Steenken, M. Tosic, B. Marjanovic and M. G. Simic, “Flavonoids as antioxidants,” J. Am. Chem. Soc., Vol. 116, June 1994, pp. 4846-4851.
- G. C. Jagetia, V. A. Venkatesha and T. K. Reddy, “Naringin, a citrus flavonone, protects against radiationinduced chromosome damage in mouse bone marrow,” Mutagenesis, Vol. 18, July 2003, pp. 337-343.
- R. Schindler and R. Mentlein, “Flavonoids and Vitamin E Reduce the Release of the Angiogenic Peptide Vascular Endothelial Growth Factor from Human Tumor Cells,” J. Nutr., Vol. 136, June 2006, pp. 1477-1482.
- M. J. Martin, E. Marhuenda, C. Perez-Guerrero and J. M. Franco, “Antiulcer Effect of Naringin on Gastric Lesions Induced by Ethanol in Rats,” Pharmacology, Vol. 49, February 1994, pp. 144-150.
- Y. F. Ueng, Y. L. Chang, Y. Oda, S. S. Park, J. F. Liao, M. F. Lin and C. F. Chen, “In vitro and in vivo effects of naringin on cytochrome p-450-depenent monooxygenase in mouse liver,” Life Sci., Vol. 65, November 1999, pp. 2591-2602.
- H. Lee, T. S. Jeong, Y. K. Choi, B. H. Hyun, G. T. Oh, E. H. Kim, J. R. Kim, J. I. Han and S. H. Bok, “Anti-atherogenic effect of citrus flavonoids, naringin and naringenin, associated with hepatic ACAT and aortic VCAM-1 and MCP-1 in high cholesterol-fed rabbits,” Biochem. Biophys. Res. Commun., Vol. 284, June 2001, pp. 681-688.
- R. R. da Silva, T. T. de Oliveira, T. J. Nagem, A. S. Pinto, L. F. Albino, M. R. de Almeida, G. H. de Moraes and J. G. Pinto, “Hypocholesterolemic effect of naringin and rutin flavonoids,” Arch. Latinoam. Nutr., Vol. 51, September 2001, pp. 258-264.
- P. B. Gordon, I. Holen and P. O. Seglen, “Protection, by naringin and some other flavonoids, of hepatocytic autophagy and endocytosis against inhibition by okadaic acid,” J. Biol. Chem., Vol. 270, March 1995, pp. 5830-5838.
- S. Kanno, A. Shouji, K. Asou and M. Ishikawa, “Effect of naringin on hydrogen peroxide-induced cytotoxicity and apoptosis in P 388 cells,” J. Pharmacol. Sci., Vol. 92, June 2003, pp. 166-170.
- H. Zhang, C. W. Wong, P. F. Coville and S. Wanwimolruk, “Effect of the grapefruit flavonoid naringin on pharmacokinetics of quinine in rats,” Drug Metabol. Drug Interact., Vol. 17, 2000, pp. 351-363.
- G. Colmenarejo, “In silico prediction of drug-binding strengths to human serum albumin,” Med. Res. Rev., Vol. 23, May 2003, pp. 275-301.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, “Crystal structure of human serum albumin at 2.5 Å resolution,” Protein Eng., Vol. 12, June 1999, pp. 439-446.
- A. Molla, S. Vasavanonda, G. Kumar, H. L. Sham, M. Johnson, B.Grabowski, J. F. Denissen, W. Kohlbrenner, J. J. Plattner, J. M. Leonard, D. W. Norbeck and D. J. Kempf, “Human serum attenuates the activity of protease inhibitors toward wild-type and mutant human deficiency virus,” Virology, Vol. 250, October 1998, pp. 255-262.
- C. Carter and J. X. Ho, “Structure of serum albumin,” Adv. Protein Chem., Vol. 45, 1994, pp. 153-203.
- C. Carter, B. Chang, J. X. Ho, K. Keeling and Z. Krishnasami, “Preliminary crystallographic studies of four crystal forms of serum albumin,” Eur. J. Biochem., Vol. 226, October 1994, pp. 1049-1052.
- K. F. Brown and M. J. Crooks, “Displacement of tolbutamide, glibencalmide and chroproparmide from serum albumin by anionic drugs,” Biochem. Pharmacol., Vol. 25, May 1976, pp. 1175-1178.
- T. Jr. Peters, “Serum albumin,” Adv. Protein Chem., Vol 37, 1985, pp. 161-245.
- U. Kragh-Hansen, “Molecular aspects of ligand binding to serum albumin,” Pharmacol. Rev., Vol. 33, March 1981, pp. 17-53.
- G. Zhang, A. Wang, T. Jiang and J. Guo, “Interaction of the irisflorentin with bovine serum albumin: A fluorescence quenching study,” J. Mol. Struct., Vol. 891, November 2008, pp. 93-97.
- L. Shang, X. Jiang and S. Dong, “In vitro study on the binding of neutral red to bovine serum albumin by molecular spectroscopy,” J. Photochem. Photobiol. A, Vol.184, November 2006, pp. 93-97.
- N. Zhou, Y. Z. Liang and P. Wang, “18β-Glycyrrhetinic acid interaction with bovine serum albumin,” J. Photochem. Photobiol. A, Vol. 185, January 2007, pp. 271-276.
- Y. P. Wang, Y. L. Wei and C. Dong, “Study on the interaction of 3,3-bis(4-hydroxy-1-naphthyl)-phthalide with bovine serum albumin by fluorescence spectroscopy,” J. Photochem. Photobiol. A, Vol. 177, January 2006, pp. 6-11.
- Bose and A. Dube, “Interaction of Chlorin p6 with bovine serum albumin and photodynamic oxidation of protein,” J. Photochem. Photobiol. B, Vol. 85, October 2006, pp. 49-55.
- Y. He, Y. W. Wang, L. F. Tang, H. Liu, W. Chen, Z. L. Zheng and G. L. Zou, “Binding of Puerarin to Human Serum Albumin: A Spectroscopic Analysis and Molecular Docking,” J. Fluoresc., Vol. 18, December 2007, pp. 433-442.
- K. Sahoo, K. S. Ghosh and S. Dasgupta, “Investigating the binding of curcumin derivatives to bovine serum albumin,” Biophys. Chem., Vol. 132, February 2008, pp. 81-88.
- B. K. Sahoo, K. S. Ghosh and S. Dasgupta, “Molecular Interactions of Isoxazolcurcumin with Human serum albumin: Spectroscopic and Molecular Modeling Studies,” Biopolymers, Vol. 91, February 2009, pp. 108-119.
- K. S. Ghosh, B. K. Sahoo, D. Jana and S. Dasgupta, “Studies on the interaction of copper complexes of (_)- epicatechin gallate and (_)-epigallocatechin gallate with calf thymus DNA,” J. Inorg. Biochem., Vol. 102, September 2008, pp. 1711-1718.
- T. K. Maiti, K. S. Ghosh and S. Dasgupta, “Interaction of (-)-Epigallocatechin-3-Gallate with Human Serum Albumin: Fluorescence, Fourier Transform Infrared, Circular Dichroism, and Docking Studies,” Proteins, vol. 64, May 2006, pp. 355-362.
- Rieutord, P. Bourget, G. Troche and J. F. Zazzo, “In vitro study of the protein binding of fusidic acid: a contribution to the comprehension of its pharmacokinetics behaviour,” Int. J. Pharm., Vol. 1, May 1995, pp. 57-64.
- Borga and B. Borga, “Serum protein binding of the nonsteroidal antiinflammatory drugs: A comparative study” J. Pharm. Biopharm., Vol. 25, January 1997, pp. 63-77.
- Y. Zhang, Y. Li, L. Dong, J. Li, W. He, X. Chen and Z. Hu, “Investigation of the interaction between naringin and human serum albumin,” J. Mol. Struct., Vol. 875, March 2008, pp. 1-8.
- Y. Sun, H. Zhang, Y. Sun, Y. Zhang, H. Liu, J. Cheng, S. Bi and H. Zhang, “Study of interaction between protein and main active components in citrus aurantium L. by optical spectroscopy,” J. Lumin., Vol. 130, February 2010, pp. 270-279.
- N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, “How to measure and predict the molar absorption coefficient of a protein,” Protein Sci., Vol. 4, November 1995, pp. 2411-2423.
- J. L. Perry, M. R. Goldsmith, T. R. Williams, K. P. Radack, T. Christensen, J. Gorham, M. A. Pasquinelli, E. J. Toone, D. N. Beratan and J. D. Simon, “Binding of warfarin influences the acid-base equilibrium of H242 in sudlow site I of human serum albumin,” Photochem. Photobiol., Vol.82, February 2006, pp. 1365-1369.
- L. Du, X. Liu, W. Huang and E. Wang, “A study on the interaction between ibuprofen and bilayer lipid membrane,” Electrochem. Acta, Vol. 51, August 2006, pp. 5754-5760.
- J. H. Tang, F. Luan and X. G. Chen, “Binding analysis of glycyrrhetinic acid to human serum albumin: Fluorescence spectroscopy, FTIR, and molecular modeling,” Bioorg. Med. Chem., Vol. 14, May 2006, pp. 3210-3217.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Third ed. (Springer, New York, 2006).
- L. Gelamo, C. H. T. P. Silva, H. Imasato, M. Tabak, “Interaction of bovine (BSA) and human (HSA) serum albumins with ionic surfactants: spectroscopy and modeling,” Biochim. Biophys. Acta, Vol. 1594, January 2002, pp. 84-99.
- M. Jiang, M. X. Xie, D. Zheng, Y. Liu, X. Y. Li and X. Cheng, “Spectroscopic studies on the interaction of cinnamic acid and its hydroxyl derivatives with human serum albumin,” J. Mol. Struct., Vol. 692, April 2004, pp. 71-80.
- P. D. Ross and S. Subramanian, “Thermodynamics of protein association reactions: Forces contributing to stability,” Biochemistry, Vol. 20, May 1981, pp. 3096- 3102.
- J. N. Miller, “Recent advances in molecular luminescence analysis,” Proc. Anal. Doc. Chem. Soc. Vol. 16, July 1979, pp. 203-208.
- H. G. Mahesha, S. A. Singh, N. Srinivasan and A. G. A. Rao, “A spectroscopic study of the interaction of isoflavones with human serum albumin,” FEBS J., Vol. 273, February 2006, pp. 451-467.
- G. Weber and L. B. Young, “Fragmentation of bovine serum albumin by pepsin. I. The origin of the acid expansion of the albumin molecule,” J. Biol. Chem., Vol. 239, May 1964, pp. 1415-1423.
- P. Heller and C. L. Greenstock, “Fluorescence lifetime analysis of DNA intercalated ethidium bromide and quenching by free dye,” Biophys. Chem., Vol. 50, June 1994, pp. 305-312.
- Y. H. Chen, J. T. Yang and H. M. Martinez, “Determination of the Secondary Structures of Proteins by Circular Dichroism and Optical Rotary Dispersion,” Biochemistry, Vol. 11, October 1972, pp. 4120-4131.
- Hong, L. Lei, Q. Kong, X. Chen and Z. Hu, “The study on the interaction between human serum albumin and a new reagent with antitumour activity by spectrophotometric methods,” J. Photochem. Photobiol. A, Vol. 167, October 2004, pp. 213-221.
- M. Byler and H. Susi, “Examination of the secondary structure of proteins by deconvolved FTIR spectra,” Biopolymers, Vol. 25, March 1986, pp. 469-487.
- M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, “The Protein Data Bank,” Nucleic Acids Res., vol. 28, January 2000, pp. 235-242.
- K. Karplus, S. Katzman, G. Shackleford, M. Koeva, J. Draper, B. Barnes, M. Soriano and R. Hughey, “SAMT04: what is new in protein-structure prediction for CASP6,” Proteins, Vol. 61, September 2005, pp. 135- 142.
- R. Karchin, M. Cline and K. Karplus, “Evaluation of local structure alphabets based on residue burial,” Proteins, Vol. 55, May 2004, pp. 508-518.
- R. Karchin, M. Cline, Y. Mandel-Gutfreund and K. Karplus, “Hidden Markov models that use predicted local structure for fold recognition: alphabets of protein backbone geometry,” Proteins, Vol. 51, June 2003, pp. 504-514.
- R. Karchin, J. Draper, J. Casper, Y. Mandel-Gutfreund, M. Diekhans and R. Hughey, “Combining local structure, fold-recognition, and new-fold methods for protein structure prediction,” Proteins, Vol. 53, March 2003, pp. 491-496.
- K. Karplus, R. Karchin, C. Barrett, S. Tu, M. Cline, M. Diekhans, L. Grate, J. Casper and R. Hughey, “What is the value added by human intervention in protein structure prediction?” Proteins, Vol. 45, January 2002, pp. 86-91.
- W. L. Delano, The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos CA, USA, 2004. http://pymol.sourceforge.net/
- S. J. Hubbard and J. M. Thornton, “‘NACCESS’, Computer Program,” Department of Biochemistry and Molecular Biology, University College, London, 1993.
- S. Y. Bi, D. Q. Song, Y. Tian, X. Zhou, Z. Y. Liu and H. Q. Zhang, “Molecular spectroscopic study on the interaction of tetracyclines with serum albumins,” Spectrochim. Acta Part A, Vol. 61, February 2005, pp. 629-636.
- Chen, X. Z. Huang, J. G. Xu, Z. Z. Zheng and Z. B. Wang, “Methods of fluorescence analysis,” Second ed., Science Press, Beijing, 1990.
- Matulis and R. Lovrien, “1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation,” Biophys. J., Vol. 74, January 1998, pp. 422-429.
- B. Luis, C. K. Silvia, A. Felipe, A. S. Marco, S. Patricio and D. F. Gerardo, “Two distinguishable fluorescent modes of 1-anilino-8-naphthalenesulfonate bound to human albumin,” J. Fluoresc., Vol. 6, January 1996, pp. 33-40.
- L. Cui, J. Fan, J. P. Li and Z. Hu, “Interactions between 1-benzoyl-4-p-chlorophenyl thiosemicarbazide and serum albumin: investigation by fluorescence spectroscopy,” Bioorg. Med. Chem., Vol. 12, January 2004, pp. 151-157.
- Susi and D. M. Byler, “Resolution-enhanced fourier transform infrared spectroscopy of enzymes,” Methods Enzymol., Vol. 130, 1986, pp. 290-311.
- S. Krimm and J. Bandekar, “Vibrational spectroscopy and conformation of peptides, polypeptides and proteins,” Adv. Protein Chem., Vol. 38, 1986, pp. 181-364.
Supplementary Information
A spectroscopic study of the interaction of antioxidant naringin with bovine serum albumin
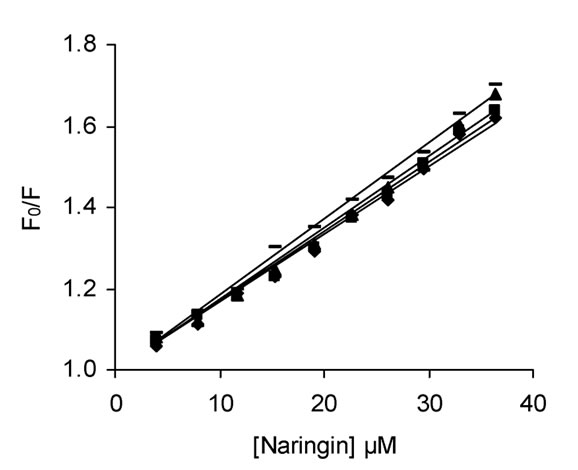
Figure S1. Stern-Volmer plots for the interaction of Nringin with BSA at different temperatures in 20 mM phosphate buffer of pH 7.0; (♦) 293 K, (▬) 299 K (▲) 305 K (■) 311 K, λex = 295 nm.
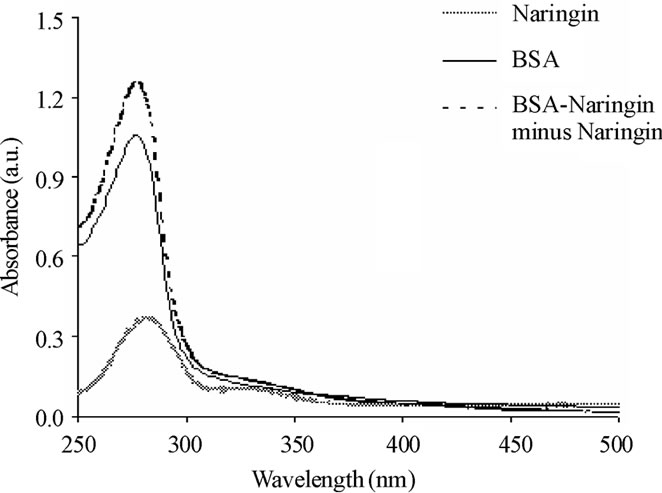
Figure S2. The UV-Vis spectra of BSA, naringin and the difference spectra of (BSA-Naringin) minus naringin system. [BSA] = [Naringin] = 25 μM.
 (a)
(a) (b)
(b)
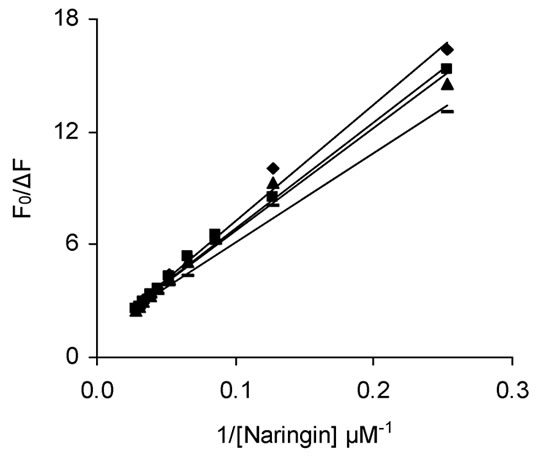
Figure S3. (a) Modified Stern-Volmer plot of BSA (2 μΜ) with increasing concentration of naringin (0 to 36 μΜ) and (b) Double logarithm plot for the interaction of naringin (0 to 36 μΜ) with BSA (2 μΜ). (♦) 293 K, (▬) 299 K (▲) 305 K (■) 311 K. λex = 295 nm.
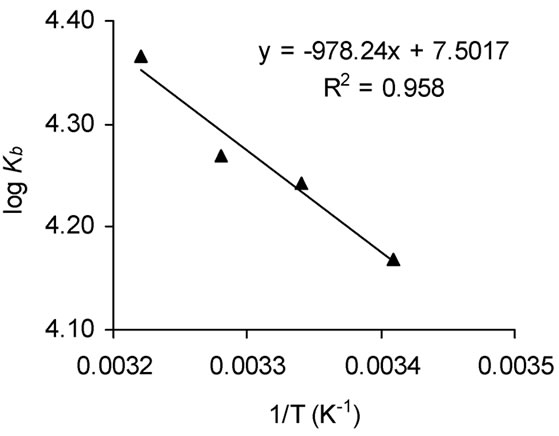
Figure S4. The van’t Hoff plot and (b) the bar diagram for interaction of naringin (0 to 36 μΜ) with BSA (2 μΜ) in 20 mM phosphate buffer of pH 7.0. λex = 295 nm.
 (a)
(a)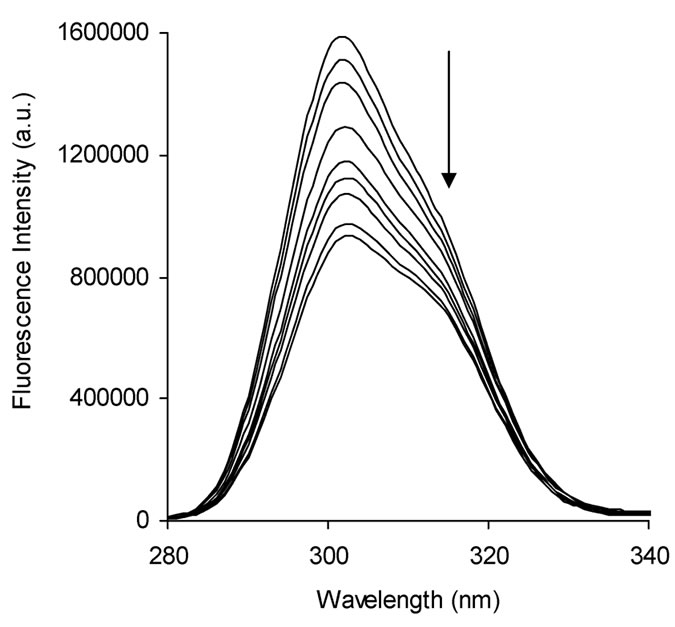 (b)
(b)
Figure S5. Synchronous fluorescence spectra of BSA when (a) ∆λ = 60 nm and (b) ∆λ = 15 nm in absence (top line) and presence (bottom lines) of naringin (0 to 14 μΜ) in 20 mM phosphate buffer of pH 7.0 at 299 K. Arrows indicate the progress of titration.

Figure S6: Displacement of bound ANS from BSA-ANS complex by naringin (0 to 21 μΜ); [BSA] = 2 μΜ, [ANS] = 6 μΜ; λex: 375 nm. Arrows indicate the progress of titration.

Figure S7. Displacement of bound warfarin from BSAwarfarin complex by naringin (0 to 40 μM); [BSA] = [warfarin] = 2 μΜ; λex: 308 nm. Arrows indicate the progress of titration.

Figure S8. Displacement of bound ibuprofen from BSAibuprofen complex by naringin (0 to 21 μΜ); [BSA] = 2 μΜ; [ibuprofen] = 1 μΜ; λex: 265 nm. Arrows indicate the progress of titration.
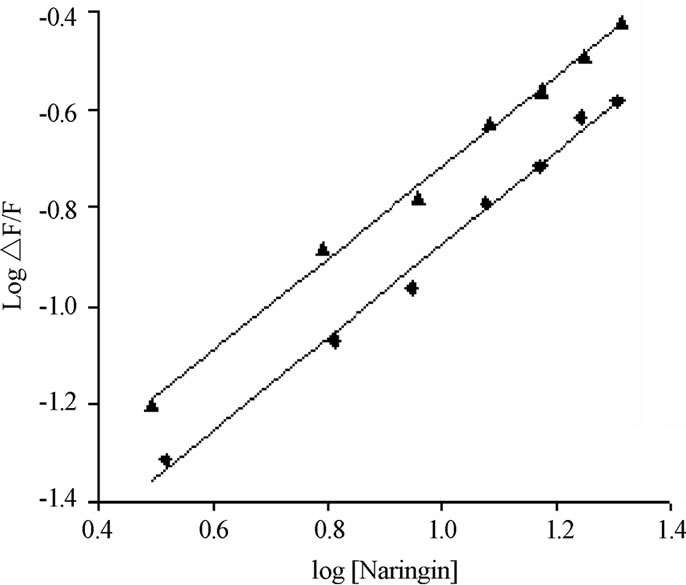
Figure S9. The effect of site marker probe on the binding of naringin with BSA in 20 mM phosphate buffer of pH 7.0 at 299 K. (▲) BSA-ibuprofen (♦) BSA-warfarin complex; λex: 265 and 308 nm respectively.

Figure S10. Curve-fitted amide I (1700-1600 cm-1) regions of free BSA and BSA-naringin complexes (molar ratios 1:1, 1:2 and 1:3) in 20mM phosphate buffer of pH 7.0.