Graphene
Vol.04 No.04(2015), Article ID:59480,6 pages
10.4236/graphene.2015.44009
Mono-Vacancy and B-Doped Defects in Carbon Heterojunction Nanodevices
Ahlam A. El-Barbary1,2*, Mohamed A. Kamel1, Khaled M. Eid1,3, Hayam O. Taha1, Mohamed M. Hassan1
1Physics Department, Faculty of Education, Ain Shams University, Cairo, Egypt
2Physics Department, Faculty of Science, Jazan University, Jazan, KSA
3Bukairiayh for Science, Qassim University, Qassim, KSA
Email: *ahla_eg@yahoo.co.uk
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 30 July 2015; accepted 6 September 2015; published 9 September 2015
ABSTRACT
We present a detailed theoretical study of the behavior of mono-vacancy and B-doped defects in carbon heterojunction nanodevices. We have introduced a complete set of formation energy and surface reactivity calculations, considering a range of different diameters and chiralities of combined carbon nanotubes. We have investigated three distinct combinations of carbon heterojunctions using density functional theory (DFT) and applying B3LYP/3-21g: armchair-armchair herteojunctions, zigzag-zigzag heterojunctions, and zigzag-armchair heterojunctions. We have shown for first time a detailed study of formation energy of mono-vacancy and B-doped defects of carbon heterojunction nanodevices. Our calculations show that the highest surface reactivity is found for the B-doped zigzag-armchair heterojunctions and it is easier to remove the carbon atom from the network of heterojunction armchair-armchair CNTs than the heterojunction zigzag-armchair and zigzag-zigzag CNTs.
Keywords:
Band Gaps, Carbon Heterojunctions, DFT, Mono-Vacancy Defects, Boron Doping

1. Introduction
A heterojunction can be defined as the interface between two dissimilar regions with unequal band gaps in one-dimension. Another definition for a heterojunction is the interface between any two solid state materials. The formation of one-dimensional heterojunctions combines the properties of two materials and makes the heterojunctions have unique properties and many applications [1] -[4] . There are a few studies on heterojunctions and fewer on heterojunction CNTs. The axial vibration of heterojunctions CNTs using nonlocal rod theory on CNTs with different lengths, chirality and diameters in the heterojunctions is investigated [5] [6] and it is found that by joining CNTs good vibrational properties are obtained by suitable selection of parameters. Also, the thermal conductivity of single-wall CNTs with linear, “Y” and “X” hetero-junctions using non equilibrium molecular dynamics method is studied using three types of junctions [7] and it is found that when the defects form an interface, they can cause a larger decrease in the thermal conductivity of CNT than when they are distributed dispersedly and also it is found that the number of defects contained in the nanojunction determines the thermal conductivity to a great extent more than the geometrical forms of the nanojunctions. The mechanical and electronic properties of the heterojunction formed by (4,4) CNT and (4,4) silicon carbide nanotube (SiCNT) by using first-principles density functional theory are investigated [8] and it shows that band structure of (4,4) SiC/CNT heterojunction is a semiconductor with direct band gap and both valence band maximum and conduction band minimum originate from CNT segment. In addition, the effect of CNT and SiCNT length on the electronic structures of the NT heterojunction is also studied. The band alignment at AlN/diamond heterojunctions is investigated [9] and it is found a weak dependence of the valence-band offset on strain and on the diamond orientation. Theoretical estimation of energy levels and energy gaps of conjugated polymers representing new donor systems is reported and the criteria and relationships for the prediction of energy data from theoretical ones are provided [10] . Different heterojunction interfaces by means of a self-consistent tight-binding approach are analyzed [11] and are found that the deposition of different intralayers introduced changes in the band offsets of the heterojunction depending on the intralayer electronegativity. A supercell containing a heterojunction ZnO/Cu2O is built and is studied the structural properties and the discontinuity of the valence band (band offset) from a semiconductor to another [12] .
2. Computational Methods
All our ab-initio calculations were performed with the Density Functional Theory (DFT) as implemented within G03W package [13] -[20] , using B3LYP exchange-functional [21] [22] and applying basis set 3 - 21 g. All obtained structures are fully optimized under spin average. Thirty six different geometries of nanodevice carbon heterojunctions were considered; eighteen structures of mono-vacancy defected carbon heterojunctions and eighteen structures of boron doped carbon heterojunctions. The studied heterojunctions are created from CNTs with equal lengths and different chiralities and diameters. To create the mono-vacancy defects, one carbon atom is individually removed at different two positions: from pristine heterojunction CNTs of smaller diameter (V1), or from pristine heterojunction CNTs of larger diameter (V2), there will be two mono-vacancy defected heterojunction structures. Also, the B-doped heterojunction CNTs are formed by individually replacing one carbon atom by boron atom at different two positions: from pristine heterojunction CNTs of smaller diameter (B1), or from pristine heterojunction CNTs of larger diameter (B2). The formation energies for mono-vacancy and B-doped defects are calculated from the following relations [23] [24] :
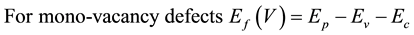
where 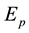 is the total energy of pure heterojunction CNTs,
is the total energy of pure heterojunction CNTs, 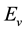 is the total energy of mono-vacancy defected heterojunction CNTs and
is the total energy of mono-vacancy defected heterojunction CNTs and  is the chemical potential of carbon atom.
is the chemical potential of carbon atom.
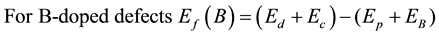
where  is the total energy of B-doped heterojunction CNTs and
is the total energy of B-doped heterojunction CNTs and  is the chemical potential of carbon and boron atoms.
is the chemical potential of carbon and boron atoms.
3. Results and Discussion
The configuration structures of mono-vacancy defects and B-doped defects of armchair-armchair, zigzag-arm- chair and zigzag-zigzag heterojunction CNTs are shown in Table 1. Also, the positions of mono-vacancy defects and B-doped defects of armchair-armchair, zigzag-armchair and zigzag-zigzag heterojunction CNTs are shown in Figure 1. As shown in Table 1 and Figure 1, there are many varieties of combinations between CNTs to form heterojunction CNTs in order to study the effects of chirality and size on the formation energy and surface reactivity of heterojunction CNTs.

Table 1. The studied nanodevices of heterojunction CNTs.
 (a) (b)
(a) (b) (c) (d)
(c) (d)
Figure 1. Positions of mono-vacancy defects (a) V1, (b) V2 and boron doped defects (c) B1, (d) B2 for zigzag-armchair (7,0)-(7,7) heterojunction CNTs. Carbon atom (gray), boron atom (pink) and hydrogen atom (white). The big circles refer to positions of the mono-vacancy defects.
3.1. Formation Energy of Mon-Vacancy and B-Doped Defects of Heterojunction CNTs
The formation energy of mono-vacancy and B-doped defects of armchair-armchair, zigzag-armchair and zigzag- zigzag heterojunction CNTs are calculated and are shown in Table 2. The formation energies of mono-vacancy defects are defined as the difference between the energy of the pure heterojunction CNT and the sum of energies of mono-vacancy defected heterojunction CNT and the chemical potential of carbon atom. From Table 2, one can report that for zigzag-zigzag and zigzag-armchair CNTs the formation energies of V1-defected heterojunctions are always higher than the formation energies of V2-defected heterojunctions and vice versa for armchair- armchair heterojunction CNTs. The highest formation energy 7.13 eV is found to be for V1-defected heterojunction zigzag-zigzag (7,0)-(9,0) CNT, however the lowest formation energy 4.25 eV is found to be for V1-defected heterojunction armchair-armchair (4,4)-(7,7) CNT. Also, one can report that the formation energy of mono- vacancy defect is independent on the size of heterojunction CNT, however it is found that it is easier to remove
Table 2. The calculated formation energies of V1-, V2-mono-vacancy and B1-, B2-doped nanodevices of heterojunction CNTs. All energies are given by eV.
the carbon atom from the network of heterojunction armchair-armchair CNTs than the heterojunction zigzag- armchair CNTs and the formation energy of the latter is lower than the heterojunction zigzag-zigzag CNTs.
The formation energies of B-doped defects are defined as the difference between the sum of B-doped heterojunction and the chemical potential of carbon atom and the sum of energies of the pure heterojunction CNT and the chemical potential of boron atom. From Table 2, one can report that for zigzag-zigzag, zigzag-armchair and armchair-armchair CNTs the formation energies of B2-doped heterojunctions are always lower than the formation energies of B1-doped heterojunctions CNTs. The highest formation energy 7.44 eV is found to be for B1-doped heterojunction zigzag-zigzag (7,0)-(9,0) CNT, however the lowest formation energy 5.14 eV is found to be for B2-doped heterojunction zigzag-armchair (7,0)-(6,6) CNT. Also, one can report that the formation energy is independent on the size of heterojunction CNT, however it is found that it is easier to doped boron atom into the network of heterojunction armchair-armchair CNTs than the heterojunction zigzag-armchair CNTs and the formation energy of the latter is lower than the heterojunction zigzag-zigzag CNTs. Finally, it is found that the formation energies of V1-, V2-defects and B1-, B2-doped are independent on the size of the heterojunction CNTs, however the formation energies are dependent on thechirality of heterojunction CNTs.
3.2. Surface Reactivity of Mon-Vacancy and B-Doped Defects of Heterojunction CNTs
The surface reactivity of mono-vacancy and B-doped defects of armchair-armchair, zigzag-armchair and zigzag-zigzag heterojunction CNTs are calculated as function of dipole moment and are shown in Table 3. It was reported that the higher the dipole moment the higher surface reactivity [25] . From Table 3, one can report that for zigzag-zigzag and armchair-armchair CNTs the surface reactivity of V1-defected heterojunctions are always higher than the surface reactivity of V2-defected heterojunctions and vice versa for zigzag-armchair heterojunction CNTs. The highest surface reactivity 9.13 Debye is found to be for V2-defected heterojunction zigzag- armchair (7,0)-(6,6) CNT, however the smallest surface reactivity 1.82 Debye is found to be for V2-defected heterojunction armchair-armchair (4,4)-(6,6) CNT. Also, one can report that the surface reactivity of mono- vacancy defect is dependent on the size of heterojunction CNT, especially for V2-defected heterojunction CNTs. Also, it is found that the surface reactivity of mono-vacancy defected is dependent on the chirality of heterojunction CNTs. The highest surface reactivity is found for the zigzag-armchair heterojunctions, followed by armchair-armchair heterojunctions and the smallest surface reactivity is for zigzag-zigzag heterojunction CNTs.
From Table 3, it is found that the surface reactivity of B1-doped zigzag-zigzag, armchair-armchair and zigzag-armchair heterojunction CNTs are always higher than the surface reactivity of B2-doped heterojunction CNTs. The highest surface reactivity 12.58 Debye is found to be for B1-doped heterojunction zigzag-armchair (7,0)-(7,7) CNT, however the smallest surface reactivity 1.66 Debye is found to be for B2-doped heterojunction armchair-armchair (4,4)-(6,6) CNT. Also, one can report that the surface reactivity is dependent on the size of
Table 3. The calculated surface reactivity of V1-, V2-mono-vacancy and B1-, B2-doped nanodevices of heterojunction CNTs. All dipole moments are given by Debye.
B-doped heterojunction CNT, especially for B2-doped heterojunction CNTs. Also, it is found that the surface reactivity is dependent on the chirality of heterojunction CNTs. The highest surface reactivity is found for the B- doped zigzag-armchair heterojunctions, followed by B-doped armchair-armchair heterojunctions and the smallest surface reactivity is for B-doped zigzag-zigzag heterojunction CNTs. Finally, it is found that the surface reactivity of V1-, V2-defects and B1-, B2-doped are dependent on the chirality and the size of the heterojunction CNTs.
4. Conclusion
The behavior of mono-vacancy and B-doped defects in nanodevice carbon heterojunctions is investigated for thirty-six different heterojunction CNTs. The formation energy and surface reactivity calculations, considering a range of different diameters and chiralities of combined carbon nanotubes are considered using density functional theory (DFT) and applying B3LYP/3-21g. Our calculations show that the highest surface reactivity is found for the B-doped zigzag-armchair heterojunctions, followed by B-doped armchair-armchair heterojunctions and the smallest surface reactivity is for B-doped zigzag-zigzag heterojunction CNTs. Also, it is found that the surface reactivity of V1-, V2-defects and B1-, B2-doped is dependent on the chirality and the size of the heterojunction CNTs. For mono-vacancy defect, it is found that the formation energy of mono-vacancy defect is independent on the size of heterojunction CNT and it is easier to remove the carbon atom from the network of heterojunction armchair-armchair CNTs than the heterojunction zigzag-armchair CNTs and the formation energy of the latter is lower than the heterojunction zigzag-zigzag CNTs. Finally, it is found that for the V1-, V2-defects and B1-, B2-doped heterojunction CNTs the formation energies are dependent on the chirality of heterojunction CNTs and are independent on the size.
Cite this paper
Ahlam A.El-Barbary,Mohamed A.Kamel,Khaled M.Eid,Hayam O.Taha,Mohamed M.Hassan,11, (2015) Mono-Vacancy and B-Doped Defects in Carbon Heterojunction Nanodevices. Graphene,04,84-90. doi: 10.4236/graphene.2015.44009
References
- 1. Ruppalt, L.B. and Lyding, J.W. (2007) Metal-Induced Gap States at a Carbon-Nanotube Intramolecular Heterojunction Observed by Scanning Tunneling Microscopy. Small, 3, 280-284. http://dx.doi.org/10.1002/smll.200600343
- 2. Hashimoto, A., Suenaga, K., Gloter, A., Urita, K. and Iijima, S. (2004) Direct Evidence for Atomic Defects in Graphene Layers. Nature, 430, 870-873. http://dx.doi.org/10.1038/nature02817
- 3. Lu, J.Q., Wu, J., Duan, W. and Gu, B.L. (2004) Effects of Finite Deformed Length in Carbon Nanotubes. Applied Physics Letters, 84, 4203. http://dx.doi.org/10.1063/1.1751608
- 4. Triozon, F., Lambin, P. and Roche, S. (2005) Electronic Transport Properties of Carbon Nanotube Based Metal/Semi- conductor/Metal Intramolecular Junctions. Nanotechnology, 16, 230. http://dx.doi.org/10.1088/0957-4484/16/2/008
- 5. Filiz, S. and Aydogdu, M. (2010) Axial Vibration of Carbon Nanotube Heterojunctions Using Nonlocal Elasticity. Computational Materials Science, 49, 619-627. http://dx.doi.org/10.1016/j.commatsci.2010.06.003
- 6. Yengejeh, S.I., Zadeh, M.A. and Ochsner, A. (2014) Numerical Modeling of Eigenmodes and Eigenfrequencies of Hetero-Junction Carbon Nanotubes with Pentagon-Heptagon Pair Defects. Computational Materials Science, 92, 76-83. http://dx.doi.org/10.1016/j.commatsci.2014.05.015
- 7. Ren, C., Xu, Z., Zhang, W., Li, Y., Zhu, Z. and Huai, P. (2010) Theoretical Study of Heat Conduction in Carbon Nanotube Hetero-Junctions. Physics Letters A, 374, 1860-1865. http://dx.doi.org/10.1016/j.physleta.2010.02.028
- 8. Xu, B., Ouyang, J., Xu, Y., Wu, M.S., Liu, G. and Ouyang, C.Y. (2013) Electronic Transport Characteristic of an Individual CNx/C Nanotube Schottky Junction. Computational Materials Science, 68, 367-370. http://dx.doi.org/10.1016/j.commatsci.2012.11.013
- 9. Silvestri, L., Cervenka, J., Prawer, S. and Ladouceur, F. (2013) First Principle Study of Valence-Band Offsets at AlN/ Diamond Heterojunctions. Diamond and Related Materials, 31, 25-29. http://dx.doi.org/10.1016/j.diamond.2012.10.010
- 10. Longo, L., Carbonera, C., Pellegrino, A., Perin, N., Schimperna, G., Tacca, A. and Po, R. (2012) Comparison between Theoretical and Experimental Electronic Properties of Some Popular Donor Polymers for Bulk-Heterojunction Solar Cells. Solar Energy Materials & Solar Cells, 97, 139-149. http://dx.doi.org/10.1016/j.solmat.2011.09.035
- 11. Munoz, A., Pérez, R., Durán, J.C. and Flores, F. (1989) A Theoretical Analysis of the Heterojunction Band Offsets as Controlled by Interlayer Deposition. Surface Science, 211-212, 503-510. http://dx.doi.org/10.1016/0039-6028(89)90807-8
- 12. Zemzemi, M. and Alaya, S. (2013) Band Offset of the ZnO/Cu2O Heterojunction from ab Initio Calculations. Superlattices and Microstructures, 64, 311-318. http://dx.doi.org/10.1016/j.spmi.2013.09.041
- 13. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Zakrzewski, V.G., Montgomery, J.A., Stratmann, R.E., Burant, J.C., Dapprich, S., Millam, J.M., Daniels, A.D., Kudin, K.N., Strain, M.C., Farkas, O., Tomasi, J., Barone, V., Cossi, M., Cammi, R., Mennucci, B., Pomelli, C., Adamo, C., Clifford, S., Ochterski, J., Petersson, G.A., Ayala, P.Y., Cui, Q., Morokuma, K., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Cioslowski, J., Ortiz, J.V., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Gomperts, R., Martin, R.L., Fox, D.J., Keith, T., Al-Lamham, M.A., Peng, C.Y., Nanayakkara, A., Gonzalez, C., Challacombe, M., Gill, P.M.W., Johnson, B.G., Chen, W., Wong, M.W., Andres, J.L., Head-Gordon, M., Replogle, E.S. and Pople, J.A. (2004) Gaussian Inc., Wallingford.
- 14. El-Barbary, A.A., Lebda, H.I. and Kamel, M.A. (2009) The High Conductivity of Defect Fullerene C40 Cage. Computational Materials Science, 46, 128-132. http://dx.doi.org/10.1016/j.commatsci.2009.02.034
- 15. Hindi, A. and El-Barbary, A.A. (2015) Hydrogen Binding Energy of Halogenated C40 Cage: An Intermediate between Physisorption and Chemisorption. Journal of Molecular Structure, 1080, 169-175. http://dx.doi.org/10.1016/j.molstruc.2014.09.034
- 16. El-Barbary, A.A. (2015) 1H and 13C NMR Chemical Shift Investigations of Hydrogenated Small Fullerene Cages Cn, CnH, CnHn and CnHn+1: n = 20, 40, 58, 60. Journal of Molecular Structure, 1097, 76-86. http://dx.doi.org/10.1016/j.molstruc.2015.05.015
- 17. El-Barbary, A.A., Eid, K.M., Kamel, M.A. and Hassan, M.M. (2013) Band Gap Engineering in Short Heteronanotube Segments via Monovacancy Defects. Computational Materials Science, 69, 87-94. http://dx.doi.org/10.1016/j.commatsci.2012.10.035
- 18. El-Barbary, A.A., Ismail, G.H. and Babeer, A.M. (2013) Effect of Monovacancy Defects on Adsorbing of CO, CO2, NO and NO2 on Carbon Nanotubes: First Principle Calculations. Journal of Surface Engineered Materials and Advanced Technology, 3, 287-294. http://dx.doi.org/10.4236/jsemat.2013.34039
- 19. El-Barbary, A.A., Eid, K.M., Kamel, M.A., Osman, H.M. and Ismail, G.H. (2014) Effect of Tubular Chiralities and Diameters of Single Carbon Nanotubes on Gas Sensing Behavior: A DFT Analysis. Journal of Surface Engineered Materials and Advanced Technology, 4, 66-74. http://dx.doi.org/10.4236/jsemat.2014.42010
- 20. El-Barbary, A.A., Eid, K.M., Kamel, M.A., Osman, H.M. and Ismail, G.H. (2015) Adsorption of CO, CO2, NO and NO2 on Carbon Boron Nitride Hetero Junction: DFT Study. Journal of Surface Engineered Materials and Advanced Technology, 5, 169-176. http://dx.doi.org/10.4236/jsemat.2015.54019
- 21. Becke, A.D. (1993) Density-Functional Thermochemistry. III. The Role of Exact Exchange. Journal of Chemical Physics, 98, 5648. http://dx.doi.org/10.1063/1.464913
- 22. Becke, A.D. (1998) Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Physical Review A, 38, 3098-3100. http://dx.doi.org/10.1103/PhysRevA.38.3098
- 23. Chang, H., Lee, J.D. Lee, S.M. and Lee, Y.H. (2001) Adsorption of NH3 and NO2 Molecules on Carbon Nanotubes. Applied Physics Letters, 79, 3863. http://dx.doi.org/10.1063/1.1424069
- 24. Garcia, A.L.E., Baltazar, S.E., Romero, A.H., Perez Robles, J.F. and Rubio, A. (2008) Influence of S and P Doping in a Graphene Sheet. Journal of Computational and Theoretical Nanoscience, 5, 1-9. http://dx.doi.org/10.1166/jctn.2008.1123
- 25. El-Barbary, A.A. (2015) The Surface Reactivity and Electronic Properties of Small Hydrogenation Fullerene Cages. Journal of Surface Engineered Materials and Advanced Technology, 5, 162-168. http://dx.doi.org/10.4236/jsemat.2015.53018
NOTES

*Corresponding author.