Journal of Diabetes Mellitus
Vol.1 No.4(2011), Article ID:8417,17 pages DOI:10.4236/jdm.2011.14012
Obesity and type 2 diabetes
![]()
Section of Endocrinology and Metabolism, Stratton VA Medical Center, Albany, USA; Subhashini.Yaturu@yahoo.com
Received 17 August 2011; revised 20 September 2011; accepted 30 September 2011.
Keywords: Diabetes; Obesity; Body Mass Index; Impaired Glucose Tolerance; Insulin Resistance Syndrome
ABSTRACT
Obesity and type 2 diabetes (T2DM) are public health problems, with health consequences and economic costs that have raised concern worldwide. The increase in the prevalence of diabetes parallels that of obesity. Some experts call this dual epidemic “diabesity”. Elevated body mass index (BMI) and waist circumference (WC) were significantly associated T2DM. One consequence of obesity is an increased risk of developing T2DM. There is evidence that the prenatal, early childhood, and adolescent periods are critical in the development of obesity. Most obese individuals have elevated plasma levels of free fatty acids (FFA), which are known to cause peripheral (muscle) insulin resistance. Weight loss either with lifestyle modification, pharmacotherapy or bariatric surgery improves glycemic control and metabolic parameters that are related to cardiovascular disease. Pharmacotherapy for glycemic control with metformin or GLP-1 agonists and DPP-4 inhibitors help in weight reduc- tion.
1. INTRODUCTION
Obesity and diabetes are emerging pandemics in the 21st century. Both are major public health problems throughout the world and are associated with significant, potentially life-threatening co-morbidities and enormous economic costs. The prevalence of overweight (body mass index (BMI) between 25 and 30 kg/m2) [1] and obesity (BMI of 30 kg/m2 or higher) [1] is increasing rapidly worldwide, especially in developing countries. There is a strong association between obesity and type 2 diabetes. Meta-analysis of studies of association of these two conditions showed higher relative risk with BMI as well as waist circumference in both men and women [2]. Not all subjects with type 2 diabetes (T2DM) are obese and many obese subjects do not have diabetes, but most of the subjects with T2DM are overweight or obese. Significant numbers of obese individuals have diabetes. Overweight, obesity and T2DM are largely preventable with change in life style and avoidance of sedentary habits and over-consumption of energy. Both obesity and T2DM, feature insulin resistance and atherogenic lipid profiles such as increased triglycerides and decreased HDL-C. The genetic basis of human obesity that predisposes to insulin resistance and T2DM is multigenic rather than monogenic. Current clinical guidelines acknowledge the therapeutic strength of exercise intervention for prevention and treatment of diabetes.
2. PREVALENCE
Data from the Third National Health and Nutrition Examination Survey (NHANES III) indicate that twothirds of adults, both men and women, had BMI values >27 kg/m2 [3]. The prevalence of T2DM parallels the increasing prevalence of obesity. The World Health Organization (WHO) projects that there are currently 2.3 billion overweight people aged 15 years and above, and that there will be over 700 million obese people worldwide in 2015 [http://www.who.int/mediacentre/factsheets/fs311/en/]. The prevalence of diabetes is increasing in the United States, and the diagnosed diabetes increased from 0.9% in 1958 to 6.3% in 2008. In 2008, 18.8 million people had diagnosed diabetes, compared to only 1.6 million in 1958 [http://www.cdc.gov/diabetes/st atistics/slides/long_term_trends.pdf.]. According to the International Diabetes Federation (IDF), it is estimated that approximately 285 million people worldwide, or 6.6%, in the age group 20 - 79, will have diabetes in 2010 [http: //www.diabetesatlas.org/map], some 70% of whom live in lowand middle-income countries. This number is expected to increase by more than 50% in the next 20 years if preventive programs are not put in place. By 2030, some 438 million people, or 7.8% of the adult population, are projected to have diabetes. T2DM is the predominant form of diabetes worldwide and constitutes 85% - 95% of all diabetes. Obesity and overweight currently affect 15% and 20% of Spanish children [4], respectively. The NHANES study noted that with increasing overweight and obesity class, there is an increase in the prevalence of diabetes, from 2.4% for normal weight to 14.2% for obesity class 3. With normal weight individuals as a reference, individuals in obesity class 3 had an adjusted odds ratio of 5.1 (95% CI 3.7 to 7.0) for diabetes [5] .
Diagnostic Criteria and Definitions
WHO defines “overweight” [1] as a BMI equal to or more than 25, and “obesity” [1] as a BMI equal to or more than 30. BMI, calculated by weight (kg)/height (m2) and adjusted for height, is used as a measure of weight standards. The criteria for diagnosis of diabetes mellitus as recommended by the American Diabetes Association7 include: 1. A1C ≥ 6.5% or fasting plasma glucose [FPG] value after an 8-hour fast ≥126 mg/dL, or 2-hour post load glucose (PG) ≥200 mg/dL (11.1 mmol/L) during an OGTT, or symptoms of diabetes mellitus and a random plasma glucose concentration ≥200 mg/dl (11.1 mmol/ L). Insulin resistance [6] is defined as a failure of target organs to respond normally to the action of insulin. Insulin resistance syndrome [7] (IRS) refers to the cluster of abnormalities that occur more commonly in insulin resistant individuals. Metabolic syndrome (MS), as defined by the National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) [8], is a cluster of metabolic abnormalities with insulin resistance as a major characteristic. The presence of any three of the five components is sufficient for diagnosis. The components of metabolic syndrome include: 1) abdominal obesity (waist circumference > 102 cm [40] in men, >88 cm [35] in women); 2) hypertriglyceridemia (≥150 mg/dL); 3) low HDL-C (<40 mg/dL in men, <50 mg/dL in women); 4) high blood pressure (≥130/85 mm Hg); and 5) high fasting glucose.
3. Impact of Obesity on Type 2 Diabetes
The relative risk of T2DM increases as BMI increases above 23 [9], and the association was found to be stronger in younger age groups in this study from the Asia-Pacific region [10]. Weight gain in early adulthood is related to a higher risk and earlier onset of type 2 diabetes than is weight gain between 40 and 55 years of age [11] . The risk of diabetes increases linearly with BMI; the prevalence of diabetes increased from 2% in those with a BMI of 25 to 29.9 kg/m2, to 8% in those with a BMI of 30 to 34.9 kg/m2 , and finally to 13% in those with a BMI greater than 35 kg/m2 [12]. Although the prevalence of diagnosed diabetes has increased significantly over the last decade, the prevalence of undiagnosed diabetes and impaired fasting glucose (IFG) has remained relatively stable [13] . More than generalized obesity, the risk of central obesity increases with increase in waist circumference (WC), waist-to-hip ratio, visceral adiposity, or abdominal obesity [14-17] . In a review of 17 prospective and 35 cross-sectional studies in adults aged 18 - 74 years, either BMI or WC predicted or was associated with T2DM independently [18]. Increase in BMI is a better predictor of diabetes than increase in weight.
Prospective studies in non-diabetic overweight adults noted a 49% increase in the incidence of diabetes in 10 years for every 1 kg/year increase in body weight and similarly each kg of weight lost annually over 10 years was associated with a 33% lower risk of diabetes in the subsequent 10 years. Similar studies in Pima Indians reported that weight gain was significantly related to diabetes incidence only in those who were not initially overweight (BMI less than 27.3 kg/m2) [19,20] . Similarly, in the Behavioral Risk Factor Surveillance System (BRFSS) for 1991-1998, Mokdad et al. [21] reported that every 1 kg increase in average self-reported weight was associated with a 9% increase in the prevalence of diabetes [23]. Visceral fat seems to be strongly associated with an abnormal metabolic profile rather than upper body subcutaneous fat. The National Institutes of Health uses WC to identify those at increased risk [22] . Though both visceral adiposity (VAT) and subcutaneous fat are associated with adverse cardio metabolic risk factors, VAT remains more strongly associated with these risk factors [23].
3.1. Impact of Childhood Obesity on T2DM
Obesity now affects 15% of children and adolescents in the United States. BMI in childhood changes substantially with age and is not applicable when defining childhood obesity [24-26] . In the United States, the 85th and 95th percentiles of body mass index for age and sex based on nationally representative survey data have been recommended as cut off points to identify overweight and obesity [27]. T2DM in children and adolescents is an important public health problem directly related to the epidemic of childhood obesity. The increasing rates of youth T2DM parallel the escalating rates of obesity, which is the major risk factor affecting insulin sensitivity [28]. Altered glucose metabolism, manifested as impaired glucose tolerance (IGT), appears early in obese children and adolescents. Obese young people with IGT are characterized by marked peripheral insulin resistance and a relative beta-cell failure [29]. The prevalence of the metabolic syndrome is considerable among obese adolescents [30,31] and the diseases that are associated with obesity in children include T2DM, hypertension, hyperlipidemia, gallbladder disease, nonalcoholic steatohepatitis [32,33] sleep apnea, and orthopedic complications [34,35] .
3.2. Neonatal Size and Impact of Catch-Up Growth
Low birth weight predicts central obesity. High BMI at birth and low socio-economic status were shown to be independent determinants for overweight [36]. Premature infants are at increased risk for persistent growth failure, hypertension, and diabetes [37]. The risk of T2DM is high among adults with low birth weight (small for gestational age) [38] and is further increased by high growth rates after 7 years of age [39]. Accelerated childhood growth is another risk factor for adiposity and insulin resistance, especially in children with low birth weight [40]. These children with low birth weight and catch up growth are at risk for diabetes, obesity, and cardiovascular disease [41]. Prevention of early catch-up growth reversed the development of glucose intolerance and obesity was shown in a mouse model of low birth weight associated diabetes [42]. Crossing into higher categories of BMI after the age of two years is also associated with these disorders [42].
4. PATHOPHYSIOLOGY
4.1. Insulin Resistance in T2DM and Obesity
Normal glucose homeostasis is maintained by a delicate balance between insulin secretion by the pancreatic β-cells and insulin sensitivity of the peripheral tissues (muscle, liver and adipose tissue). Insulin resistance is a key feature of the metabolic syndrome and often progresses to T2DM. Decreased insulin sensitivity and impaired β-cell function are the two key components in T2DM pathogenesis based on long-term experience in adults [43-46] . The major link between obesity and T2DM is insulin resistance. In the natural history of diabetes, obesity and insulin resistance precede abnormal glucose. Insulin resistance in both of these conditions is manifested by decreased insulin-stimulated glucose transport and metabolism in adipocytes and skeletal muscle and by impaired suppression of hepatic glucose output [47].
4.2. Visceral Adiposity, Obesity, Insulin Resistance, T2DM
In obesity the initial deposition of triglycerides occurs in subcutaneous adipose tissue and as this increases in size insulin resistance will rise and limit further subcutaneous lipid accumulation. Triglycerides will then be diverted to the visceral fat depot as well as to ectopic sites. This leads to a substantial rise in insulin resistance and the prevalence of its associated disorders. Evidence supporting this hypothesis includes studies showing that in lean subjects the prime determinant of insulin resistance is BMI, that is, subcutaneous fat, whilst in overweight and obese subjects, it is waist circumference and visceral adiposity. It has also been shown that the metabolic syndrome suddenly increases in prevalence at high levels of insulin resistance and it is suggested that this is due to the diversion of lipids from the subcutaneous to the visceral depot [48]. Accumulation of fat in abdomen regions has major implications for metabolism and particularly for insulin sensitivity [49-51] . This high prevalence of co-morbidities relates more to waist circumference than to BMI.
Many studies have pointed to an association between insulin resistance and intra-abdominal fat accumulation (visceral obesity) [52]. The look AHEAD (Action for Health in Diabetes) Trial of patients with T2DM demonstrated adipose tissue distribution that was significantly altered, with more visceral adipose tissue and intermuscular adipose tissue, depots known to exacerbate insulin resistance, and less subcutaneous adipose tissue in people with diabetes than in healthy control subjects [53]. A high prevalence of obesity and abdominal obesity in Mexicans is associated with a markedly increased incidence of diabetes and hypertension [54] . Visceral adiposity is considered a risk factor for insulin resistance metabolic syndrome [55] and T2DM in adults [56] , as well as in first degree relatives of patients with T2DM with normal glucose levels [57] . Adipocytokines, hormones secreted by the visceral adipocytes, generate the insulin resistant state and the chronic inflammatory profile that frequently goes along with visceral obesity [58].
4.3. Visceral Adiposity and Children
As defined by a BMI greater than the 95th percentile for age and gender from the revised National Center for Health Statistics growth charts, 10% - 15% of 6- to 17- year-old children and adolescents are overweight in the United States [3] and worldwide [59]. Visceral adiposity is considered a risk factor for insulin resistance in children [29,60-65] . Children with central adiposity can develop the metabolic syndrome with insulin resistance, hypertension, and dyslipidemia [66]. The dynamics of glucose tolerance status in these youngsters seems to be more rapid than in adults, thus representing a narrow window of opportunity for successful intervention to prevent diabetes [67]. White adolescent subjects with obesity without DM were noted to have approximately 50% lower levels of adiponectin [68,69] . In addition, the study noted that hypoadiponectinemia was a strong and independent correlate of insulin resistance, β-cell dysfunction, and increased abdominal adiposity [69]. It is hypothesized that abdominal obesity leads to insulin resistance partly through decreased adiponectin [70] .
FFA, Free Fatty Acids Visceral fat exhibits accelerated lipolytic activity with increased release of free fatty acids (FFA), which can adversely affect insulin action and glucose disposal in several tissues [71-75] . Conversely, declines in visceral adiposity and reduced FFA levels following weight-loss diets have been associated with enhanced insulin sensitivity [76,77] . A strong correlation between intramyocellular triacylglycerol concentrations and the severity of insulin resistance has been found, which led to the assumption that lipid oversupply to skeletal muscle contributes to reduced insulin action [71,78] . These increases in circulating FFA levels may also result in the development of triglyceride reservoirs in both muscle and liver, depressing the actions of insulin and increasing hepatic very-low-density lipoprotein output [79-81] . In most obese subjects, plasma FFA levels are increased. FFAs have been shown to have an important contributing role in the pathogenesis of insulin resistance in human obesity [82]. The mechanism involves intramyocellular accumulation of diacylglycerol and activation of protein kinase C. FFAs cause hepatic insulin resistance by inhibiting insulin-mediated suppression of glycolgenolysis [82-85] . Physiologic increases in plasma FFA levels cause insulin resistance in both diabetic and nondiabetic subjects by producing several metabolic defects: 1) FFAs inhibit insulin-stimulated glucose uptake at the level of glucose transport or phosphorylation (or both); 2) FFAs inhibit insulin-stimulated glycogen synthesis; and 3) FFAs inhibit insulin-stimulated glucose oxidation (this last-mentioned defect probably does not contribute to insulin resistance). Studies have shown that both obesity and T2DM impair insulin-induced suppression of glycogenolysis and gluconeogenesis, and that the degree of impairment correlates with plasma FFA concentrations [86,87] . The stimulatory effect of FFAs on hepatic glucose production (HGP) would then become unchecked, resulting in hyperglycemia. Hence, continuously elevated levels of plasma FFAs may play a key role in the pathogenesis of T2DM in predisposed individuals by impairing peripheral glucose utilization and by promoting hepatic glucose overproduction [86-90] . Potential mechanisms of this increased risk with VAT may include increased free fatty acid release and alterations in adipokines. Evidence from studies that manipulate FFA concentrations suggests that a number of these metabolic abnormalities are caused by elevated FFAs, because the most consistent abnormality in FFA metabolism is failure to normally suppress FFA in response to insulin/meal ingestion [91]. In people with visceral obesity, omental and mesenteric fat may play a special role in delivering both excess FFA and IL-6 to the liver [91]. Atherogenic lipid abnormalities with lower levels of high-density liporotein (HDL) and overproduction of large very lowdensity lipoprotein (VLDL) particles precede the diagnosis of T2DM by several years [92] . In obesity and T2DM, the altered communication between adipose tissue and the liver results in the altered regulation of VLDL production. A number of studies indicate that adipocytokines, in particular adiponectin, may be seminal players in the regulation of fat metabolism in the liver [93]. VLDL assembly in the liver is catalyzed by microsomal triglyceride transfer protein (MTP). A study by Wolfrum and Stoffel [94] showed that the forkhead protein Foxa2 stimulates hepatic VLDL production in concert with the coactivator PGC-1beta and that insulin inhibits this process by inactivating Foxa2. It appears that excessive VLDL production associated with insulin resistance is caused by the inability of insulin to regulate FoxO1 transcriptional activation of MTP [95,96] .
4.4. Visceral Fat Tissue as a Pro-Inflammatory Tissue
Adipose tissue is a highly active metabolic and endocrine organ. During the progression from normal weight to obesity and then to overt diabetes, adipocyte-derived factors contribute to the occurrence and development of β-cell dysfunction and type 2 diabetes [97]. Adipocytes secrete a variety of products known as “adipokines”, including leptin, adiponectin, resistin and visfatin, as well as cytokines and chemokines such as TNF-α, IL-6, and monocyte chemoattractant protein-1 [98] all of which also play important roles in the pathogenesis of diabetes, dyslipidemia, inflammation, and atherosclerosis [98-100] . The release of adipokines by either adipocytes or adipose tissue-infiltrated macrophages leads to a chronic sub inflammatory state that could play a central role in the development of insulin resistance and T2DM, and the increased risk of cardiovascular disease associated with obesity [98].
In addition to inducing insulin resistance in insulinresponsive tissues, adipocyte-derived factors play an important role in the pathogenesis of β-cell dysfunction. Leptin, free fatty acids, adiponectin, TNF-α and IL-6 are all produced and secreted by adipocytes, and may directly influence aspects of β-cell function, including insulin synthesis and secretion, insulin cell survival and apoptosis [97]. Leptin is almost exclusively expressed and produced by white adipose tissue—specifically, by differentiated adipocytes. Subcutaneous fat is responsible for 80% of total leptin production. This was shown in cultures ex vivo where the production of leptin was higher in subcutaneous adipocytes than in those of deeper origin [101] . Leptin improves insulin sensitivity through activation of AMP protein kinase (AMPK), which controls cellular concentrations of malonyl-CoA, thereby inhibiting acetyl-CoA carboxy-lase (the enzyme involved in malonyl-CoA transformation) [102] . While a deficiency of leptin is very likely to contribute to insulin resistance when adipose tissue is lacking and leptin resistance is considered as a main feature of human obesity.
TNF-α and IL-6 are expressed in adipose tissues, with visceral fat responsible for more TNF-α production than subcutaneous fat. These cytokines inhibit insulin signaling and TNF-α may play a crucial role in the systemic insulin resistance of T2DM [103,104] . Interleukin-6 (IL- 6), one of the adipokines, has emerged as one of the potential mediators linking obesity-derived chronic inflammation with insulin resistance. Adipose tissue contributes to up to 35% of circulating IL-6, the systemic effects of which have been best demonstrated in the liver, where a STAT3-SOCS-3 pathway mediates IL-6 impairment of insulin actions. In contrast to its role in liver, IL- 6 is believed to be beneficial for insulin-regulated glucose metabolism in muscle [105].
TNF-α is a pro-inflammatory cytokine produced by numerous cells, but adipocytes also produce TNF-α. TNF- α is overexpressed in adipose tissue from obese animals and humans, and obese mice lacking either TNF-α or its receptor show protection against developing insulin resistance. TNF-α induces a state of insulin resistance in terms of glucose uptake in myocytes and adipocytes that impair insulin signalling at the level of the insulin receptor substrate (IRS) proteins. The mechanism involves Ser phosphorylation of IRS-2 mediated by TNF-α activation of MAPKs [106]. TNF-α inhibits tyrosine kinase phosphorylation of the insulin receptor, resulting in defects in insulin signaling and ultimately leading to insulin resistance and impaired glucose transport [107, 108] . An imbalance in favor of pro-inflammatory cytokines from adipose tissue (hypoadiponectinemia and increased levels of IL-6) and other sources may trigger CRP secretion. CRP levels are related strongly to insulin resistance and adiposity, and elevated levels correlate with impaired endothelial dysfunction (ED) and CAD. This in turn can exacerbate mild insulin resistance and accentuate other metabolic abnormalities that together constitute metabolic syndrome. Thus, insulin resistance, itself, appears to be an endothelial dysfunction risk equivalent. The pathways are clearly intertwined with and sparked by obesity, in large part by excess adipokine production. The association of endothelial dysfunction with insulin resistance in the absence of overt diabetes or metabolic syndrome provides evidence that the atherosclerosis may actually begin earlier in the spectrum of insulin resistance, ultimately resulting in a progression of metabolic syndrome to prediabetes and then to T2DM [109,110] .
Adiponectin is a protein highly expressed in adipose tissue. Like leptin, adiponectin enhances insulin sensitivity through activation of AMPK [111] . Functional analyses including generation of adiponectin transgenic or knockout mice have revealed that adiponectin serves as an insulin-sensitizing adipokine. Obesity-linked downregulation of adiponectin is a mechanism that explains how obesity could cause insulin resistance and diabetes [112]. Adiponectin also affects hepatic glucose production by decreasing the mRNA expression of two essential gluconeogenesis enzymes: phosphoenolpyruvate carboxykinase and glucose-6-phosphatase. It appears that high-molecular-weight adiponectin may be the most insulin-sensitizing [112]. Adiponectin correlates with blood concentrations of free fatty acids and reduced body mass index or body weight, and may be the molecular link between obesity and insulin resistance, and may serve as a biomarker for the metabolic syndrome [113]. Interventions to reduce insulin resistance by increasing adiponectin concentrations may be effective particularly in obese, insulin-resistant individuals [113]. Plum treatment (plum juice) significantly increased plasma adiponectin concentrations and PPAR-γ mRNA expression in adipose tissue from Wistar fatty rats [114] . Decreased plasma adiponectin and insulin resistance coexist in subjects with prediabetes, diabetes and atherosclerosis [115].
Visfatin (also known as pre-B cell colony-enhancing factor or PBEF) is a newly discovered adipocyte hormone, highly expressed in visceral fat, with a direct relationship existing between plasma visfatin levels and T2- DM. Visfatin binds to the insulin receptor at a site distinct from that of insulin and causes hypoglycemia by reducing glucose release from liver cells and stimulating glucose utilization in adipocytes and myocytes [116]. Serum visfatin levels were reported to be elevated in type 2 diabetes independent of insulin resistance [117]. Increased visfatin levels have been shown to be associated with BMI and insulin resistance in obese children [118]. Visfatin is not related to insulin resistance either as assessed by a homeostasis model assessment or during lipid infusion [119] .
Lipocalin (LCN) 2 belongs to the lipocalin subfamily of low-molecular mass-secreted proteins that bind small hydrophobic molecules. LCN2 has been characterized recently as an adipose-derived cytokine, and its expression is upregulated in adipose tissue. LCN2 has been shown to play a critical role in the regulation of body fat mass, lipid metabolism, and insulin resistance, especially as it attenuates diet-induced insulin resistance [120] . LCN2 is associated with MMP-2 and MMP-9 activities as well as with pro-inflammatory markers, suggesting its potential involvement in the low-grade chronic inflamemation accompanying obesity [121] .
4.5. Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is characterized by hepatic steatosis in the absence of a history of significant alcohol use or other known liver diseases. Primary NAFLD emerges due to the metabolic syndrome. Compared with healthy controls, risk for steatosis is increased 4.6-fold in obese subjects. Subjects with NAFLD have a 2-fold greater risk of diabetes [122]. Free fatty acids (FFAs) play a pivotal role in the development of simple hepatic steatosis. The development of NAFLD is closely linked to an excess flow of FFAs arising from visceral adipose tissue. Multiple mechanisms including pro-inflammatory cytokines and pathways have been implicated in the pathogenesis of NAFLD. Understanding the role of obesity and lipotoxicity in patients with liver disease as part of a broader metabolic disorder is likely to improve the management of these challenging diseases. In a 11 year follow-up study, NAFLD with elevated aminotransaminase (ALT) levels was a risk factor for incident diabetes or the metabolic syndrome (MS) [123] . Insulin resistance is the basis of both NAFLD and metabolic syndrome [124,125] . Obesity results in marked enlargement of the intra-abdominal visceral fat depots. The development of insulin resistance leads to continuous lipolysis within these depots, releasing fatty acids into the portal circulation, where they are rapidly translocated to the liver and reassembled into triglycerides. Reactive oxygen species, generated in the liver from oxidation of fatty acids, are precipitating factors in the cascade of events leading from simple steatosis to NASH [32] . Circulating free fatty acids may be cytotoxic by inducing lipid peroxidation and hepatocyte apoptosis. Insulin resistance is often associated with chronic low-grade inflammation, and numerous mediators released from immune cells and adipocytes may contribute liver damage and liver disease progression [125].
5. BENEFITS OF WEIGHT LOSS IN TYPE 2 DIABETES AND OBESITY
Weight loss of 5% to 10% can significantly improve risk factors for obesity-related diseases [126]. Weight loss has been shown to improve glycemic control and cardio-vascular risk factors in obese individuals with type 2 diabetes. Clinical studies demonstrate that the therapeutic benefit rises with increasing weight loss, but that losses as low as 0.45 - 4 kg (1 - 9 lb) have positive effects on metabolic control, cardiovascular risk factors and mortality rates. The current data support a continued focus on weight loss, including moderate weight loss, as a key component of good care for overweight patients with type 2 diabetes [127]. The list of benefits is shown in Table 1. The most effective interventions include comprehensive behavioral management, dietary modifycation, exercise, pharmacotherapy and bariatric surgery. The most widely investigated drugs, sibutramine and orlistat, result in modest weight loss with demonstrable improvements in co-morbidities, among which is type 2 diabetes.
Nutrition in the management of obesity and type 2 diabetes: Observational and interventional studies have clearly shown that type 2 diabetes can be prevented by lifestyle measures, including reduced energy intake to induce a modest but sustained weight reduction, together with changes in diet composition [128]. Even in elderly individuals, diet induced weight loss results in improved insulin sensitivity and improved β-cell function [129]. In short-term [130] as well as long-term studies [131], use of a low carbohydrate diet in obese subjects with type 2 diabetes has improved glucose profiles, insulin sensitiveity and decreased plasma triglyceride and cholesterol levels.
5.1. Prevention of Type 2 Diabetes by Weight Reduction
Several interventional studies have demonstrated that significant weight reduction could lead to a decreased incidence of progression to type 2 diabetes.
Finnish study [132]: This study from Finland included middle-aged obese subjects with impaired glucose tolerance (IGT) who were randomized to receive either
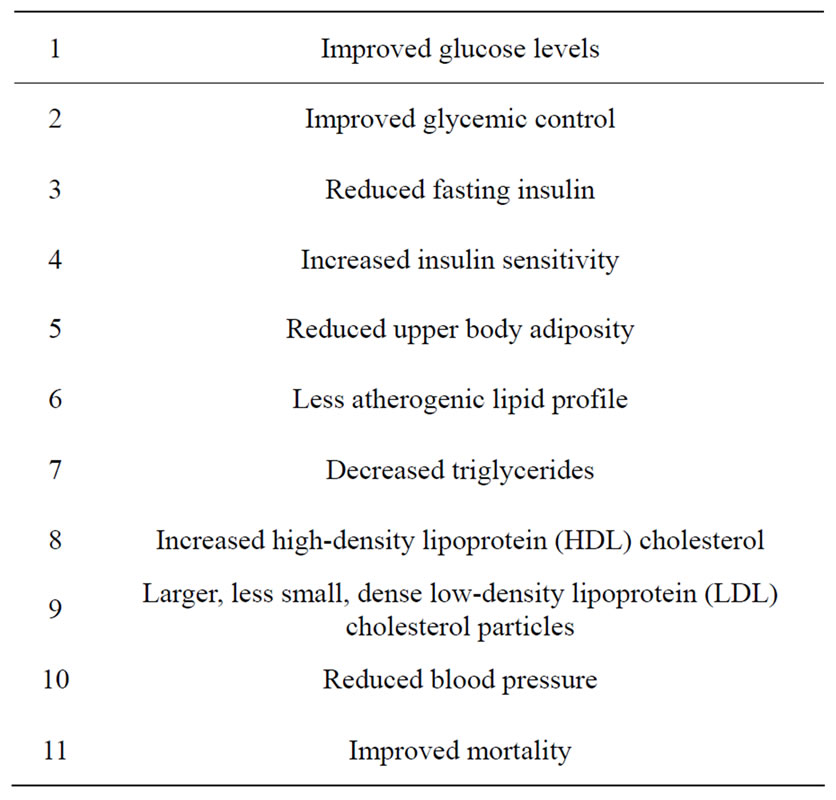
Table 1. Benefits of weight loss in type 2 diabetes.
brief diet and exercise counseling (control group) or intensive individualized instruction on weight reduction, food intake, and guidance on increasing physical activity (intervention group). After an average follow-up of 3.2 years, there was a 58% relative reduction in the incidence of diabetes in members of the intervention group compared with the control subjects.
Diabetes Prevention Program (DPP) [133]: This multicenter study enrolled subjects with impaired glucose tolerance who were slightly younger and more obese. Approximately 45% of study subjects were recruited from minority groups (e.g., African American, Hispanic) and 20% of subjects were aged ≥ 60 years. Subjects were randomized to one of three intervention groups: the intensive nutrition and exercise counseling (“lifestyle”) group, or either of two masked medication treatment groups, the metformin group or the placebo group. Partipants in both the placebo group and the metformin group received standard diet and exercise recommendations. After an average follow-up of 2.8 years, compared with the control group, a 58% relative reduction in the progression to diabetes was observed in the lifestyle group and a 31% relative reduction in the metformin group. On an average, 50% of the subjects in the lifestyle group achieved the goal of ≥7% weight reduction and about 74% maintained at least 150 min/week of moderately intense activity. During follow-up after DPP at 10 years, incidences in placebo and metformin groups fell to equal those in the former lifestyle group, but the cumulative incidence of diabetes remained lowest in the lifestyle group, indicating that prevention or delay of diabetes with lifestyle intervention or metformin can persist for at least 10 years [134] . Progression to diabetes is more common in women with a history of gestational diabetes (GDM) compared with those without a history of GDM, despite equivalent degrees of impaired glucose tolerance (IGT) at baseline. Both intensive lifestyle changes and metformin use are highly effective in delaying or preventing diabetes in women with IGT and a history of GDM [135] .
Da Qing study [136]: this study from China included subjects with IGT determined by oral glucose tolerance tests, who were randomized by clinic to a control group or to one of three active treatment groups: diet only, exercise only, or diet plus exercise. Subjects were followed biannually. After an average of 6 years’ follow-up, the diet, exercise, and diet plus exercise interventions were associated with 31%, 46% and 42% reductions in risk of developing type 2 diabetes, respectively.
Xenical in the prevention of Diabetes in Obese Subjects (XENDOS) study [137]: this study addressed the benefit of weight reduction achieved by the addition of orlistat to lifestyle change, to delay the progression of type 2 diabetes in a group with BMI ≥ 30 kg/m2 with or without IGT. After 4 years of treatment, the effect of orlistat addition corresponded to a 45% reduction in risk factors in the IGT group, with no effect observed in those without IGT [137].
Look AHEAD (Action for Health in Diabetes) [138] : This was a multicenter randomized study of 5145 participants randomized between 2001 and 2004, whose ethnicities paralleled the ethnic distribution of DM in the National Health and Nutrition Examination Survey (NHANES) 1999-2000 survey, with a mean age of 59 +/– 6.8 years (mean +/– SD), and of whom 60% were women. Furthermore, 65.0% of participants had a firstdegree relative with diabetes. Overall, BMI averaged 36 +/– 5.9 kg/m2 at baseline, with 83.6% of the men and 86.1% of women having a BMI > 30 kg/m2 and 17.9% of men and 25.4% of women having a BMI > 40 kg/m2. Intensive lifestyle intervention (ILI) participants had a greater percentage of sustained weight loss (–6.15% vs. –0.88%; P < 0.001) and improvements in fitness, glycemic control, and CVD risk factors in individuals with type 2 diabetes [139] .
In a recent Japanese randomized control trial to test the feasibility and effectiveness of a lifestyle intervenetion program in the primary care setting, in 30-60-year old subjects and BMI > 22.5 kg/m2, a significant improvement in insulin sensitivity was observed representing a significant reduction in the cumulative incidence of progression to diabetes [140].
5.2. Pharmacotherapy for Obesity and Improvement in Metabolic Risk Factors and Diabetes
Orlistat, a gastrointestinal lipase inhibitor drug, has been used effectively and safely in the treatment of obesity [141]. Orlistat significantly reduces body weight, and improves glycemic control and several cardiovascular risk factors in overweight and obese subjects with type 2 diabetes [142,143]. In type 2 diabetic patients, orlistat also attenuates postprandial increases in triglycerides, remnant-like particles, cholesterol, and free fatty acids [144]. The anti-hyperglycemic effect of orlistat has been attributed to a weight loss-associated decrease in insulin resistance [145] and augmentation of the postprandial increases in plasma levels of glucagon-like peptide 1 (GLP-1) [146].
RIO-Europe study [147]: rimonabant is a selective cannabinoid-1 receptor blocker with both central and peripheral actions [148]. A 20 mg/day dose of rimonabant, along with a low calorie diet, resulted in significant weight reduction and improvement in cardiovascular risk factors such as waist circumference, HDL cholesterol, triglycerides, insulin resistance and the incidences of metabolic syndrome.
Rimonabant in obesity-lipids study [149]: it has shown to reduce body weight and improve cardiovascular risk factors in obese patients, such as a reduction in waist circumference, increase in HDL cholesterol and reduction in triglycerides. In addition, rimonabant use at a daily dose of 20 mg also resulted in an increase in plasma adiponectin levels that was partly independent of weight loss alone.
Sibutramine: sibutramine is an anti-obesity drug that induces satiety and thermogenesis [150]. Sibutramine use has been shown to reduce weight, lower the levels of nonesterified fatty acids, decrease hyperinsulinemia, and reduce insulin resistance. It has been used as an effective adjunct to oral hypoglycemic therapy in obese subjects with type 2 diabetes [151]. However, the magnitude of weight loss was modest, and the long-term health benefits and safety remain unclear [152].
Pharmacotherapy for diabetes and weight reduction: Weight loss is an important therapeutic objective for individuals with type 2 diabetes [153]. Both short [154] and long-term [155] weight loss in overweight or obese type 2 diabetic subjects on very low calorie diets was shown to decrease insulin resistance, improve measures of glycemic control, improve lipid abnormalities and lower blood pressure [154,156]. Metformin, an oral hypoglycemic agent, decreases calorie intake in a dosedependent manner and leads to a reduction in body weight in subjects with type 2 diabetes and obesity [157- 159] . Exenatide: exenatide is a member of a new class of agents known as incretin mimetics currently in development for the treatment of type 2 diabetes. In shortterm studies, Exenatide improved glycemic control and helped to reduce body weight over 28 days in patients with type 2 diabetes treated with diet/exercise or metformin [160].
Bariatric surgery for obesity and effect on type 2 diabetes: bariatric surgery for severe obesity results in long-term weight loss, which leads to an improved lifestyle and recovery from diabetes [161,162], hypertriglyceridemia, low levels of high-density lipoprotein cholesterol, hypertension, and hyperuricemia [162-164] .
5.3. Potential Role of Adiponectin in the Treatment of Obesity, Diabetes and Insulin Resistance
Studies have shown that adiponectin administration in rodents has insulin-sensitizing, anti-atherogenic and antiinflammatory effects and under certain settings also decreases body weight. Therefore, adiponectin replacement in humans may represent a promising approach to prevent and/or treat obesity, insulin resistance and type 2 diabetes; however, clinical studies with adiponectin administration need to be conducted to confirm this hypothesis.
6. PHARMACOGENETICS: POTENTIAL ROLE IN THE TREATMENT OF DIABETES AND OBESITY
The prevalence of obesity and diabetes, which are heritable traits that arise from the interactions of multiple genes and lifestyle factors, continues to rise worldwide. Until recently, candidate gene and genome-wide linkage studies have been the main genetic epidemiological approaches to identify genetic loci for obesity and diabetes, yet progress has been slow, with limited success. Recent advances have transformed the situation and there has been progress in understanding how genetic variation predisposes individuals to diabetes and obesity, and how candidate genes may alter drug response. The discovery of causal genes includes family-based linkage analyses and focused candidate-gene studies; among them, largescale surveys of association between common DNA sequence variants and disease were most successful. The current total of approximately 40 confirmed type 2 diabetes loci includes variants in or near WFS1 (wolframin) and the hepatocyte nuclear factors HNF1A and HNF1B (genes that also harbor rare mutations responsible for monogenic forms of diabetes) [165-168] ; the melatoninreceptor gene MTNR1B (which highlights the link between circadian and metabolic regulation) [169-171] ; and IRS1 (encoding insulin-receptor substrate 1), one of a limited number of type 2 diabetes loci with a primary effect on insulin action rather than on secretion [172] . Genetic discoveries have provided a molecular basis for the clinically useful classification of monogenic forms of diabetes and obesity [173,174] . Genomewide association studies of population-based samples undertaken to examine the full range of BMI values have identified approximately 30 loci influencing BMI and the risk of obesity. The strongest signal remains the association with variants within FTO (the fat-mass and obesity–related gene) [171,175-177] . Other signals near BDNF, SH2B1, and NEGR1 (all implicated in aspects of neuronal function) reinforce the view of obesity as a disorder of hypothalamic function [178-181] . There are insufficient genetic data to support management decisions for common forms of type 2 diabetes and obesity [182]. Although the TCF7L2 genotype variants influence therapeutic response to sulfonylureas but not metformin [183], the effect is too modest to guide the care of individual patients. Three large genome-wide association studies on obesity, together involving more than 150,000 individuals, were published in Nature Genetics last year. The results suggested the involvement of a large number of genetic variants in disease susceptibility and have identified 19 loci for common obesity and 18 for common type 2 diabetes. The combined contribution of these loci to the variation in obesity and diabetes risk is small and their predictive value is typically low. One of these loci, variants in the fat-mass and obesity-associated gene (FTO), influences susceptibility to type 2 diabetes via an effect on adiposity/obesity [184]. The EPIC-Norfolk study is a population-based, ethnically homogeneous, white European cohort study of 25,631 residents living in the city of Norwich, United Kingdom, and its surrounding area. Of these, 12,201 had complete genotype data for all 12 single nucleotide polymorphisms (SNPs). The FTO locus represented the largest [185]. Variants that predispose to common obesity also result in altered susceptibility to PCOS, probably mediated through adiposity [200]. One single-nucleotide polymorphism (SNP) associated with weight is located close to monoacylglycerol acyltransferase 1 (MGAT1), the MGAT enzyme family known to be involved in dietary fat absorption [186] . Genetic studies offer two main avenues for clinical translation. First, the identification of new pathways involved in disease predisposition-for example, those influencing zinc transport and pancreatic islet regeneration in the case of type 2 diabetes-offers opportunities for development of novel therapeutic and preventive approaches. Second, with continuing efforts to identify additional genetic variants, it may become possible to use patterns of predisposition to tailor individual management of these conditions.
7. BARIATRIC SURGERY FOR OBESITY AND IMPROVEMENT IN DIABETES CONTROL
Bariatric surgery as a modality to treat obesity in the US is reserved for patients with BMI ≥ 35 kg/m2 and the presence of serious co-morbidities (T2DM, moderate or severe obstructive sleep apnea (OSA), pseudotumor cerebri, and severe steatohepatitis), or BMI > 40
kg/m2 and the presence of serious co-morbidities (T2DM, moderate or severe obstructive sleep apnea (OSA), pseudotumor cerebri, and severe steatohepatitis), or BMI > 40 kg/m2 and minor comorbidities (mild OSA, hypertension (HTN), insulin resistance, glucose intolerance, dyslipidemia, impaired quality of life, or activities of daily living). Gastric bypass surgery appears to have significant increased therapeutic potential for treating obesity and Type 2 diabetes. In view of the growing enthusiasm for surgical interventions to treat T2DM, the first diabetes surgery summit (DSS) was held in Rome in March 2007. Trends in mortality in bariatric surgery were reported by Buchwald et al. in 2007 [187] in a systemic review that included meta-analysis of 361 studies and a total of 85,048 patients with a mean BMI of 47.4
kg/m2 and minor comorbidities (mild OSA, hypertension (HTN), insulin resistance, glucose intolerance, dyslipidemia, impaired quality of life, or activities of daily living). Gastric bypass surgery appears to have significant increased therapeutic potential for treating obesity and Type 2 diabetes. In view of the growing enthusiasm for surgical interventions to treat T2DM, the first diabetes surgery summit (DSS) was held in Rome in March 2007. Trends in mortality in bariatric surgery were reported by Buchwald et al. in 2007 [187] in a systemic review that included meta-analysis of 361 studies and a total of 85,048 patients with a mean BMI of 47.4 kg/m2. The early and late mortality rates after bariatric surgery were reported as low.
kg/m2. The early and late mortality rates after bariatric surgery were reported as low.
A review by Cunneen reported that all studies reporting on co-morbidities showed significant resolution or improvement of type 2 diabetes mellitus ([T2DM] 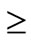 60%), hypertension (
60%), hypertension (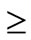 43%), and dyslipidemia (
43%), and dyslipidemia (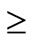 70%). One meta-analysis study reported that surgery was found to be superior to medical therapy in resolving T2DM, hypertension, and dyslipidemia. Sleep apnea was significantly resolved/improved in
70%). One meta-analysis study reported that surgery was found to be superior to medical therapy in resolving T2DM, hypertension, and dyslipidemia. Sleep apnea was significantly resolved/improved in  85% across procedures in the one meta-analysis that addressed this co-morbidity [188]. Studies have shown that those who undergo bariatric surgery for obese diabetic patients experience complete remission of diabetes, maintaining euglycemia without medications for more than 10 years [189]. Additionally, following some gastrointestinal (GI) procedures, T2DM resolves within days to weeks, long before the occurrence of major weight loss. T2DM resolution or remission has usually been defined as HbA1C values ranging from <6% to <7% in the absence of antidiabetic medications. Meta-analysis of bariatric surgery by Buchwald et al. [190] included 136 studies for a total of 22,094 patients; mean baseline BMI was 46.9
85% across procedures in the one meta-analysis that addressed this co-morbidity [188]. Studies have shown that those who undergo bariatric surgery for obese diabetic patients experience complete remission of diabetes, maintaining euglycemia without medications for more than 10 years [189]. Additionally, following some gastrointestinal (GI) procedures, T2DM resolves within days to weeks, long before the occurrence of major weight loss. T2DM resolution or remission has usually been defined as HbA1C values ranging from <6% to <7% in the absence of antidiabetic medications. Meta-analysis of bariatric surgery by Buchwald et al. [190] included 136 studies for a total of 22,094 patients; mean baseline BMI was 46.9 kg/m2 (32.3 - 68.8). The studies that reported resolution of T2DM included a total of 1846 patients. Diabetes resolution rates were 98.9% after biliopancreatic diversion (BPD), 83.7% after RYGB and 47.9% after AGB. Another systematic review by Levy et al. [191]. confirmed that bariatric surgery was highly effective in obtaining weight reduction in morbidly obese patients with losses of up to 60% of the excess weight, along with resolution of preoperative diabetes in more than 75% of the cases.
kg/m2 (32.3 - 68.8). The studies that reported resolution of T2DM included a total of 1846 patients. Diabetes resolution rates were 98.9% after biliopancreatic diversion (BPD), 83.7% after RYGB and 47.9% after AGB. Another systematic review by Levy et al. [191]. confirmed that bariatric surgery was highly effective in obtaining weight reduction in morbidly obese patients with losses of up to 60% of the excess weight, along with resolution of preoperative diabetes in more than 75% of the cases.
Pharmacotherapy for Diabetes that Helps Weight Reduction
Pramlintide is an analog of amylin, a naturally occurring hormone produced by pancreatic β-cells. The major mechanism of action appears to be inhibition of gastric emptying and suppression of glucagon release. Clinically, it also suppresses appetite in patients who receive it.
Glucagon-like peptide-1 (GLP-1) is recognized as an important regulator of glucose homeostasis. Exenatide and liraglutide are analogs of GLP-1, a naturally occurring incretin produced by the L-cells of the distal ileum. GLP-1 stimulates insulin release from the pancreatic β- cells, suppresses glucagon release from the pancreatic α- cells, slows gastric emptying, and acts on the brain to increase satiety. Increases in GLP-1 may be responsible for some of the weight loss following roux-en-Y gastric bypass surgery in patients with type 2 diabetes [192] . The improvement in overall glucose control has been modest in clinical trials, at around 0.3%. However, those using the medication have also experienced weight reduction of ~1 - 1.5 kg in patients with type 1 diabetes and ~2.0 - 2.5 kg in patients with type 2 diabetes. Administration of exenatide in patients with type 2 diabetes has similar effects. Clinically, the result is an A1C reducetion of ~1%. Preliminary studies suggest that a signifycant proportion of insulin-treated patients with type 2 diabetes may be successfully transitioned from insulin to Exenatide in addition to their oral agents. Most patients experience significant weight loss of ~2.5 kg when Exenatide is used in addition to metformin and ~1 kg when it is added to a sulfonylurea [193,194] . Exenatide was associated with a significant reduction in mean (SD) body weight from baseline (–2.1 [0.2] kg), with progressive reductions after 2 years (–4.7 [0.3] kg; P < 0.001 vs baseline) [195] .
Liraglutide works in a manner similar to that of Exenatide but has a longer half-life, which allows for once daily (rather than twice daily) dosing. Some studies, including one meta-analysis, suggest that it may have a slightly greater A1C-lowering effect than Exenatide, although more investigation is warranted to substantiate such findings. In a meta-analysis, it is noted that exenatide and liraglutide resulted in greater weight loss (from 2.3 to 5.5 kg) with improvements in HbA1C similar to that obtained with sufonylureas [196]. Neither exenatide nor liraglutide are indicated for simple weight loss.
8. MULTIPLE RISK FACTORS FOR CARDIOVASCULAR DISEASE AND DIABETES MELLITUS
The metabolic syndrome is a constellation of central adiposity, impaired fasting glucose, elevated blood pressure, and dyslipidemia (high triglyceride and low HDL cholesterol). When three of these five criteria are present, the risk of cardiovascular disease and diabetes is increased 1.5- to 2-fold [197,198] .
8.1. Obesity and Co-Morbidities
Obesity is becoming a major public health problem throughout the world and is associated with significant, potentially life-threatening co-morbidities. Either obesity itself or the co-morbidities that accompany obesity are responsible for increased cardiovascular risk. Obesity is associated with most of the components of metabolic syndrome, the leading cause of type 2 diabetes. The comorbidities of obesity and type 2 diabetes associated with insulin resistance syndrome include obstructive sleep apnea, hypertension, polycystic ovary syndrome, nonalcoholic fatty liver disease and certain forms of cancer.
8.2. Impact of Diabetes and Obesity on Cardiovascular Disease
Cardiovascular disease (CVD) is a major cause of morbidity and mortality among subjects with type 2 diabetes and is responsible for up to 75% of deaths among them. The risk of CVD mortality in type 2 diabetic patients is more than double compared with that in agematched subjects [199]. The risk of coronary artery disease (CAD) in subjects with type 2 diabetes is considered equivalent to that of nondiabetic subjects who have CAD [200,201], especially women [202]. The association of obesity with clinically significant coronary artery disease is blatant in two classical prospective studies highly consulted: the Framingham Heart Study [203] and the Nurses Health Study [204]. Elevated pro-inflammatory cytokine levels found in obese patients relate mainly to obesity rather than to T2DM. Moreover, surgery-induced weight loss reduces circulating concentrations of key pro-inflammatory factors, which contriute to the improvement in the cardiovascular co-morbidity following excess weight loss [205] . Obesity and overweight are often defined by WHO (World Health Organization) in terms of excess weight for a given height [1206] Overweight was defined as a body mass index (BMI) of 25.0 to 29.9 and obesity as a BMI of >30.0. BMI, calculated by weight (kg)/height (m2) and adjusted for height is used as a measure of weight standards.
9. CONCLUSIONS
Obesity has become an epidemic worldwide. The development of obesity and diabetes involves complex genetic and environmental factors. Diabetes is fastest growing disease in the world. The health consequences and economic costs of the overweight, obesity and type 2 diabetes epidemics are enormous. Behavioral changes leading to increased body weight is a major contributing factor to the rising incidence of diabetes.
10. ACKNOWLEDGEMENTS
Dr. Yaturu receives salary support from Veterans Health Administration.
REFERENCES
- WHO Technical Series (2000) Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. World Health Organization Technical Report Series, 894, 1-253.
- Guh, D.P., et al. (2009) The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health, 9, 88. doi:10.1186/1471-2458-9-88
- Flegal, K.M. and Troiano, R.P. (2000) Changes in the distribution of body mass index of adults and children in the US population. International Journal of Obesity and Related Metabolic Disorders, 24, 807-818. doi:10.1038/sj.ijo.0801232
- Franco, M., et al. (2010) Prevention of childhood obesity in Spain: A focus on policies outside the health sector. SESPAS Report 2010. Gaceta Sanitaria, 24, 49-55.
- Nguyen, N.T., et al. (2008) Association of hypertension, diabetes, dyslipidemia, and metabolic syndrome with obesity: Findings from the National Health and Nutrition Examination Survey, 1999 to 2004. Journal of the American College of Surgeons, 207, 928-934. doi:10.1016/j.jamcollsurg.2008.08.022
- Cefalu, W.T. (2001) Insulin resistance: Cellular and clinical concepts. Experimental Biology and Medicine (Maywood), 226, 13-26.
- Reaven, G. (2004) The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinology Metabolism Clinics of North America, 33, 283-303. doi:10.1016/j.ecl.2004.03.002
- Erkelens, D.W. (2001) Insulin resistance syndrome and type 2 diabetes mellitus. American Journal of Cardiology, 88, 38J-42J. doi:10.1016/S0002-9149(01)01883-5
- Colditz, G.A., et al. (1990) Weight as a risk factor for clinical diabetes in women. American Journal of Epidemiology, 132, 501-513.
- Ni Mhurchu, C., et al. (2006) Body mass index and risk of diabetes mellitus in the Asia-Pacific region. Asia Pacific Journal of Clinical Nutrition, 15, 127-133.
- Schienkiewitz, A., et al. (2006) Body mass index history and risk of type 2 diabetes: Results from the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. The American Journal of Clinical Nutrition, 84, 427-433.
- Harris, M.I., et al. (1998) Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988-1994. Diabetes Care, 21, 518-524. doi:10.2337/diacare.21.4.518
- Cowie, C.C., et al. (2006) Prevalence of diabetes and impaired fasting glucose in adults in the US population: National Health and Nutrition Examination Survey, 1999-2002. Diabetes Care, 29, 1263-1268. doi:10.2337/dc06-0062
- Snijder, M.B., et al. (2003) Associations of hip and thigh circumferences independent of waist circumference with the incidence of type 2 diabetes: The Hoorn study. The American Journal of Clinical Nutrition, 77, 1192-1197.
- Cassano, P.A., et al. (1992) Obesity and body fat distribution in relation to the incidence of non-insulin-dependent diabetes mellitus. A prospective cohort study of men in the normative aging study. American Journal of Epidemiology, 136, 1474-1486.
- Lundgren, H., et al. (1989) Adiposity and adipose tissue distribution in relation to incidence of diabetes in women: Results from a prospective population study in Gothenburg, Sweden. International Journal of Obesity, 13, 413- 423.
- Ohlson, L.O., et al. (1985) The influence of body fat distribution on the incidence of diabetes mellitus. 13.5 years of follow-up of the participants in the study of men born in 1913. Diabetes, 34, 1055-1058. doi:10.2337/diabetes.34.10.1055
- Qiao, Q. and Nyamdorj, R. (2010) Is the association of type II diabetes with waist circumference or waist-to-hip ratio stronger than that with body mass index? European Journal of Clinical Nutrition, 64, 30-34. doi:10.1038/ejcn.2009.93
- Aucott, L.S. (2008) Influences of weight loss on longterm diabetes outcomes. Proceedings of the Nutrition Society, 67, 54-59. doi:10.1017/S0029665108006022
- Resnick, H.E., et al. (2000) Relation of weight gain and weight loss on subsequent diabetes risk in overweight adults. Journal of Epidemiology & Community Health, 54, 596-602. doi:10.1136/jech.54.8.596
- Mokdad, A.H., et al. (2000) Diabetes trends in the US: 1990-1998. Diabetes Care, 23, 1278-1283. doi:10.2337/diacare.23.9.1278
- Janssen, I., Katzmarzyk, P.T. and Ross, R. (2002) Body mass index, waist circumference, and health risk: Evidence in support of current National Institutes of Health guidelines. Archives of Internal Medicine, 162, 2074- 2079. doi:10.1001/archinte.162.18.2074
- Liu, J., et al. (2010) Impact of abdominal visceral and subcutaneous adipose tissue on cardiometabolic risk factors: The Jackson Heart Study. The Journal of Clinical Endocrinology & Metabolism, 95, 5419-5426. doi:10.1210/jc.2010-1378
- Roelants, M., Hauspie, R. and Hoppenbrouwers, K. (2009) References for growth and pubertal development from birth to 21 years in Flanders, Belgium. Annals of Human Biology, 36, 680-694. doi:10.3109/03014460903049074
- De Onis, M., et al. (2009) WHO growth standards for infants and young children. Archives of Pediatrics & Adolescent Medicine, 16, 47-53. doi:10.1016/j.arcped.2008.10.010
- Rolland-Cachera, M.F., et al. (1982) Adiposity indices in children. The American Journal of Clinical Nutrition, 36, 178-184.
- Cole, T.J., et al. (2007) Body mass index cut offs to define thinness in children and adolescents: International survey. BMJ, 335, 194. doi:10.1136/bmj.39238.399444.55
- Tfayli, H. and Arslanian, S. (2009) Pathophysiology of type 2 diabetes mellitus in youth: The evolving chameleon. Arquivos Brasileiros de Endocrinologia & Metabologia, 53, 165-174. doi:10.1590/S0004-27302009000200008
- Weiss, R. and Caprio, S. (2005) The metabolic consequences of childhood obesity. Best Practice & Research: Clinical Endocrinology & Metabolism, 19, 405-419. doi:10.1016/j.beem.2005.04.009
- Aboul Ella, N.A., et al. (2010) Prevalence of metabolic syndrome and insulin resistance among Egyptian adolescents 10 to 18 years of age. Journal of Clinical Lipidology, 4, 185-195. doi:10.1016/j.jacl.2010.03.007
- Holst-Schumacher, I., et al. (2009) Components of the metabolic syndrome among a sample of overweight and obese Costa Rican schoolchildren. Food and Nutrition Bulletin, 30, 161-170.
- Verna, E.C. and Berk, P.D. (2008) Role of fatty acids in the pathogenesis of obesity and fatty liver: Impact of bariatric surgery. Seminars in Liver Disease, 28, 407-426. doi:10.1055/s-0028-1091985
- Hoppin, A.G. (2004) Obesity and the liver: Developmental perspectives. Seminars in Liver Disease, 24, 381-387. doi:10.1055/s-2004-860867
- Whitlock, E.P., et al. (2005) Screening and interventions for childhood overweight: A summary of evidence for the US Preventive Services Task Force. Pediatrics, 116, e125-e44. doi:10.1542/peds.2005-0242
- Barlow, S.E. and Dietz, W.H. (1998) Obesity evaluation and treatment: Expert Committee recommendations. The Maternal and Child Health Bureau, Health Resources and Services Administration and the Department of Health and Human Services. Pediatrics, 102, E29. doi:10.1542/peds.102.3.e29
- Thorn, J., et al. (2010) Overweight among four-year-old children in relation to early growth characteristics and socioeconomic factors. Journal of Obesity, Article ID: 580642. doi:10.1155/2010/580642
- Hermann, G.M., et al. (2009) Neonatal catch up growth increases diabetes susceptibility but improves behavioral and cardiovascular outcomes of low birth weight male mice. Pediatric Research, 66, 53-58. doi:10.1203/PDR.0b013e3181a7c5fd
- Bhargava, S.K., et al. (2004) Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. The New England Journal of Medicine, 350, 865-875. doi:10.1056/NEJMoa035698
- Forsen, T., et al. (2000) The fetal and childhood growth of persons who develop type 2 diabetes. Annals of Internal Medicine, 133, 176-182.
- Yajnik, C.S. (2004) Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. Journal of Nutrition, 134, 205-210.
- Jimenez-Chillaron, J.C. and Patti, M.E. (2007) To catch up or not to catch up: Is this the question? Lessons from animal models. Current Opinion in Endocrinology, Diabetes and Obesity, 14, 23-29. doi:10.1097/MED.0b013e328013da8e
- Jimenez-Chillaron, J.C., et al. (2006) Reductions in caloric intake and early postnatal growth prevent glucose intolerance and obesity associated with low birthweight. Diabetologia, 49, 1974-1984. doi:10.1007/s00125-006-0311-7
- Kaiser, N. and Leibowitz, G. (2009) Failure of beta-cell adaptation in type 2 diabetes: Lessons from animal models. Frontiers in Bioscience, 14, 1099-1115. doi:10.2741/3296
- Lowell, B.B. and Shulman, G.I. (2005) Mitochondrial dysfunction and type 2 diabetes. Science, 307, 384-387. doi:10.1126/science.1104343
- Kahn, S.E. (2001) Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. The Journal of Clinical Endocrinology & Metabolism, 86, 4047-4058. doi:10.1210/jc.86.9.4047
- DeFronzo, R.A. (1988) Lilly lecture 1987. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes, 37, 667-687.
- Reaven, G.M. (1995) Pathophysiology of insulin resistance in human disease. Physiological Reviews, 75, 473- 486.
- Ali, A.T., et al. (2011) Insulin resistance in the control of body fat distribution: A new hypothesis. Hormone metabolism Research, 43, 77-80.
- Wiklund, P., et al. (2008) Abdominal and gynoid fat mass are associated with cardiovascular risk factors in men and women. The Journal of Clinical Endocrinology & Metabolism, 93, 4360-4366. doi:10.1210/jc.2008-0804
- Yamashita, S., et al. (1996) Insulin resistance and body fat distribution. Diabetes Care, 19, 287-291. doi:10.2337/diacare.19.3.287
- Kaplan, N.M. (1989) The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Archives of Internal Medicine, 149, 1514- 1520. doi:10.1001/archinte.149.7.1514
- Frayn, K.N. (2000) Visceral fat and insulin resistance— causative or correlative? British Journal of Nutrition, 83, S71-S77. doi:10.1017/S0007114500000982
- Gallagher, D., et al. (2009) Adipose tissue distribution is different in type 2 diabetes. The American Journal of Clinical Nutrition, 89, 807-814. doi:10.3945/ajcn.2008.26955
- Sanchez-Castillo, C.P., et al. (2005) Diabetes and hypertension increases in a society with abdominal obesity: Results of the Mexican National Health Survey 2000. Public Health Nutrition, 8, 53-60. doi:10.1079/PHN2005659
- Merino-Ibarra, E., et al. (2005) Ultrasonography for the evaluation of visceral fat and the metabolic syndrome. Metabolism, 54, 1230-1235. doi:10.1016/j.metabol.2005.04.009
- Hayashi, T., et al. (2008) Visceral adiposity, not abdominal subcutaneous fat area, is associated with an increase in future insulin resistance in Japanese Americans. Diabetes, 57, 1269-1275. doi:10.2337/db07-1378
- Nyholm, B., et al. (2004) Evidence of increased visceral obesity and reduced physical fitness in healthy insulin-resistant first-degree relatives of type 2 diabetic patients. European Journal of Endocrinology, 150, 207-214. doi:10.1530/eje.0.1500207
- Vettor, R., et al. (2005) Review article: Adipocytokines and insulin resistance. Alimentary Pharmacology & Therapeutics, 22, 3-10. doi:10.1111/j.1365-2036.2005.02587.x
- Aylin, P., Williams, S. and Bottle, A. (2005) Obesity and type 2 diabetes in children, 1996-1997 to 2003-2004. BMJ, 331, 1167. doi:10.1136/bmj.331.7526.1167
- Lee, S., Guerra, N. and Arslanian, S. (2010) Skeletal muscle lipid content and insulin sensitivity in black versus white obese adolescents: Is there a race differential? The Journal of Clinical Endocrinology & Metabolism, 95, 2426-2432. doi:10.1210/jc.2009-2175
- Chiarelli, F. and Marcovecchio, M.L. (2008) Insulin resistance and obesity in childhood. European Journal of Endocrinology, 159, S67-S74. doi:10.1530/EJE-08-0245
- Bacha, F., et al. (2003) Obesity, regional fat distribution, and syndrome X in obese black versus white adolescents: Race differential in diabetogenic and atherogenic risk factors. The Journal of Clinical Endocrinology & Metabolism, 88, 2534-2540. doi:10.1210/jc.2002-021267
- Goran, M.I., Bergman, R.N. and Gower, B.A. (2001) Influence of total vs. visceral fat on insulin action and secretion in African American and white children. Obesity Research, 9, 423-431. doi:10.1038/oby.2001.56
- Caprio, S. and Tamborlane, W.V. (1999) Metabolic impact of obesity in childhood. Endocrinology and Metabolism Clinics of North America, 28, 731-747. doi:10.1016/S0889-8529(05)70099-2
- Gower, B.A., Nagy, T.R. and Goran, M.I. (1999) Visceral fat, insulin sensitivity, and lipids in prepubertal children. Diabetes, 48, 1515-1521. doi:10.2337/diabetes.48.8.1515
- Abrams, P. and Levitt Katz, L.E. (2011) Metabolic effects of obesity causing disease in childhood. Current Opinion in Endocrinology, Diabetes and Obesity, 18, 23-27. doi:10.1097/MED.0b013e3283424b37
- Weiss, R. and Caprio, S. (2006) Altered glucose metabolism in obese youth. Pediatric Endocrinology Reviews, 3, 233-238.
- Lee, S., et al. (2006) Racial differences in adiponectin in youth: Relationship to visceral fat and insulin sensitivity. Diabetes Care, 29, 51-56. doi:10.2337/diacare.29.01.06.dc05-0952
- Bacha, F., et al. (2004) Adiponectin in youth: Relationship to visceral adiposity, insulin sensitivity, and beta-cell function. Diabetes Care, 27, 547-552. doi:10.2337/diacare.27.2.547
- Rasmussen-Torvik, L.J., et al. (2009) Influence of waist on adiponectin and insulin sensitivity in adolescence. Obesity (Silver Spring), 17, 156-161. doi:10.1038/oby.2008.482
- Timmers, S., Schrauwen, P. and de Vogel, J. (2008) Muscular diacylglycerol metabolism and insulin resistance. Physiology & Behavior, 94, 242-251. doi:10.1016/j.physbeh.2007.12.002
- Boden, G. and Shulman, G.I. (2002) Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. European Journal of Clinical Investigation, 32, 14-23. doi:10.1046/j.1365-2362.32.s3.3.x
- Yu, C., et al. (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of Biological Chemistry, 277, 50230-50236. doi:10.1074/jbc.M200958200
- Mittelman, S.D., et al. (2002) Extreme insulin resistance of the central adipose depot in vivo. Diabetes, 51, 755- 761. doi:10.2337/diabetes.51.3.755
- Griffin, M.E., et al. (1999) Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes, 48, 1270-1274. doi:10.2337/diabetes.48.6.1270
- Brunzell, J.D. and Ayyobi, A.F. (2003) Dyslipidemia in the metabolic syndrome and type 2 diabetes mellitus. American Journal of Medicine, 115, 24S-28S. doi:10.1016/j.amjmed.2003.08.011
- Purnell, J.Q., et al. (2000) Effect of weight loss with reduction of intra-abdominal fat on lipid metabolism in older men. The Journal of Clinical Endocrinology & Metabolism, 85, 977-982. doi:10.1210/jc.85.3.977
- Barrett, J.F. (2001) Targeting DNA gyrase. Expert Opinion on Therapeutic Targets, 5, 531-533. doi:10.1517/14728222.5.4.531
- Koo, S.H. and Montminy, M. (2006) Fatty acids and insulin resistance: A perfect storm. Molecular Cell, 21, 449-450. doi:10.1016/j.molcel.2006.02.001
- Ginsberg, H.N. and Stalenhoef, A.F. (2003) The metabolic syndrome: Targeting dyslipidaemia to reduce coronary risk. Journal of Cardiovascular Risk, 10, 121-128. doi:10.1097/00043798-200304000-00007
- Ginsberg, H.N. (2000) Insulin resistance and cardiovascular disease. The Journal of Clinical Investigation, 106, 453-458. doi:10.1172/JCI10762
- Boden, G. (2001) Free fatty acids-the link between obesity and insulin resistance. Endocrine Practice, 7, 44-51.
- Boden, G. (2003) Effects of free fatty acids (FFA) on glucose metabolism: Significance for insulin resistance and type 2 diabetes. Experimental and Clinical Endocrinology & Diabetes, 111, 121-124. doi:10.1055/s-2003-39781
- Lam, T.K., et al. (2002) Free fatty acid-induced hepatic insulin resistance: A potential role for protein kinase C-delta. American Journal of Physiology—Endocrinology and Metabolism, 283, E682-E691.
- Boden, G., et al. (2002) FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. American Journal of Physiology―Endocrinology and Metabolism, 283, E12-E19.
- Basu, R., et al. (2005) Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes, 54, 1942-1948. doi:10.2337/diabetes.54.7.1942
- Basu, R., et al. (2005) Obesity and type 2 diabetes do not alter splanchnic cortisol production in humans. The Journal of Clinical Endocrinology & Metabolism, 90, 3919-3926. doi:10.1210/jc.2004-2390
- Boden, G. (1997) Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes, 46, 3-10. doi:10.2337/diabetes.46.1.3
- Bonadonna, R.C., et al. (1990) Obesity and insulin resistance in humans: A dose-response study. Metabolism, 39, 452-459. doi:10.1016/0026-0495(90)90002-T
- Kolterman, O.G., et al. (1980) Mechanisms of insulin resistance in human obesity: Evidence for receptor and postreceptor defects. The Journal of Clinical Investigation, 65, 1272-1284. doi:10.1172/JCI109790
- Jensen, M.D. (2008) Role of body fat distribution and the metabolic complications of obesity. The Journal of Clinical Endocrinology & Metabolism, 93, S57-S63. doi:10.1210/jc.2008-1585
- Adiels, M., et al. (2008) Overproduction of very lowdensity lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, 1225-1236. doi:10.1161/ATVBAHA.107.160192
- Taskinen, M.R. (2005) Type 2 diabetes as a lipid disorder. Current Molecular Medicine, 5, 297-308. doi:10.2174/1566524053766086
- Wolfrum, C. and Stoffel, M. (2006) Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metabolism, 3, 99-110. doi:10.1016/j.cmet.2006.01.001
- Kamagate, A. and Dong, H.H. (2008) FoxO1 integrates insulin signaling to VLDL production. Cell Cycle, 7, 3162-3170. doi:10.4161/cc.7.20.6882
- Sparks, J.D. and Sparks, C.E. (2008) Overindulgence and metabolic syndrome: Is FoxO1 a missing link? The Journal of Clinical Investigation, 118, 2012-2015.
- Zhao, Y.F., Feng, D.D. and Chen, C. (2006) Contribution of adipocyte-derived factors to beta-cell dysfunction in diabetes. The International Journal of Biochemistry & Cell Biology, 38, 804-819. doi:10.1016/j.biocel.2005.11.008
- Antuna-Puente, B., et al. (2008) Adipokines: the missing link between insulin resistance and obesity. Diabetes & Metabolism, 34, 2-11. doi:10.1016/j.diabet.2007.09.004
- Kralisch, S., et al. (2007) Adipokines in diabetes and cardiovascular diseases. Minerva Endocrinologica, 32, 161-171.
- Arner, P. (2005) Insulin resistance in type 2 diabetes—role of the adipokines. Current Molecular Medicine, 5, 333-339. doi:10.2174/1566524053766022
- Considine, R.V., et al. (1996) Serum immunoreactiveleptin concentrations in normal-weight and obese humans. The New England Journal of Medicine, 334, 292-295. doi:10.1056/NEJM199602013340503
- Minokoshi, Y., et al. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature, 415, 339-343. doi:10.1038/415339a
- Greenfield, J.R. and Campbell, L.V. (2006) Relationship between inflammation, insulin resistance and type 2 diabetes: “Cause or effect”? Current Diabetes Reviews, 2, 195-211. doi:10.2174/157339906776818532
- Hotamisligil, G.S. and Spiegelman, B.M. (1994) Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes, 43, 1271-1278. doi:10.2337/diabetes.43.11.1271
- Kim, J.H., Bachmann, R.A. and Chen, J. (2009) Interleukin-6 and insulin resistance. Vitam Horm, 80, 613-633. doi:10.1016/S0083-6729(08)00621-3
- Nieto-Vazquez, I., et al. (2008) Insulin resistance associated to obesity: The link TNF-alpha. Archives of Physiology and Biochemistry, 114, 183-194. doi:10.1080/13813450802181047
- Lorenzo, M., et al. (2008) Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. Journal of Animal Science, 86, E94-E104. doi:10.2527/jas.2007-0462
- Feinstein, R., et al. (1993) Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. The Journal of Biological Chemistry, 268, 26055-26058.
- Hartge, M.M., Unger, T. and Kintscher, U. (2007) The endothelium and vascular inflammation in diabetes. Diabetes and Vascular Disease Research, 4, 84-88. doi:10.3132/dvdr.2007.025
- Hsueh, W.A. and Quinones, M.J. (2003) Role of endothelial dysfunction in insulin resistance. American Journal of Cardiology, 92, 10J-17J. doi:10.1016/S0002-9149(03)00611-8
- Yamauchi, T., et al. (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature Medicine, 8, 1288- 1295. doi:10.1038/nm788
- Kadowaki, T. and Yamauchi, T. (2005) Adiponectin and adiponectin receptors. Endocrine Reviews, 26, 439-451. doi:10.1210/er.2005-0005
- Schondorf, T., et al. (2005) Biological background and role of adiponectin as marker for insulin resistance and cardiovascular risk. Clinical Laboratory, 51, 489-494.
- Utsunomiya, H., et al. (2005) Anti-hyperglycemic effects of plum in a rat model of obesity and type 2 diabetes, Wistar fatty rat. Biomedical Research, 26, 193-200. doi:10.2220/biomedres.26.193
- Yaturu, S., Bridges, J.F. and Subba Reddy, D.R. (2006) Decreased levels of plasma adiponectin in prediabetes, Type 2 diabetes and coronary artery disease. Medical Science Monitor, 12, CR17-CR20.
- Adeghate, E. (2008) Visfatin: Structure, function and relation to diabetes mellitus and other dysfunctions. Current Medicinal Chemistry, 15, 1851-1862. doi:10.2174/092986708785133004
- Esteghamati, A., et al. (2011) Serum visfatin is associated with type 2 diabetes mellitus independent of insulin resistance and obesity. Diabetes Research and Clinical Practice, 91, 154-158.
- Davutoglu, M., et al. (2009) Plasma visfatin concentrations in childhood obesity: Relationships with insulin resistance and anthropometric indices. Swiss Medical Weekly, 139, 22-27.
- Pagano, C., et al. (2006) Reduced plasma visfatin/pre-B cell colony-enhancing factor in obesity is not related to insulin resistance in humans. The Journal of Clinical Endocrinology & Metabolism, 91, 3165-3170. doi:10.1210/jc.2006-0361
- Guo, H., et al. (2010) Lipocalin-2 deficiency impairs thermo-genesis and potentiates diet-induced insulin resistance in mice. Diabetes, 59, 1376-1385. doi:10.2337/db09-1735
- Catalan, V., et al. (2009) Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. Journal of Molecular Medicine, 87, 803-813. doi:10.1007/s00109-009-0486-8
- Musso, G., et al. (2011) Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Annals of Medicine, 43, 617-649.
- Adams, L.A., et al. (2009) NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. The American Journal of Gastroenterology, 104, 861-867. doi:10.1038/ajg.2009.67
- Bugianesi, E., et al. (2010) Insulin resistance in nonalcoholic fatty liver disease. Current Pharmaceutical Design, 16, 1941-1951. doi:10.2174/138161210791208875
- Fan, J.G. (2008) Impact of non-alcoholic fatty liver disease on accelerated metabolic complications. Journal of Digestive Diseases, 9, 63-67. doi:10.1111/j.1751-2980.2008.00323.x
- Guidelines Committee (1998) Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults—The evidence report. National Institutes of Health. Obesity Research, 6, 51S-209S.
- Fujioka, K. (2010) Benefits of moderate weight loss in patients with type 2 diabetes. Diabetes, Obesity and Metabolism, 12, 186-194. doi:10.1111/j.1463-1326.2009.01155.x
- Riccardi, G., Capaldo, B. and Vaccaro, O. (2005) Functional foods in the management of obesity and type 2 diabetes. Current Opinion in Clinical Nutrition & Metabolic Care, 8, 630-635. doi:10.1097/01.mco.0000171126.98783.0c
- Utzschneider, K.M., et al. (2004) Diet-induced weight loss is associated with an improvement in beta-cell function in older men. The Journal of Clinical Endocrinology & Metabolism, 89, 2704-2710. doi:10.1210/jc.2003-031827
- Boden, G., et al. (2005) Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Annals of Internal Medicine, 142, 403-411.
- Stern, L., et al. (2004) The effects of low-carbohydrate versus conventional weight loss diets in severely obese adults: one-year follow-up of a randomized trial. Annals of Internal Medicine, 140, 778-785.
- Tuomilehto, J., et al. (2001) Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. The New England Journal of Medicine, 344, 1343-1350. doi:10.1056/NEJM200105033441801
- Knowler, W.C., et al. (2002) Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. The New England Journal of Medicine, 346, 393-403. doi:10.1056/NEJMoa012512
- Knowler, W.C., et al. (2009) 10-Year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet, 374, 1677-1686. doi:10.1016/S0140-6736(09)61457-4
- Ratner, R.E., et al. (2008) Prevention of diabetes in women with a history of gestational diabetes: Effects of metformin and lifestyle interventions. The Journal of Clinical Endocrinology & Metabolism, 93, 4774-4779. doi:10.1210/jc.2008-0772
- Pan, X.R., et al. (1997) Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care, 20, 537-544. doi:10.2337/diacare.20.4.537
- Torgerson, J.S., et al. (2004) XENical in the prevention of diabetes in obese subjects (XENDOS) study: A randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care, 27, 155-161. doi:10.2337/diacare.27.1.155
- Ryan, D.H., et al. (2003) Look AHEAD (Action for Health in Diabetes): Design and methods for a clinical trial of weight loss for the prevention of cardiovascular disease in type 2 diabetes. Controlled Clinical Trials, 24, 610-628. doi:10.1016/S0197-2456(03)00064-3
- Wing, R.R. (2010) Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: Four-year results of the Look AHEAD trial. Archives of Internal Medicine, 170, 1566-1575.
- Sakane, N., et al. (2011) Prevention of type 2 diabetes in a primary healthcare setting: Three-year results of lifestyle intervention in Japanese subjects with impaired glucose tolerance. BMC Public Health, 11, 40.
- Sjostrom, L., et al. (1998) Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet, 352, 167-172. doi:10.1016/S0140-6736(97)11509-4
- Shi, Y.F., et al. (2005) Orlistat in the treatment of overweight or obese Chinese patients with newly diagnosed Type 2 diabetes. Diabetic Medicine, 22, 1737-1743. doi:10.1111/j.1464-5491.2005.01723.x
- Rowe, R., et al. (2005) The effects of orlistat in patients with diabetes: Improvement in glycaemic control and weight loss. Current Medical Research and Opinion, 21, 1885-1890. doi:10.1185/030079905X74943
- Tan, K.C., et al. (2002) Acute effect of orlistat on post-prandial lipaemia and free fatty acids in overweight patients with Type 2 diabetes mellitus. Diabetic Medicine, 19, 944-948. doi:10.1046/j.1464-5491.2002.00823.x
- Hollander, P.A., et al. (1998) Role of orlistat in the treatment of obese patients with type 2 diabetes. A 1-year randomized double-blind study. Diabetes Care, 21, 1288- 1294. doi:10.2337/diacare.21.8.1288
- Damci, T., et al. (2004) Orlistat augments postprandial increases in glucagon-like peptide 1 in obese type 2 diabetic patients. Diabetes Care, 27, 1077-1080. doi:10.2337/diacare.27.5.1077
- Van Gaal, L.F., et al. (2005) Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-Year experience from the RIO-Europe study. Lancet, 365, 1389-1397. doi:10.1016/S0140-6736(05)66374-X
- Yanovski, S.Z. (2005) Pharmacotherapy for obesity― promise and uncertainty. The New England Journal of Medicine, 353, 2187-2189. doi:10.1056/NEJMe058243
- Despres, J.P., Golay, A. and Sjostrom, L. (2005) Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. The New England Journal of Medicine, 353, 2121- 2134. doi:10.1056/NEJMoa044537
- McNeely, W. and Goa, K.L. (1998) Sibutramine. A review of its contribution to the management of obesity. Drugs, 56, 1093-1124. doi:10.2165/00003495-199856060-00019
- Gokcel, A., et al. (2001) Effects of sibutramine in obese female subjects with type 2 diabetes and poor blood glucose control. Diabetes Care, 24, 1957-1960. doi:10.2337/diacare.24.11.1957
- Norris, S.L., et al. (2004) Efficacy of pharmacotherapy for weight loss in adults with type 2 diabetes mellitus: A meta-analysis. Archives of Internal Medicine, 164, 1395- 1404. doi:10.1001/archinte.164.13.1395
- National Institutes of Health (1987) Consensus development conference on diet and exercise in non-insulin-dependent diabetes mellitus. Diabetes Care, 10, 639-644.
- Hughes, T.A., et al. (1984) Effects of caloric restriction and weight loss on glycemic control, insulin release and resistance, and atherosclerotic risk in obese patients with type II diabetes mellitus. American Journal of Medicine, 77, 7-17. doi:10.1016/0002-9343(84)90429-7
- Redmon, J.B., et al. (2005) Two-year outcome of a combination of weight loss therapies for type 2 diabetes. Diabetes Care, 28, 1311-1315. doi:10.2337/diacare.28.6.1311
- Henry, R.R., et al. (1986) Metabolic consequences of very-low-calorie diet therapy in obese non-insulin-dependent diabetic and nondiabetic subjects. Diabetes, 35, 155-164. doi:10.2337/diabetes.35.2.155
- Lee, A. and Morley, J.E. (1998) Metformin decreases food consumption and induces weight loss in subjects with obesity with type II non-insulin-dependent diabetes. Obesity Research, 6, 47-53.
- Genuth, S. (2000) Implications of the United Kingdom prospective diabetes study for patients with obesity and type 2 diabetes. Obesity Research, 8, 198-201. doi:10.1038/oby.2000.22
- Greenway, F. (1999) Obesity medications and the treatment of type 2 diabetes. Diabetes Technology & Therapeutics, 1, 277- 287. doi:10.1089/152091599317198
- Poon, T., et al. (2005) Exenatide improves glycemic control and reduces body weight in subjects with type 2 diabetes: A dose-ranging study. Diabetes Technology & Therapeutics, 7, 467-477. doi:10.1089/dia.2005.7.467
- Pories, W.J., et al. (1992) Is type II diabetes mellitus (NIDDM) a surgical disease? Annals of Surgery, 215, 633-643. doi:10.1097/00000658-199206000-00010
- O’Leary, J.P. (1980) Overview: Jejunoileal bypass in the treatment of morbid obesity. The American Journal of Clinical Nutrition, 33, 389-394.
- Sjostrom, L., et al. (2004) Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. The New England Journal of Medicine, 351, 2683-2693. doi:10.1056/NEJMoa035622
- Aucott, L., et al. (2004) Weight loss in obese diabetic and non-diabetic individuals and long-term diabetes outcomes—a systematic review. Diabetes, Obesity and Metabolism, 6, 85-94. doi:10.1111/j.1462-8902.2004.00315.x
- Franks, P.W., et al. (2008) Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologia, 51, 458-463. doi:10.1007/s00125-007-0887-6
- Gudmundsson, J., et al. (2007) Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nature Genetics, 39, 977-983. doi:10.1038/ng2062
- Sandhu, M.S., et al. (2007) Common variants in WFS1 confer risk of type 2 diabetes. Nature Genetics, 39, 951-953. doi:10.1038/ng2067
- Winckler, W., et al. (2007) Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes, 56, 685-693. doi:10.2337/db06-0202
- Bouatia-Naji, N., et al. (2009) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nature Genetics, 41, 89-94. doi:10.1038/ng.277
- Lyssenko, V., et al. (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nature Genetics, 41, 82-88. doi:10.1038/ng.288
- Prokopenko, I., et al. (2009) Variants in MTNR1B influence fasting glucose levels. Nature Genetics, 41, 77-81. doi:10.1038/ng.290
- Rung, J., et al. (2009) Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nature Genetics, 41, 1110-1115. doi:10.1038/ng.443
- O’Rahilly, S. (2009) Human genetics illuminates the paths to metabolic disease. Nature, 462, 307-314. doi:10.1038/nature08532
- Jafar-Mohammadi, B. and McCarthy, M.I. (2008) Genetics of type 2 diabetes mellitus and obesity—A review. Annals of Medicine, 40, 2-10. doi:10.1080/07853890701670421
- Scuteri, A., et al. (2007) Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genetics, 3, e115. doi:10.1371/journal.pgen.0030115
- Dina, C., et al. (2007) Variation in FTO contributes to childhood obesity and severe adult obesity. Nature Genetics, 39, 724-726. doi:10.1038/ng2048
- Frayling, T.M., et al. (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science, 316, 889-894. doi:10.1126/science.1141634
- Speliotes, E.K., et al. (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nature Genetics, 42, 937-948. doi:10.1038/ng.686
- Willer, C.J., et al. (2009) Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature Genetics, 41, 25-34. doi:10.1038/ng.287
- Thorleifsson, G., et al. (2009) Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nature Genetics, 41, 18-24. doi:10.1038/ng.274
- Loos, R.J., et al. (2008) Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nature Genetics, 40, 768-775. doi:10.1038/ng.140
- Tong, Y., et al. (2009) Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: A large Human Genome Epidemiology (HuGE) review and meta-analysis. BMC Medical Genetics, 10, 15. doi:10.1186/1471-2350-10-15
- Pearson, E.R., et al. (2007) Variation in TCF7L2 influences therapeutic response to sulfonylureas: A GoDARTs study. Diabetes, 56, 2178-2182. doi:10.2337/db07-0440
- Lindgren, C.M. and McCarthy, M.I. (2008) Mechanisms of disease: Genetic insights into the etiology of type 2 diabetes and obesity. Nature Clinical Practice Endocrinology & Metabolism, 4, 156-163. doi:10.1038/ncpendmet0723
- Lillioja, S., et al. (1987) Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. The Journal of Clinical Investigation, 80, 415-424. doi:10.1172/JCI113088
- Johansson, A., et al. Linkage and genome-wide association analysis of obesity-related phenotypes: Association of weight with the MGAT1 gene. Obesity (Silver Spring), 18, 803-808.
- Buchwald, H., et al. (2007) Trends in mortality in bariatric surgery: A systematic review and meta-analysis. Surgery, 142, 621-632. doi:10.1016/j.surg.2007.07.018
- Cunneen, S.A. (2008) Review of meta-analytic comparisons of bariatric surgery with a focus on laparoscopic adjustable gastric banding. Surgery for Obesity and Related Diseases, 4, S47-S55. doi:10.1016/j.soard.2008.04.007
- Buchwald, H., et al. (2004) Bariatric surgery: A systematic review and meta-analysis. Journal of the American Medical Association, 292, 1724-1737. doi:10.1001/jama.292.14.1724
- Buchwald, H., et al. (2009) Weight and type 2 diabetes after bariatric surgery: Systematic review and metaanalysis. American Journal of Medicine, 122, 248-256 e5.
- Levy, P., et al. (2007) The comparative effects of bariatric surgery on weight and type 2 diabetes. Obesity Surgery, 17, 1248-1256. doi:10.1007/s11695-007-9214-z
- Laferrere, B., et al. (2007) Incretin levels and effect are markedly enhanced 1 month after Roux-en-Y gastric bypass surgery in obese patients with type 2 diabetes. Diabetes Care, 30, 1709-1716. doi:10.2337/dc06-1549
- Kendall, D.M., et al. (2005) Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care, 28, 1083-1091. doi:10.2337/diacare.28.5.1083
- Buse, J.B., et al. (2004) Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylureatreated patients with type 2 diabetes. Diabetes Care, 27, 2628-2635. doi:10.2337/diacare.27.11.2628
- Buse, J.B., et al. (2007) Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: An interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clinical Therapeutics, 29, 139-153. doi:10.1016/j.clinthera.2007.01.015
- Shyangdan, D.S., et al. (2010) Glucagon-like peptide analogues for type 2 diabetes mellitus: Systematic review and meta-analysis. BMC Endocrine Disorders, 10, 20. doi:10.1186/1472-6823-10-20
- Smith Jr., S.C. (2007) Multiple risk factors for cardiovascular disease and diabetes mellitus. American Journal of Medicine, 120, S3-S11. doi:10.1016/j.amjmed.2007.01.002
- Bray, G.A. and Bellanger, T. (2006) Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine, 29, 109-117. doi:10.1385/ENDO:29:1:109
- Laakso, M. (2010) Cardiovascular disease in type 2 diabetes from population to man to mechanisms: The Kelly West Award Lecture 2008. Diabetes Care, 33, 442-449.
- Haffner, S.M., et al. (1998) Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England Journal of Medicine, 339, 229-234. doi:10.1056/NEJM199807233390404
- James I. and Cleeman, M.D. (2001) Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). Journal of the American Medical Association, 285, 2486-2497. doi:10.1001/jama.285.19.2486
- Juutilainen, A., et al. (2005) Type 2 diabetes as a “coronary heart disease equivalent”: An 18-year prospective population-based study in Finnish subjects. Diabetes Care, 28, 2901-2907. doi:10.2337/diacare.28.12.2901
- Manson, J.E., et al. (1990) A prospective study of obesity and risk of coronary heart disease in women. The New England Journal of Medicine, 322, 882-889. doi:10.1056/NEJM199003293221303
- Manson, J.E., et al. (1995) Body weight and mortality among women. The New England Journal of Medicine, 333, 677-685. doi:10.1056/NEJM199509143331101
- Catalan, V., et al. (2007) Proinflammatory cytokines in obesity: Impact of type 2 diabetes mellitus and gastric bypass. Obesity Surgery, 17, 1464-1474. doi:10.1007/s11695-008-9424-z
- WHO (1995) Physical status: The use and interpretation of anthropometry. Report of a WHO Expert Committee. World Health Organization Technical Report Series, 854, 1-452.