Journal of Biomaterials and Nanobiotechnology
Vol.06 No.01(2015), Article ID:52929,11 pages
10.4236/jbnb.2015.61002
Synthesis, Characterization, and Study of PLGA Copolymer in Vitro Degradation
Anamaria Teodora Coêlho Rios Silva, Barbara Camilla Oliveira Cardoso, Maria Elisa Scarpelli Ribeiro e Silva, Roberto Fernando Souza Freitas, Ricardo Geraldo Sousa*
Polymer Science and Technology Laboratory, Chemical Engineering Department, School of Engineering, Federal University of Minas Gerais, Belo Horizonte, Brazil
Email: *sousarg@ufmg.br
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 20 November 2014; revised 18 December 2014; accepted 26 December 2014
ABSTRACT
The poly(lactic-co-glycolic acid), known as PLGA, is one of the main bioreabsorbable polymers used in the field of medicine today. This copolymer is widely applied in sutures, devices geared toward the controlled release of medication, and the guided regeneration of bone tissue as it presents a short degradation time. This work aimed to synthesize the 82/18 PLGA (expressed by the mass ratio of D,L-lactide and glycolide, respectively), to characterize and study the in vitro degradation in the form of rods in phosphate buffer solution (PBS). The copolymer was synthesized by opening the cyclic dimer rings of the monomers D,L-lactide and glycolide, in the presence of the tin octanoate initiator and of the lauryl alcohol co-initiator. The characterization of the copolymer and the follow-up of its in vitro degradation were studied using: Differential Scanning Calorimetry (DSC), Thermogravimetry (TG), Infrared Molecular Absorption Spectroscopy with Fourier Transform (FTIR), Rheometry, and Scanning Electron Microscopy (SEM). Through these characterization techniques, it was possible to obtain the glass transition temperature, thermal stability, chemical composition, morphology, and molar mass of both the synthesized and the degraded copolymer. The molar mass of the synthesized copolymer was, approximately, 106 g∙mol−1. The degradation rate of PLGA significantly increased from the 19th to the 28th day in PBS. After 28 days in PBS, the glass transition temperature and the molar mass reduced from 45˚C to 17˚C and from 1.5 × 106 g∙mol−1 to 7.5 × 104 g∙mol−1, respectively. The pH of the medium has a significant influence on the copolymer degradation profile. When it diminishes, it accelerates the degradation process, resulting in smaller PLGA polymer chains. This pH dependent degradation can be useful for drug release systems.
Keywords:
Synthesis, Characterization, In Vitro Degradation, PLGA

1. Introduction
Many types of surgically implantable devices, which serve a temporary function within the body, can be obtained through polymers that, upon undergoing degradation, generate products that are fully metabolized in the physiological medium [1] - [6] .
One major advantage of these polymers, which are used as bioreabsorbable materials, is that, before being eliminated, they fulfill the initial purpose of tissue (or organ) recovery, or release of a drug [3] [7] . Although the first applications of these polymers have been as materials used for surgical sutures, other applications, such as implants to repair fractures and drug release devices are already commonplace in medical procedures [8] - [10] . Great effort has been made to improve the profiles of drug release systems, aimed at diminishing the effects of therapy and treatment time [11] [12] .
Due to their diverse applications and functionalities, especially in drug release therapies, the polymers are among the most widely used excipients when obtaining pharmaceutical structures [3] [13] . During drug release, the polymer is used as an active protection agent [14] - [16] .
The first copolymer (Vicril®), made up of units from lactic and glycolic acid, the poly(lactic co-glycolic acid), known as PLGA, was commercialized in the 1970’s. The great interest in this copolymer stems from the fact that its mechanical and degradation time properties can be controlled through monomer ratios [10] [17] -[22] . Figure 1 presents the chemical structure of the lactide and glycolide dimers and the reaction of copolymerization, with the polymer structures.
These copolymers can present linear and saturated chains, a constant rate of biodegradation, mechanical resistance, crystallinity, hydrophobicity, regular geometry of individual chains, and thermoplastic behavior. They possess an asymmetric center, presented by the lactic acid (PLA) in a racemic mixture (D,L-PLA) or by one of its forms―levogyrous (L-PLA) or dextrogyrous (D-PLA)―alternately connected to the glycolic acid polymer (PGA) [18] [22] [23] .
The biodegradation of PLGA occurs through bulk erosion (cleavage of polymer chains by hydrolysis), resulting in free lactic and glycolic acids. These acids, as they are natural metabolites from the body, are eliminated by the Kreb’s cycle in the form of carbon dioxide (CO2) and water (H2O) [24] -[27] , as shown in Figure 2.
The main advantage of PLGA over other bioreabsorbable polymers is that this copolymer requires a shorter time for its complete degradation, implying a lower probability of adverse reactions, which often results from the crystalline fragments released by polymers whose degradation time is excessively long. As its polymer chain possesses the mers from glycolic acid, which presents a weaker barrier against attacks from H2O molecules when compared to lactic acid, the chemical structure of PLGA is more susceptible to hydrolysis reaction, when compared to PLLA, whose polymer chain is made up of meres only from lactic acid [29] - [33] .
Polymers derived from lactic and glycolic acids have received great attention in the research on alternative biodegradable polymers, as they have already received approval from the Food and Drug Administration (FDA)
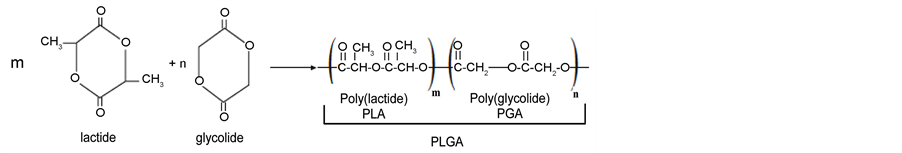
Figure 1. Chemical structure of the dimers, polymers, and copolymerization reaction.
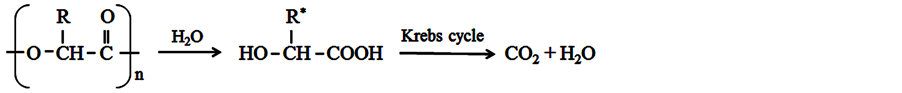 R = H (glycolic acid) or R = CH3 (lactic acid). *Degradation products: 1) lactic acid (if the polymer is PLA); 2) glycolic acid (PGA polymer source); 3) both (source PLGA polymer).
R = H (glycolic acid) or R = CH3 (lactic acid). *Degradation products: 1) lactic acid (if the polymer is PLA); 2) glycolic acid (PGA polymer source); 3) both (source PLGA polymer).
Figure 2. Reaction of PGA/PLA/PLGA degradation [28] .
for use as drug release systems. A wide range of studies has been published demonstrating their low toxicity [3] [9] [34] .
For the diverse application of PLGA, it is necessary to follow up on the degradation process. Many studies have developed methods for monitoring the stages of copolymer degradation [19] . The present work aimed to synthesize, characterize, and study the PLGA copolymer in vitro degradation in the form of a rod placed in phosphate buffer solution (PBS) (pH 7.4). The following characterization techniques were applied: Infrared Molecular Absorption Spectroscopy with Fourier Transform (FTIR), Differential Scanning Calorimetry (DSC), Thermogravimetry (TG), Scanning Electron Microscopy (SEM), and Rheometry analysis.
2. Materials and Methods
2.1. PLGA Copolymer Synthesis
The copolymer was synthesized by opening the cyclic dimer rings of the monomers D,L-lactide and glycolide, in the presence of a tin octanoate initiator (0.02% over the total mass of monomers) and of the lauryl alcohol co- initiator (0.01% over the total mass of monomers). The polymerization occurred in a glass flask-ampoule, where D,L-lactide and glycolide were added in the mass proportions of 82/18, respectively. After adding the initiator and co-initiator, the flask was closed, vacuumed sealed in the ampoule, and immersed in a silicone oil bath at 175˚C. After two hours of polymerization, the flask was removed from the bath and dichloromethane was added to dissolve the PLGA. Next, anhydrous methanol was added for PLGA precipitation. The copolymer was then kept in a heated vacuum oven at 60˚C, for 24 hours. The efficient vacuum control, the absence of humidity in the reactional medium, coupled with adequate stirring, were keys to achieving a successful synthesis [34] . The synthesized PLGA copolymer was then placed in hermetically sealed flasks in a refrigerator.
The tin octanoate initiator was used in the PLGA copolymerization reaction, for being quite reactive, of great use in the food and pharmaceuticals industries, and recognized by the FDA for this very use. The co-initiator, lauryl alcohol, and the synthesis route for the copolymerization of the monomers of cyclic diesters from lactic and glycolic acids, through the opening in the rings of the lactide and glycolide, were chosen aiming to produce copolymers with a greater molar mass for their use in drug release systems.
2.2. Study of in Vitro Hydrolytic Degradation
The PLGA rods were molded using a spatula and a non-stick surface over a steel plate heated to ±80˚C. The average mass of the produced rods was 203 ± 4 mg. The PLGA copolymer rods were immersed in closed test tubes, containing phosphate buffer solution (PBS), in a metabolic bath at 37˚C, under stirring, at 30 rpm. At determined period of times: 0, 6, 12, 19, 28, 40, and 55 days, the rods were removed, rinsed in distilled water (80 mL), dried in a heated vacuum oven at 60˚C for 48 hours, and characterized by DSC, TG, rheometry, FTIR, and SEM. After having removed the rods from the medium, the pH of the solutions was measured, by using a Denver Instrument, model 220, pH meter. All analyses were carried out in triplicate and the analysis conditions were as follows.
2.3. Fourier Transform Infrared Spectroscopy (FTIR)
The absorption spectra in the infrared region for the samples, within the range of 4000 cm−1 to 650 cm−1, were obtained in a Nicolet 6700 FTIR spectrometer (Thermo Fisher SCIENTIFIC), in the Attenuated Total Reflection―ATR mode, with 64 scans and a resolution of 4 cm−1. The spectra were obtained at room temperature (±20˚C) with the sample added directly into the device, with no prior treatment.
2.4. Thermogravimetry (TG)
The TG analysis was performed to verify the mass variation with increasing temperature, the thermal stability, and the maximum degradation temperature of the samples. Using a Shimadzu model TGA-50, the test was conducted at a heating ratio of 10˚C∙min−1, from room temperature (±20˚C) up to 400˚C, in a platinum sample holder and in a nitrogen atmosphere (N2), at a flow rate of 50 mL∙min−1. The mass of the analyzed samples was 5 ± 1 mg.
2.5. Differential Scanning Calorimetry (DSC)
The DSC equipment (Shimadzu model DSC-60) used in this study was programmed to heat the samples in two distinct runs. The first run was performed from room temperature (±20˚C) up to 150˚C, at a heating ratio of 20˚C∙min−1, intended to erase the thermal history of the material. The second run was performed from −50˚C (cooled with liquid N2) up to 200˚C, at a heating ratio of 20˚C∙min−1. An aluminum sealed sample holder was used, with a hole punctured on the cover. Nitrogen was used as the flow gas, at a rate of 50 mL∙min−1. The mass of the analyzed samples was 10 ± 1 mg.
2.6. Rheometry Analysis
The rheometric tests were conducted in a rotational rheometer, model AR-G2 (TA Instruments), using an 8 mm plate to plate geometry, heated by the equipment environmental test chamber (ETC). All rheological tests were conducted at room temperature. Through a strain sweep test, at a constant frequency of 1 Hz, curves for the elastic (G’) and viscous (G”) moduli were obtained as a function of a wide strain range (0.01% to 100%), in order to determine the strain value to be applied in the rheological tests (region of linear viscoelasticity). Frequency sweep test was carried out in order to determine the average molar mass variation of the samples. The frequency varied from 0.8 rad∙s−1 to 400 rad∙s−1 (0.1 Hz to 64 Hz), with a strain of 5%, which falls into the linear viscoelasticity range. The tests were conducted at a temperature of 80˚C (melt state).
2.7 Scanning Electron Microscopy (SEM)
A scanning electron microscope model TM3000 tabletop microscope (Hitachi), at a voltage of 5.0 kV and at a working distance between 3 mm and 10 mm, was used for the SEM tests, and the obtained images were processed using the TM3000 software. All samples were analyzed at room temperature (±20˚C). There was no need for sample coating with gold, as this analysis was conducted under low vacuum.
3. Results and Discussion
PLGA was synthesized with a yield close to 90% and the average molar mass of the samples was approximately 106 g∙mol−1. The studied samples were identified according to the in vitro time of degradation and are presented in Table 1.
The FTIR spectrum of the synthesized PLGA sample can be seen in Figure 3. In Figure 4, it’s shown the FTIR spectra of the synthesized PLGA sample and of all the copolymer samples removed from the phosphate buffer solution at the pre-determined time intervals, during the in vitro degradation study.
In the synthesized PLGA sample spectrum, it is possible to observe a thick band in the range between 1760 cm−1 and 1750 cm−1, a characteristic range of carbonyl (C=O), present in the two monomers. Also, a grouping band (C-O), between 1300 cm−1 and 1150 cm−1, can be observed, characteristic of ester groups. The absorption bands that are characteristic of the functional groups present in the PLGA copolymer can be observed in the spectrum and are presented in Table 2. The FTIR spectra, shown in Figure 4, are quite similar, which indicates that there was no significant modification on the chemical groups of PLGA, due to degradation, during the investigated period.

Table 1. Identification of the studied samples according to the in vitro time of degradation.
Table 2. Absorption bands concerning the PLGA and it attributions (adapted from [34] ).
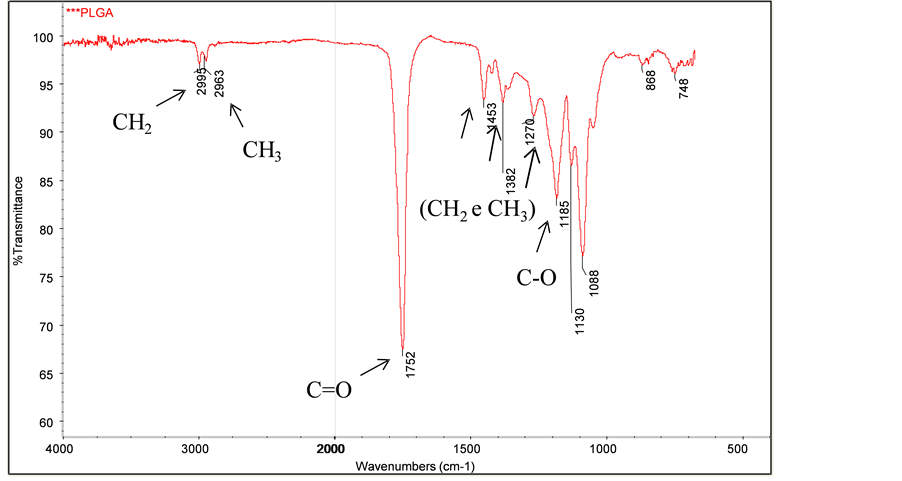
Figure 3. FTIR spectrum of the synthesized PLGA sample.
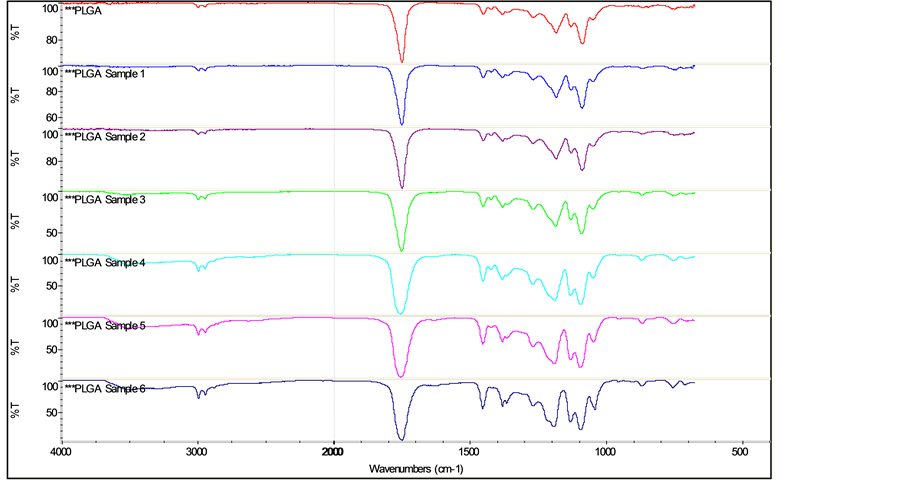
Figure 4. FTIR Spectra of synthesized PLGA and of PLGA samples degraded in vitro in PBS.
In Figure 5, it’s shown the TG curves for the synthesized PLGA sample and for all the copolymer samples during the degradation study in PBS.
In the TG curves, it is possible to observe that the values of Tonset (onset temperature of the thermal event, measured by the software), Ti (initial temperature of the thermal event) and of Tdeg.max. (temperature where the decomposition rate of the thermal event reaches its maximum level?this temperature was obtained by the TG curve) diminished as the time of degradation increased. This is most likely due to the fact that, as the PLGA copolymer begins to degrade over time, it loses thermal stability. The profiles of the PLGA decomposition curves, samples 5 and 6, correspond to the degradation times of 40 and 55 days, and proved to be quite similar, indicating that these samples are, apparently, at the same stage of degradation. Moreover, these two samples present a stage of degradation that is more pronounced than the others, given that their Tonset, Ti, and Tdeg.max. temperatures are lower than those observed for the other samples. The values of these temperatures are summarized in Table 3.
The obtained results from thermogravimetry, within the studied temperature interval, indicate that the PLGA samples decomposed almost completely and in a single stage.
The decomposition of the polymer chains occurs mainly between 180˚C and 340˚C. Samples 4, 5 and 6 presented a slight loss in mass when they reached 100˚C, which may be related to the presence of moisture in these samples or, most probably, to the beginning of polymer degradation, since those are the samples which were

Figure 5. TG curves of synthesized PLGA and of PLGA samples degraded in vitro in PBS.
Table 3. Results of the TG analyses for the PLGA in vitro degradation study.
kept longer at PBS (greater time of degradation). It is important to register that the PLGA samples studied are thermally stable until approximately 100˚C, which means that, with respect to such aspect, they are adequate for being used in drug release systems.
The DSC curves for both the PLGA and the PLGA samples tested in vitro in PBS are presented in Figure 6. The glass transition temperature values (Tgs), obtained through these curves, are shown in Table 4. The synthesized PLGA sample presented a glass transition temperature (Tg) of 45˚C, which is in agreement with the values (between 40˚C and 60˚C) found in literature [29] .
During the in vitro degradation process, there was a significant decrease of the glass transition temperature, which changed from 45˚C, at time 0, to 17˚C, after 28 days of the degradation process. From 28 to 55 days, the Tg stabilized at around 18.5˚C ± 1.5˚C. The slow start of the degradation process, observed through the relatively small decrease in Tg during the first 19 days, may well be associated with the plasticizer effect caused by the absorption of H2O by the PLGA rods [35] . The increase in the copolymer degradation rate, shown by the sharp fall in Tg, may be due to the reduction in the pH of the medium where the PLGA undergoes the degradation process (Table 4). As of 28 days of degradation, the pH level of the medium, where the PLGA samples can be found, undergo an abrupt reduction, going from 6.9 to approximately 2.4. With the PLGA degradation, the tendency of the medium’s pH level is to diminish due to the release of lactic and glycolic acid. During this reduction in pH, the Tg of the PLGA samples undergoes a pronounced decrease, thus demonstrating the increase in their degradation.
Through the strain sweep test, the linear viscoelasticity range could be determined as being between 0.2% and
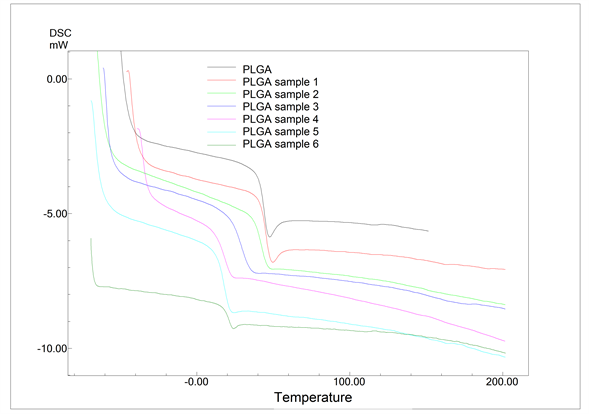
Figure 6. DSC curves of synthesized PLGA and of PLGA samples degraded in vitro in PBS.
Table 4. Results of Tgs and pHs for the PLGA in vitro degradation study.
10% of the strain. The strain value chosen for the frequency sweep test was 5%. The obtained result by the frequency sweep rheological test, for the synthesized PLGA sample, i.e., before the in vitro degradation study, is presented in Figure 7. The curves generated by the software show G’ (elastic module, in red) and G” (viscous module, in blue) as a function of angular frequency.
Through the curves shown Figure 7, one can observe the predominance of the viscous behavior over the elastic one (G” > G’) up to the angular frequency of approximately 10 rad∙s−1, with the inversion of these behaviors occurring when this frequency is reached. The curves of the elastic and viscous moduli, obtained for the PLGA samples that underwent in vitro degradation in PBS, are similar to those presented in Figure 7. The average molar mass of each sample was estimated based on the results from the frequency sweep test and in models that describe the relation of rheological data and the relaxation times with molar mass distribution [36] -[38] . The results of the average molar mass of the PLGA samples, in the in vitro degradation study, obtained indirectly through rheometry, are shown in Figure 8. Table 5 shows the average molar mass obtained by the frequency sweep tests for the PLGA samples that had been degraded in vitro in PBS.
Figure 8 shows that the synthesized PLGA copolymer, with an average molar mass in the order of 106 g∙mol−1,
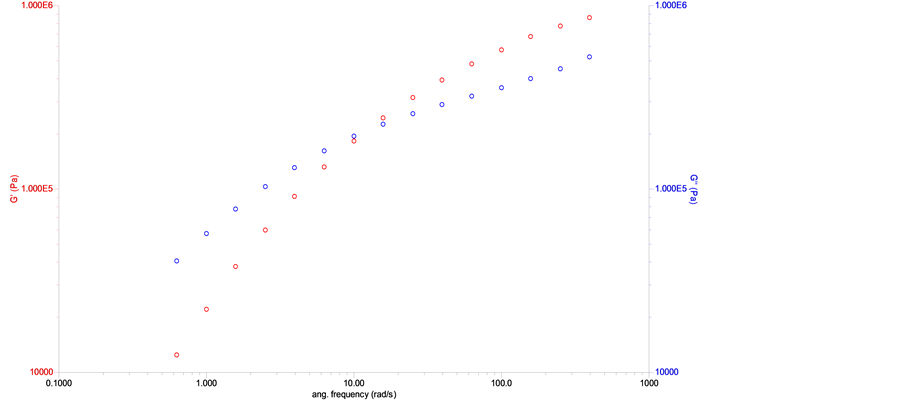
Figure 7. Curves for the elastic and viscous moduli as a function of angular frequency for synthesized PLGA sample.
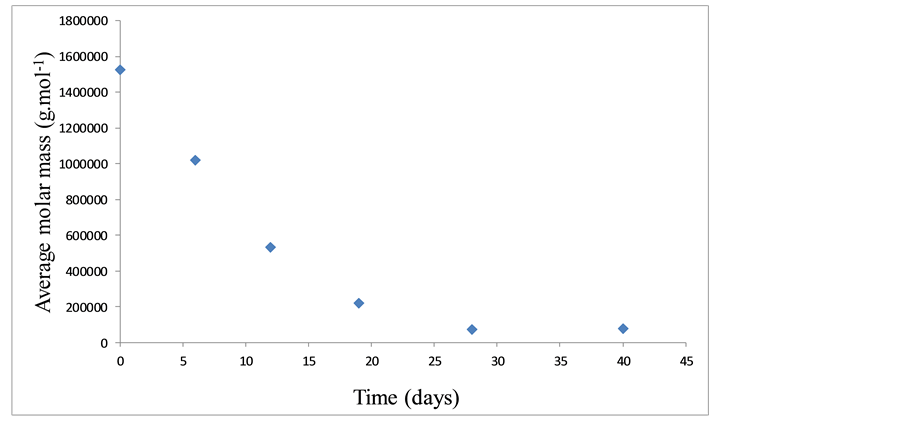
Figure 8. Average molar mass for the PLGA samples with respect to degradation time.
Table 5. Results of the rheometric analysis of the study of PLGA degradation in vitro.
during the in vitro degradation process in PBS, presented a reduction in its average molar mass to around 104 g∙mol−1, within 28 days (PLGA sample 4), which corresponds to a decrease of approximately 95%. That is, the average molar mass of the PLGA sample after 28 days in PBS is approximately 5% of the average molar mass of the synthesized PLGA sample. Before reaching this value, a sharp decrease in the PLGA’s molar mass, in direct proportion with degradation time, could be observed, particularly between the 19th and the 28th day in PBS.
Due to the PLGA sample’s stage of degradation after 55 days, it was impossible to perform a rheometric analysis to obtain the average molar mass of this sample, given that the sample size was insufficient to perform an effective rheometric test.
In Figures 9(a), it can be seen the micrograph obtained for the synthesized PLGA sample. In Figure 9(b), Figure 9(c), and Figure 9(d), the micrographs for the PLGA samples 1, 2 and 3, respectively, are shown. The morphology of the surface of the PLGA rods was followed up during the degradation process until the 19th day. In the subsequent samples, the SEM analysis of the sample surfaces became unfeasible, due to the samples’ stage of degradation. These samples, upon being inserted into the equipment, had their degradation processes accelerated due to the presence of the electron beam, rendering it impossible to obtain the micrographs of these samples.
The micrograph from Figure 9(a) shows the PLGA rod’s surface before beginning the degradation process. It can be noted that it presents integrity and a non-porous appearance. After 6 days of degradation (Figure 9(b)), it could be observed that the aspect of the sample continued the same (integral and non-porous), with no visible beginning of degradation. After 12 days of immersion in PBS, the micrograph of the PLGA rod (Figure 9(c)) showed some small cracks on the rod’s surface, most likely due to the beginning of PLGA degradation. This phenomenon becomes even clearer and more explicit when one looks at the PLGA sample micrograph, after 19 days of degradation (Figure 9(d)). It can be observed a surface with many fractures and cracks, indicating a more pronounced intensity of degradation. This behavior corroborates the one observed for the glass transition temperature, shown in Table 4, where one can observe the occurrence of the first significant reduction in Tg value, from 42˚C to 31˚C, within the interval of 12 and 19 days in PBS.
Through the results presented in this study, one can clearly see a sharp decrease in the values of Tg, molar mass, and pH of the medium from the 19th to the 28th day in PBS. This decrease, observed in these properties, may be due to the presence of the PLGA chains with a reduced molar mass, stemming from the in vitro degradation process. These chains can bring about a plasticizer effect within the samples, thus justifying the reported decrease. In this study, it was also observed that, when handling the samples, they become more and more brittle and fragile when reaching this period of in vitro degradation in PBS.
4. Conclusion
The synthesized PLGA copolymer (82/12) in this work had a yield of nearly 90%, presented an average molar mass in the order of 106 g∙mol−1, and showed a glass transition temperature of 45˚C. The study of the PLGA copolymer in vitro degradation in PBS indicated that, after 28 days of immersion in this solution, the degradation became much more pronounced. This behavior was observed due to the reduction in the glass transition temperature values, from 45˚C to 17˚C; of the average molar mass, from 1.5 × 106 g∙mol−1 to 7.5 × 104 g∙mol−1; and of

 (a) (b)
(a) (b)
 (c) (d)
(c) (d)
Figure 9. Micrographs of the: (a) PLGA rods before the in vitro degradation process; (b) PLGA rods, sample 1; (c) PLGA rods, sample 2; and (d) PLGA rods, sample 3.
the pH of the medium, from 7.4 to 2.5. The degradation rate of PLGA significantly increased from the 19th to the 28th day in PBS. The pH of the medium has a significant influence on the copolymer degradation profile. When it diminishes, it accelerates the degradation process, resulting in smaller PLGA polymer chains. This pH dependent degradation can be useful for drug release systems.
Acknowledgements
The authors acknowledge the financial support of FAPEMIG, CEMIG, PETROBRAS, CAPES and CNPq.
References
- Ratner, B.D. and Hoffman, A.S. (1996) Biomaterials Science―An Introduction to Materials in Medicine. Elsevier, New York.
- Langer, R. and Peppas, N.A. (2003) Advances in Biomaterials, Drug Delivery, and Bionanotechnology. Alche Journal, 49, 2990-3006. http://dx.doi.org/10.1002/aic.690491202
- Stevanović, M., Maksin, T., Petković, J, Filipić, M. and Uskoković, D. (2009) An Innovative, Quick and Convenient Labeling Method for the Investigation of Pharmacological Behavior and the Metabolism of Poly(DL-lactide-coglyco- lide) Nanospheres. Nanotechnology, 20, 1-12. http://dx.doi.org/10.1088/0957-4484/20/33/335102
- Hoffman, A.S. (2008) The Origins and Evolution of “Controlled” Drug Delivery Systems. Journal of Controlled Release, 132, 153-163. http://dx.doi.org/10.1016/j.jconrel.2008.08.012
- Park, J.H., Lee, S., Kim, J.H., Park, K., Kim, K. and Kwon, I.C. (2008) Polymeric Nanomedicine for Cancer Therapy. Progress in Polymer Science, 33, 113-137. http://dx.doi.org/10.1016/j.progpolymsci.2007.09.003
- Pamula, E. and Menaszak, E. (2008) In Vitro and in Vivo Degradation of Poly(L-lactide-co-glycolide) Films and Scaffolds. Journal of Materials Science: Materials in Medicine, 19, 2063-2070. http://dx.doi.org/10.1007/s10856-007-3292-2
- Duarte, M.A.T., Duek, E.A.R. and Motta, A.C. (2014) In Vitro Degradation of Poly (L-co-D,L lactic acid) Containing PCL-T. Polímeros, 24, 1-8. http://dx.doi.org/10.4322/polimeros.2014.056
- Xiaoling, L. and Haskara, R.J. (2006) Design of Controlled Release Drug Delivery Systems. MacGraw-Hill, New York.
- Stevanović, M. and Skoković, D. (2009) Poly(lactide-co-glycolide)-Basedmicro and Nanoparticles for the Controlled Drug Delivery of Vitamins. Current Nanoscience, 5, 1-14.
- Stevanović, M., Radulović, A., Jordović, B. and Uskoković, D. (2008) Poly(DL-lactide-co-glycolide) Nanospheres for the Sustained Release of Folic Acid. Journal of Biomedical Nanotechnology, 4, 349-358. http://dx.doi.org/10.1166/jbn.2008.021
- Liang, R.C., Li, X., Shi, Y., Wang, A., Sun, K., Liu, W.H. and Li, Y.X. (2013) Effect of Water on Exenatide Acylation in Poly(lactide-co-glycolide) Microspheres. International Journal of Pharmaceutics, 454, 344-353. http://dx.doi.org/10.1016/j.ijpharm.2013.07.012
- Meng, Z.X., Zheng, W., Li, L. and Zheng, Y.F. (2011) Fabrication, Characterization and in Vitro Drug Release Behavior of Electrospun PLGA/Chitosan Nanofibrous Scaffold. Materials Chemistry and Physics, 125, 606-611. http://dx.doi.org/10.1016/j.matchemphys.2010.10.010
- Rios, M. (2005) Polymers for Controlled Release: Formulation Follows Function. Pharmaceutical Technology, 29, 42-50.
- Mundargi, R.C., Babu, V.R., Rangaswamy, V., Patel, P. and Aminabhavi, T.M. (2008) Nano/Micro Technologies for Delivering Macromolecular Therapeutics Using Poly(D,L-lactide-co-glycolide) and Its Derivatives. Journal of Controlled Release, 125, 193-209. http://dx.doi.org/10.1016/j.jconrel.2007.09.013
- Cohen-sela, E., Chorny, M., Koroukhov, N., Danenberg, H.D. and Golomb, G. (2009) A New Double Emulsion Solvent Diffusion Technique for Encapsulating Hydrophilic Molecules in PLGA Nanoparticles. Journal of Controlled Release, 133, 90-95. http://dx.doi.org/10.1016/j.jconrel.2008.09.073
- Bae, S.E., Son, J.S., Park, K. and Han, D.K. (2009) Fabrication of Covered Porous PLGA Microspheres Using Hydrogen Peroxide for Controlled Drug Delivery and Regenerative Medicine. Journal of Controlled Release, 133, 37-43. http://dx.doi.org/10.1016/j.jconrel.2008.09.006
- Jain, R.A. (2000) The Manufacturing Techniques of Various Drug Loaded Biodegradable Poly(lactídeo-co-glicolídeo) (PLGA) Devices. Biomaterials, 21, 2475-2490. http://dx.doi.org/10.1016/S0142-9612(00)00115-0
- Soares, A.Q., Oliveira, L.F., Rabelo, D. and Souza, A.R. (2005) Polímeros Biodegradáveis: Novas Perspectivas para as Ciências Farmacêuticas. Revista Eletrônica de Farmácia, 2, 202-205.
- Ji, W., Yang, F., Seyednejad, H., Chen, Z., Hennink, W.E., Anderson, J.M., Beucken, J.J.J.P. and Jansen, J. (2012) Biocompatibility and Degradation Characteristics of PLGA-Based Electrospun Nanofibrous Scaffolds with Nanoapatite Incorporation. Biomaterials, 33, 6604-6614. http://dx.doi.org/10.1016/j.biomaterials.2012.06.018
- Kang, S.W., Yang, H.S., Seo, S.W., Han, D.K. and Kim, B.S. (2008) Apatite-Coated Poly(lactic-coglycolic Acid) Microspheres as an Injectable Scaffold for Bone Tissue Engineering. Journal of Biomedical Materials Research Part A, 85, 747-756. http://dx.doi.org/10.1002/jbm.a.31572
- Qi, F., Wu, J., Yang, T., Ma, G. and Su, Z.G. (2014) Mechanistic Studies for Monodisperse Exenatide-Loaded PLGA Microspheres Prepared by Different Methods Based on SPG Membrane Emulsification. Acta Biomaterialia, 10, 4247- 4256. http://dx.doi.org/10.1016/j.actbio.2014.06.018
- Sun, L., Xie, Z., Zhao, Y., Wei, H.M. and Gu, Z.Z. (2013) Optical Monitoring the Degradation of PLGA Inverse Opal Film. Chinese Chemical Letters, 24, 9-12. http://dx.doi.org/10.1016/j.cclet.2013.01.012
- Khare, V., Kour, S., Alam, N., Dubey, R.D., Saneja, A., Koul, M., Gupta, A.P., Singh, D., Singh, S., Saxena, A.K. and Gupta, P.N. (2014) Synthesis, Characterization and Mechanistic-Insight into the Antiproliferative Potential of PLGA- Gemcitabine Conjugate. International Journal of Pharmaceutics, 470, 51-62. http://dx.doi.org/10.1016/j.ijpharm.2014.05.005
- Athanasiou, K.A., Niederauer, G.G. and Agrawal, C.M. (1996) Sterilization, Toxicity, Biocompatibility and Clinical Applications of Polylactic Acid/Polyglycolic Acid Copolymers. Biomaterials, 17, 93-102. http://dx.doi.org/10.1016/0142-9612(96)85754-1
- Lanao, R.P.F., Leeuwenburgh, S.C.G., Wolke, J.G.C. and Jansen, J.A. (2011) Bone Response to Fast-Degrading, Injectable Calcium Phosphate Cements Containing PLGA Microparticles. Biomaterials, 32, 8839-8847. http://dx.doi.org/10.1016/j.biomaterials.2011.08.005
- Lanao, R.P.F., Leeuwenburgh, S.C.G., Wolke, J.G.C. and Jansen, J.A. (2011) In Vitro Degradation Rate of Apatitic Calcium Phosphate Cement with Incorporated PLGA Microspheres. Acta Biomaterialia, 7, 3459-3468. http://dx.doi.org/10.1016/j.actbio.2011.05.036
- Li, H. and Chang, J. (2005) pH-Compensation Effect of Bioactive Inorganic Fillers on the Degradation of PLGA. Composites Science and Technology, 65, 2226-2232. http://dx.doi.org/10.1016/j.compscitech.2005.04.051
- Merkli, A., Tabatabay, C., Gurny, R. and Heller, J. (1998) Biodegradable Polymers for the Controlled Release of Ocular Drug. Progress in Polymer Science, 23, 563-580. http://dx.doi.org/10.1016/S0079-6700(97)00048-8
- Astete, C.E. and Sabliov, C.M. (2006) Synthesis and Characterization of PLGA Nanoparticles. Journal of Biomaterials Science, Polymer Edition, 17, 247-289. http://dx.doi.org/10.1163/156856206775997322
- Houchin, M.L. and Topp, E.M. (2008) Chemical Degradation of Peptides and Proteins in PLGA: A Review of Reactions and Mechanisms. Journal of Pharmaceutical Sciences, 97, 2395-2404. http://dx.doi.org/10.1002/jps.21176
- Tracy, M.A., Ward, K.L., Firouzabadian, L., Wang, Y., Dong, N., Qian, R. and Zhang, Y. (1999) Factors Affecting the Degradation Rate of Poly(lactide-co-glycolide) Microspheres in Vivo and in Vitro. Biomaterials, 20, 1057-1062. http://dx.doi.org/10.1016/S0142-9612(99)00002-2
- Tsukadaa, Y., Haraa, K., Bandoa, Y., Huangb, C.C., Kousakaa, Y., Kawashimac, Y., Morishitad, R. and Tsujimotoa, H. (2009) Particle Size Control of Poly(DL-lactide-co-glycolide) Nanospheres for Sterile Applications. International Journal of Pharmaceutics, 370, 196-201. http://dx.doi.org/10.1016/j.ijpharm.2008.11.019
- Xiao, D., Liu, Q., Wang, D., Xie, T., Guo, T., Duan, K. and Weng, J. (2014) Room-Temperature Attachment of PLGA Microspheres to Titanium Surfaces for Implant-Based Drug Release. Applied Surface Science, 309, 112-118. http://dx.doi.org/10.1016/j.apsusc.2014.04.195
- Erbetta, C.D.C., Alves, R.J., Resende, J.M., Freitas, R.F.S. and Sousa, R.G. (2012) Synthesis and Characterization of Poly(D,L-lactide-co-glycolide) Copolymer. Journal of Biomaterials and Nanobiotechnology, 3, 208-225. http://dx.doi.org/10.4236/jbnb.2012.32027
- kasperczyk, J. (1996) Microstructural Analysis of Poly(l-lactide)-co-(glycolide) by 1H and13 C n.m.r. Spectroscopy. Polymer, 37, 201-203.
- Maier, D., Eckstein, A., Friedrich, C. and Honerkamp, J. (1998) Evaluation of Models Combining Rheological Data with the Molecular Weight Distribution. Journal of Rheology, 42, 1153-1173. http://dx.doi.org/10.1122/1.550952
- Thimm, W.B., Friedrich, C., Marth, M. and Honerkamp, J. (1999) An Analytical Relationship between Relaxation Time Spectrum and Molecular Weight Distribution. Journal of Rheology, 43, 1663-1672. http://dx.doi.org/10.1122/1.551066
- Thimm, W.B., Friedrich, C., Marth, M. and Honerkamp, J. (1999) On the Rouse Spectrum and the Determination of the Molecular Weight Distribution from Rheological Data. Journal of Rheology, 44, 429-438. http://dx.doi.org/10.1122/1.551094
NOTES

*Corresponding author.