Pharmacology & Pharmacy
Vol.3 No.3(2012), Article ID:20639,12 pages DOI:10.4236/pp.2012.33035
Method Development for the Detection of Basic/Weak Basic Drugs in Hair by LCMSMS: Comparison between Methanolic and Alkaline Extraction on Real Samples
![]()
1The William Harvey Research Institute, Barts & The London School of Medicine & Dentistry, Queen Mary University of London, London, UK; 2Department of Pharmacology and Toxicology, Faculty of Medicine, Kuwait University, Jabriya, Kuwait; 3Analytical Services International Ltd., St. George’s University of London, London, UK.
Email: j.aljaber@qmul.ac.uk
Received March 4th, 2012; revised April 20th, 2012; accepted May 12th, 2012
Keywords: Hair Analysis; Basic Drugs; LCMSMS; Benzodiazepines; Method Development; LLE
ABSTRACT
Detection of drugs in hair has become popular in recent years. The significantly long drug detection window (months) in hair has allowed the retrospective investigation and measurement of past consumption of drug. As the majority of drugs are basic, an extraction method was developed based on a methanolic solution for detection of basic/weak basic drugs in hair. It was compared with alkaline digestion (NaOH) followed by LLE. A filtration step with filtration vials was added and their materials were compared. After filtration, extracts were injected directly onto a C18 column coupled to Sciex ABI 2000 MSMS. The mobile phase was 50% methanol, 0.1% formic acid and 2 mM ammonium acetate (isocratic). Both methods were compared by applying them to real samples. Results showed that calibration was linear with r2 of 0.991 - 0.999 for 20 tested analytes. The matrix effect was assessed to be between 91.4% - 110.2% for 18 analytes. PTFE filter material showed better recoveries over the GMF and PVDF based filters. Stability of analytes during extraction in general was better with methanolic incubation than alkaline digestion. With regard to real sample recovery, 6 out of 10 analytes recovered better with alkaline digestion. In conclusion, the methanolic method is capable of extracting most basic drugs in hair samples but only part of the total incorporated drug. Therefore, these results suggest that a combination of both methods (methanolic and alkaline extractions) in hair sample processing for general detection of basic and weak basic drugs may produce better results. However, not all basic drugs are stable under alkaline digestion.
1. Introduction
Drug analysis in hair has grabbed the attention of toxicology analysts and researchers in recent years. This is mainly because it has provided some ability of proving drug ingestion when conventional samples could not. Hair differs from other traditional biological samples used for human toxicological analysis such as urine, blood, liver or saliva with its significantly longer detection window (months) allowing retrospective investigation and measurement of drug consumption. Hair analysis is becoming accepted in many developed countries for substance consumption related issues in a wide range of sectors; the medico-legal sector, workplace testing, treatment monitoring, schools, forensics, research, insurance companies, environmental biomonitoring and driving licensing [1-4].
Extraction of drugs from hair is considered one of the most important steps in hair analysis. Apart from external drug deposition on hair, drugs are mainly enclosed tightly in the hair shaft and to a certain extent maybe bound to proteins, melanin or lipids of the cell membrane complex. Therefore, hair matrix type, structure of the drug, method and duration of extraction, and solvent used are all important factors affecting the final extraction yield [5].
There are numerous reports of screening strategies published for analysing different forensic basic drugs groups. Hypnotic drugs such as benzodiazepines are at the top of the list of drug facilitated crimes. These are weak basic drugs and have been reported to be extracted from hair by different methods, with phosphate buffers, methanol or digestive enzymes [6-8]. Other basic drug groups like antipsychotics, antidepressants and amfetamines have frequently been reported to be extracted with alkaline digestion (NaOH) [9-12]. However, recently there has been some focus on using methanolic solutions as an extraction medium consisting of varying ratios of methanol, acetonitrile and formate buffer for extraction of several groups of basic drugs simultaneously [13-15]. However, the extent of their efficiency was not examined most of the time. Therefore, in the present study the aim was to develop a method to enable the detection and quantification of basic and weak basic drugs in hair simultaneously based on the methanolic solution extraction technique and to compare its efficiency with the alkaline digestion technique which was followed by liquid-liquid extraction (LLE).
2. Materials
2.1. Chemicals
All chemicals were of HPLC or analytical grade. Methanol, acetonitrile, acetone, propanol, methyl-tert-butylether (MTBE), citalopram, clobazam, clonazepam, cocaine, codeine, desmethyldiazepam, 3,4-methylenedioxyamphetamine (MDA), 3,4-methylenedioxy-N-ethylamphetamine (MDEA), 3,4-Methylenedioxyme-thamphetamine (MDMA), midazolam, sulpiride, zaleplon and zolpidem were purchased from Sigma-Aldrich Co, Dorset UK. 7-aminoclonazepam, amfetamine, amfetamineD11, benzoylecgonine-D8, benzoylecognine, codeine-D3, diazepam, diazepam-D5, ecognine methyl ester, medazepam, oxazepam and temazepam were purchased from LGC standards, Middlesex UK. Other prescription drug standards were supplied from their manufacturer. Deionised water was prepared on site (ELGA Limited). Dichloromethane, 7.5 M ammonium acetate solution and all consumables (tubes, etc) are from VWR International Ltd, Lutterworth UK. The rest of the chemicals used in the solutions below were from Sigma-Aldrich Co, Dorset UK.
2.2. Solutions
2.2.1. Standard Solutions
1000 mg/L or 100 mg/L working standards made with MS grade methanol of each analyte and internal standard. All stored at –20˚C and diluted to the required concentration prior to the experiment.
2.2.2. 0.1 M Phosphate Solution
One litre of solution was prepared by dissolving 1.7 g of anhydrous disodium orthophosphate and 12.14 g of sodium dihydrogen orthophosphate monohydrate in 800 mL of de-ionized water. The pH was adjusted to 6 with 1 M potassium hydroxide and the solution made up to volume with de-ionized water.
2.2.3. 0.1% Formic Acid (v/v)
100 µL of concentrated formic acid (98/100) was mixed with 99.9 mL of de-ionized water, mixed well and stored at room temperature.
2.2.4. Methanolic Extraction Solution
60 mL of methanol, 10 mL of acetonitrile and 30 mL of 0.1% formic acid were measured into measuring cylinder, mixed well and stored at room temperature.
2.2.5. 1 M NaOH Solution
40 g of NaOH (MW 40.0) pellets were measured into a 1000 mL volumetric flask and made up to the 1000 mL mark with de-ionized water. This was mixed thoroughly and stored at room temperature.
2.2.6. 2 M Ammonium Acetate
266.6 mL of 7.5 M ammonium acetate solution were measured into a 1000 mL volumetric flask and made up to the 1000 mL mark with de-ionized water.
3. Methods and Results
3.1. Hair Preparation
The decontamination process was by using three washing steps with two solvents and one aqueous solution: 0.01 M phosphate solution (pH 6), dichloromethane and propanol/acetone (1:1). The uncut hair was put in a 6 ml glass tube, then 2 mL of the first solution added and vortex mixed for 1 minute. After that the solvent was decanted and then the next solvent was added. After the last washing step (2 mL propanol/acetone) the hair was left to dry at room temperature or the process was speeded up with the speed-vac. This decontamination method was found to be quick and very efficient in cleaning postmortem hair samples.
After the hair had dried segments of ~0.6 cm each were cut into small pieces (1 - 3 mm) with scissors and ideally 25 mg were weighed directly. If this was not possible any amount between 10 and 80 mg was considered and placed into a 2 mL glass tube with a screw cap.
3.2. Methanolic Extraction
Five hundred micro-litres of the methanolic extraction solution (described earlier) was added to the hair in each tube and left in a sonicator for 16 hours at 40˚C. In the case of calibrators, the required concentrations were made based on adding 25 μL of drugs standard (e.g. 1 mg/L in methanol) to the calibrators’ tubes containing 25 mg blank hair each and those 25 μL were deducted from the extraction solution (475 μL). Also 25 μL of internal standards (bromperidol and the dueterated standards amfetamine-D11, benzoylecgonine-D8, codeine-D3 and diazepam-D5) were added as well at a concentration similar to the middle calibrator. After centrifugation for 10 minutes the extracted solutions were transferred to autosampler vials and evaporated to dryness followed by reconstitution with 125 μL of 25% methanol and filtered with the syringeless mini-uniprep filters (Whatman GE). Finally 20 μL of this filtrate was injected into the LCMSMS system.
3.3. Alkaline Digestion
Twenty five milligrams of washed blank/patient hair was incubated with 0.5 mL of 1 M NaOH for 3 hours in a water bath (50˚C) to allow for digestion of the hair. Samples were then extracted after digestion with 2 mL MTBE by 30 minutes rotary mixing and 10 minutes centrifugation. Solvent layers were transferred into clean tubes containing 150 µL 1% formic acid, then mixed on a rotary mixer for 20 minutes and removed after 10 minutes of centrifugation by aspiration. From the remaining aqueous layer 20 µL was injected into the LCMSMS system.
3.4. Method Development with Some Validation for Selected Analytes
3.4.1. Instrumentation and Ion Identification
The LCMSMS analysis was carried out with an HPLC system consisting of a Perkin Elmer PE200 series autosampler, pump and column oven. The MSMS part was a SCIEX API 2000 Triple Quadrupole MS-MS instrument (Applied Biosystems) equipped with an electrospray interface (Turbo Ion Spray) set on the positive mode. Ion spray voltage was set to 5500 V. Nitrogen was used as the nebulizer gas-GS1 (30 psi), auxiliary gas-GS2 (20 psi heated to 300˚C), curtain gas (20 psi) and as the CAD gas (set on 3).
Analytes were infused at 0.25 mL/minute to the LCMSMS system at a concentration of 1 mg/L in 50% methanol/2mM ammonium acetate solution. The precursor and product ion identification and parameter tuning were completed by automatic and manual tuning. Optimum tuning parameters, precursor and product quantitation ions are shown in Table 1. Mobile phase was 50% methanol, 0.1% formic acid and 2 mM ammonium acetate (isocratic). Separation with Altech Alltima C18 column (15 cm × 2.1 mm, 5 μm) maintained at 50˚C.
3.4.2. Comparison of Mini-Uniprep Filter Media
Three filter materials were compared by using spiked blank hair samples at concentrations of 0.5 ng/mg hair. 25 mg of blank hair were weighted out in glass tubes. Then the whole extraction/incubation method was applied to all the samples. The three materials were 0.45 µm pore size Glass Microfiber (GMF), 0.2 µm pore size Polytetrafluroethylene (PTFE) and 0.2 µm pore size Polyvinylidenefluoride (PVDF) used in manufacturing Whatman Mini-UniPrep Syringeless Filters. The filters efficiency was calculated by comparing the percentage of extraction recovery through their peak area ratio of product ion for each drug after filtration with unfiltered calibrators.
Table 2 shows clearly that the PTFE filter medium has the greatest recovery (mean = 100%) of all the analytes while the GMF and PVDF filters (mean = 89% and 72%, respectively) reduced significantly the response of amisulpride and citalopram. Also the responses of medazepam, midazolam and zolpidem were considerably reduced by the PVDF filter.
3.4.3. Linearity
The linearity study reflects the relationship between change of spiked analyte concentration and detector response to that change [16]. It was attained by spiking hair samples with 50, 75, 100, 500, 1000 and 2000 pg/mg of each analyte. The linear correlation coefficients (r2) values were better than 0.99 for all analytes.
3.4.4. LOD and LOQ
Limit of detection and limit of quantification were assessed by spiking blank hair with the analytes in decreasing concentrations. The LOD and LOQ were calculated at a signal to noise ratio of 3 and 10, respectively. LOD values for all analytes were 0.5 - 23 pg/mg and LOQ values were 1.7 - 76.7 pg/mg (Table 3).
3.4.5. Selectivity
Selectivity in drug analysis is the ability of an analytical method to differentiate and quantify the analyte of interest in the presence of other components in the sample [17]. In order to achieve that a low standard (0.1 ng/mg) of each drug from the group was injected individually into the LCMSMS system and all the other drug detection windows and retention times were observed to see if there were any interfering peaks or false positive results that might appear in the same retention time and same window as the injected analyte. No interference was found on the same detection window and retention time for any of the analytes. Also drug-free hair samples showed no false positive results.
3.4.6. Matrix Effect
The matrix effect (ME) is basically the ion suppression or enhancement of the analyte peak caused by co-eluting compounds from the biological matrix (urine, blood, hair, etc). The aim of this experiment was to acquire more accurate data on the extent of matrix influence on the results when matrices come from different types of hair. As proposed by Matuszewski et al. the ME was assessed by comparing the mean peak area of product ion of ex-
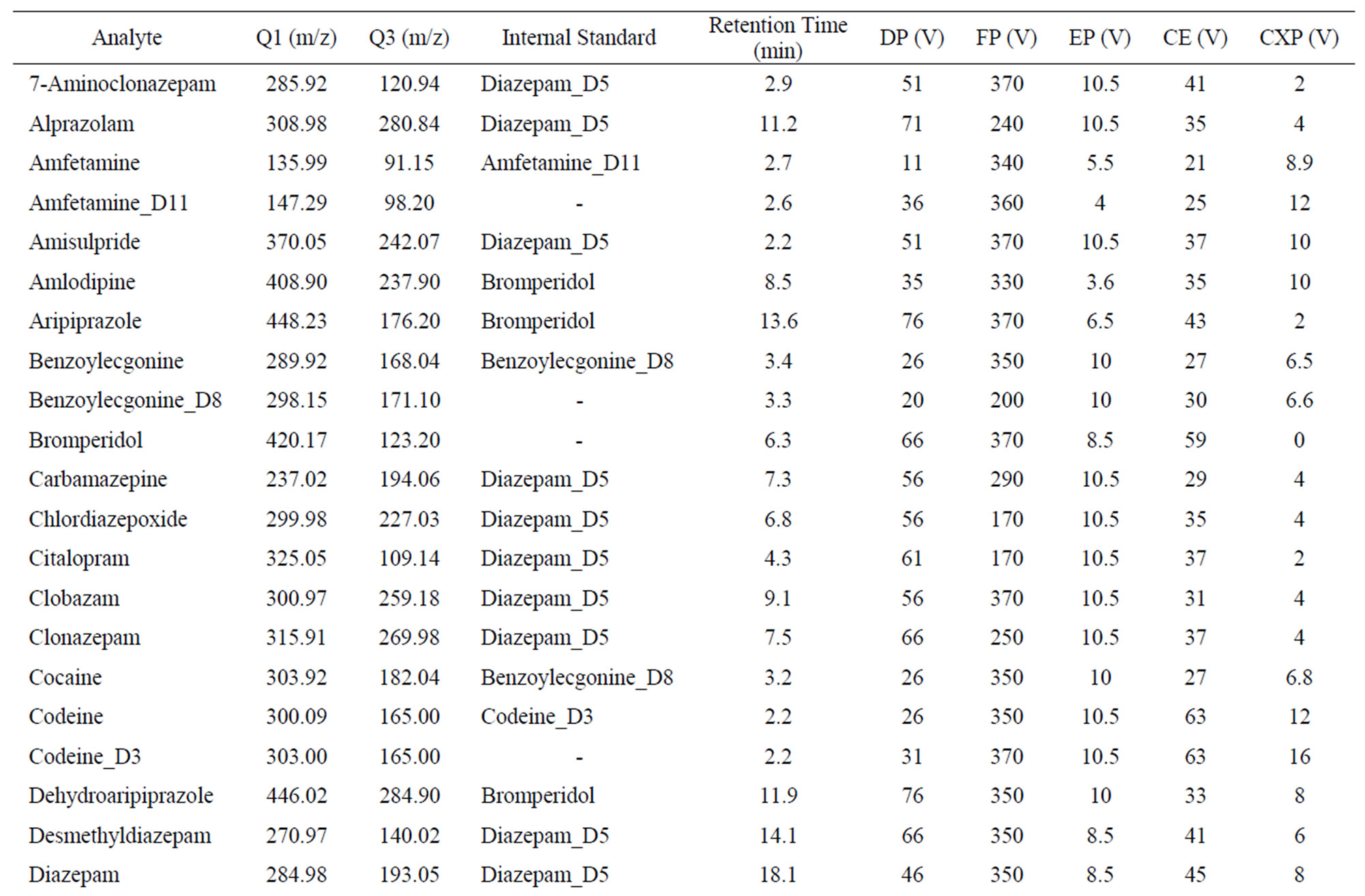
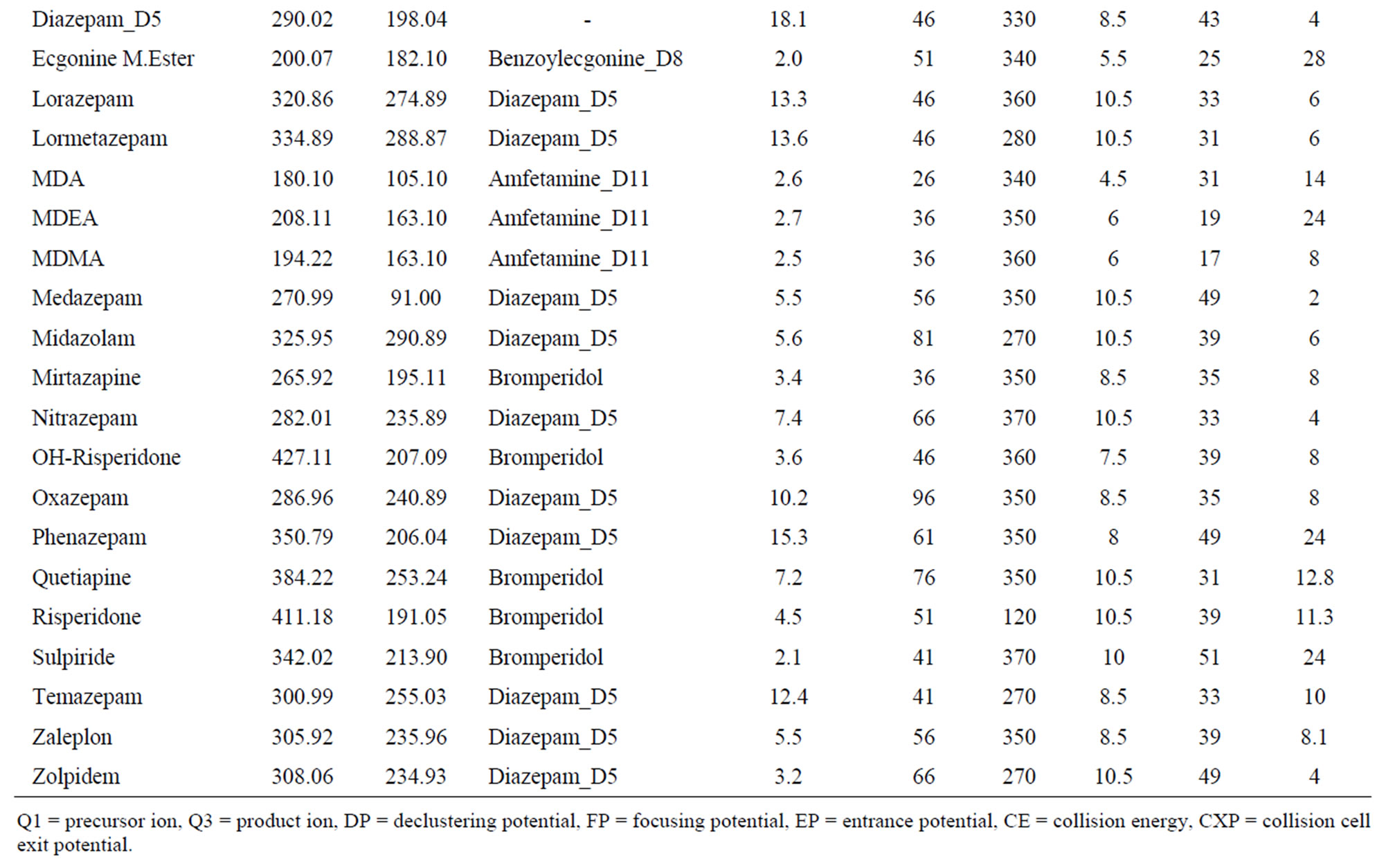
Table 1. Optimized MRM and MS/MS parameters for analytes and internal standards.
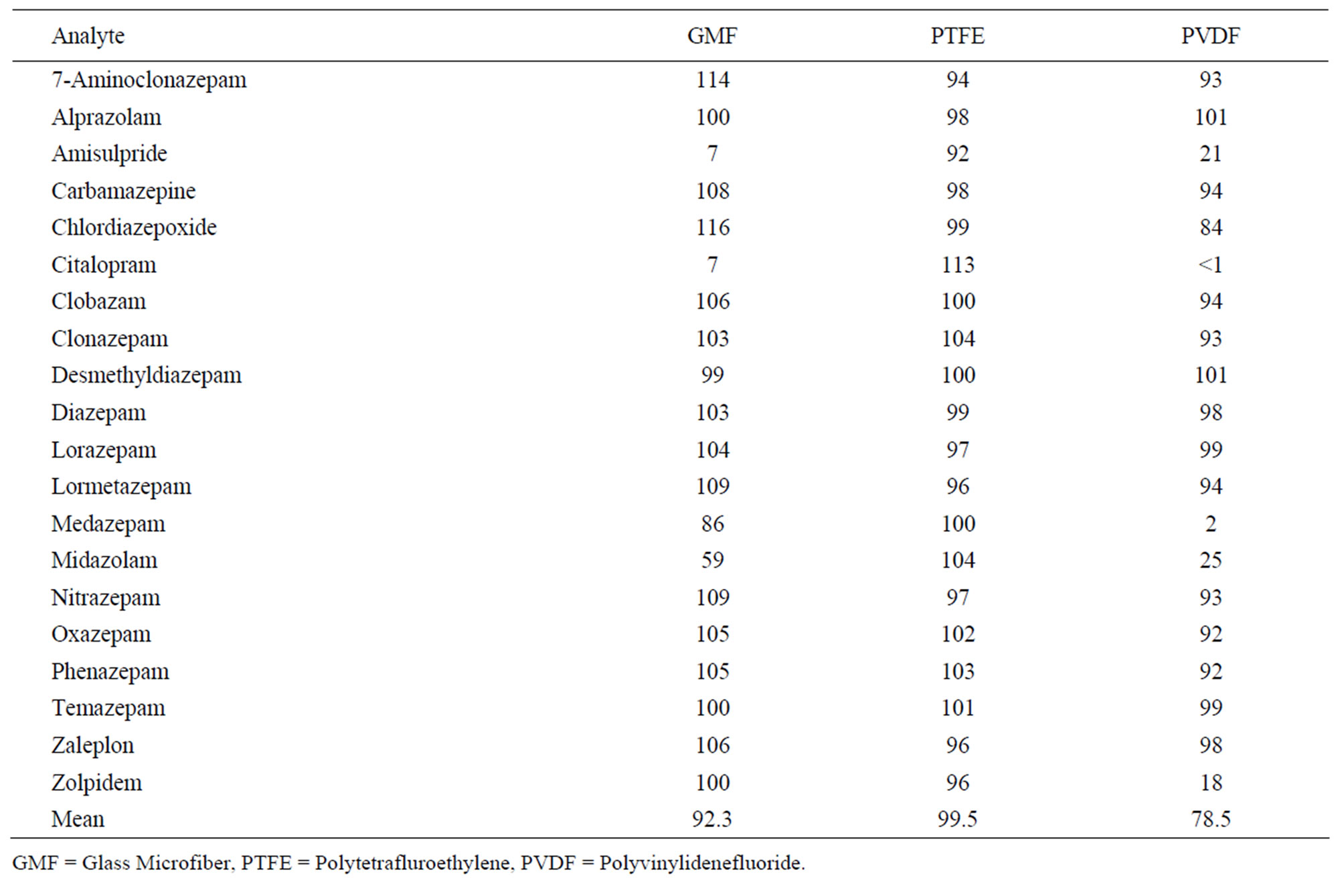
Table 2. Mini-UniPrep filter media: comparison of results for % recovery of 0.5 ng/mg spiked analytes.
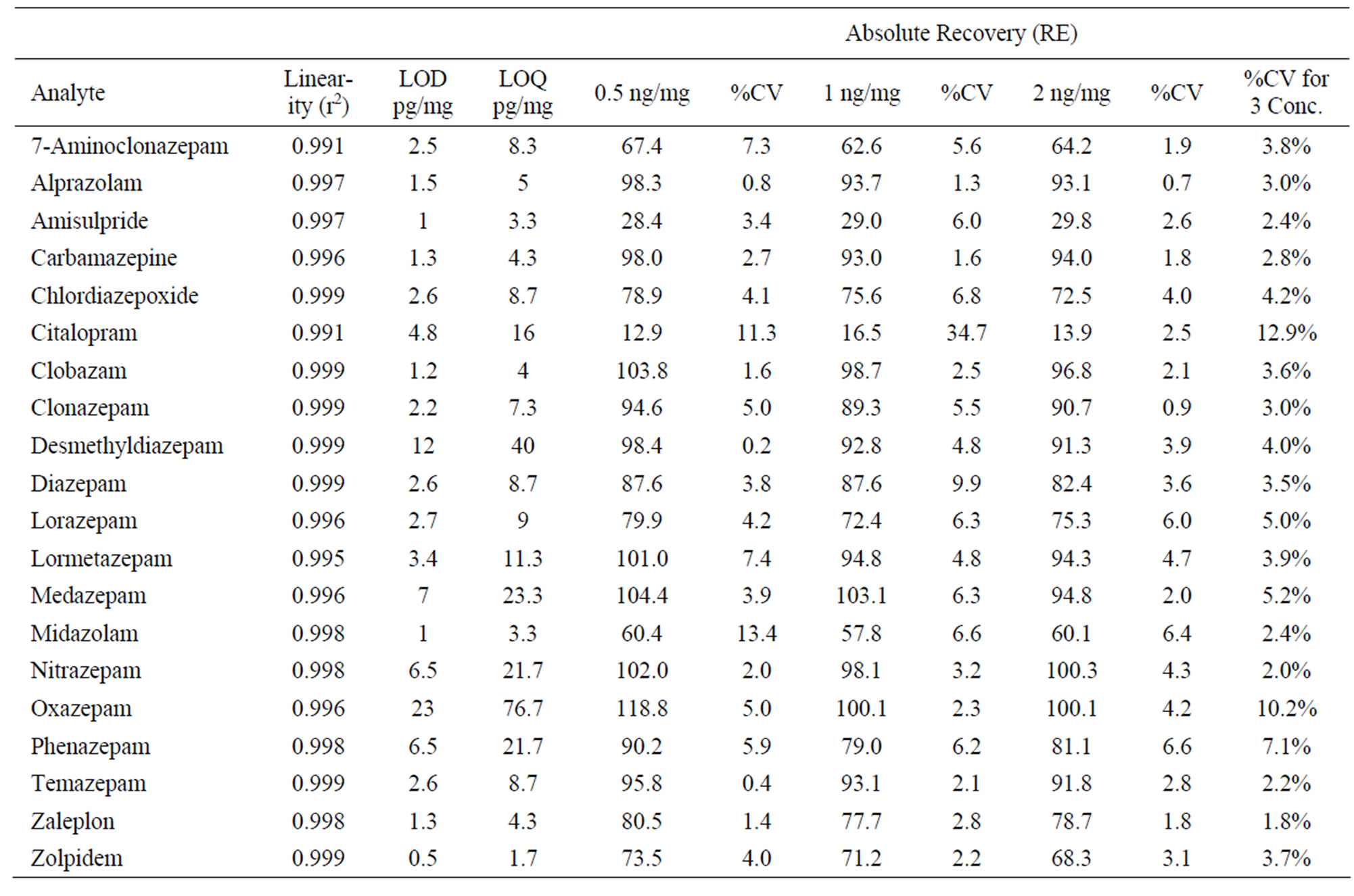
Table 3. Linearity, LOD, LOQ and Absolute Recovery (RE) results for 0.5, 1 and 2 ng/mg of analytes (n = 3).
tracted drug-free hair samples from 5 different sources (black, baby-brown, black dyed, white and blonde) spiked with 0.2 ng/mg low standard solution following filtration (i.e. after extraction/incubation) with the mean peak area of product ion of neat unextracted standards prepared in the mobile phase [18]. The percentage of the ME was calculated by dividing the mean of the extracted samples by the mean of unextracted ones according to Equation (1).
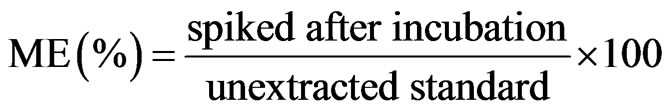 (1)
(1)
This percentage represents the absolute ME (both ion suppression and ion enhancement).
The ME results shown in Table 4 indicate that there was no significant ion suppression or enhancement affecting the analytes, except with amisulpride and the metabolite of clonazepam the 7-amino. Some of the analytes showed slight ion enhancement such as diazepam and clobazepam, but the rest of the drugs were within 91.4 - 110.2% ME. Also the %CV was in the range of 3.2% - 11.1% showing a good agreement level between the different hair samples.
3.4.7. Absolute Recovery with Methanolic Incubation
Recoveries were estimated by determining peak areas of analytes obtained when the drugs spiked with blank hair (in extraction solution) at three concentrations 0.5, 1, and 2 ng/mg (n = 3) where compared with unextracted/unfiltered standards. Therefore, this recovery takes into account all the extraction steps including filtration, incubation and matrix effect (absolute). The recovery was calculated as a percentage by dividing the average peak area ratio of the extracted standards over the average peak area ratio of the unextracted standards and multiplied by 100.
The results in Table 3 show that all the analytes had good recoveries (>58%) at all three concentrations with %CV below 13.4%, except for 2 analytes which gave low recoveries (12.9% - 29.8%).
3.4.8. Stability during Methanolic Extraction
All analytes were prepared at 0.5 mg/L (~= ng/mg) of the extraction solution (mentioned in solutions section) and incubated/sonicated with slightly harsher conditions at 50˚C for 18 h without the blank hair. Then the mean peak area ratio of product ion/internal standard was compared
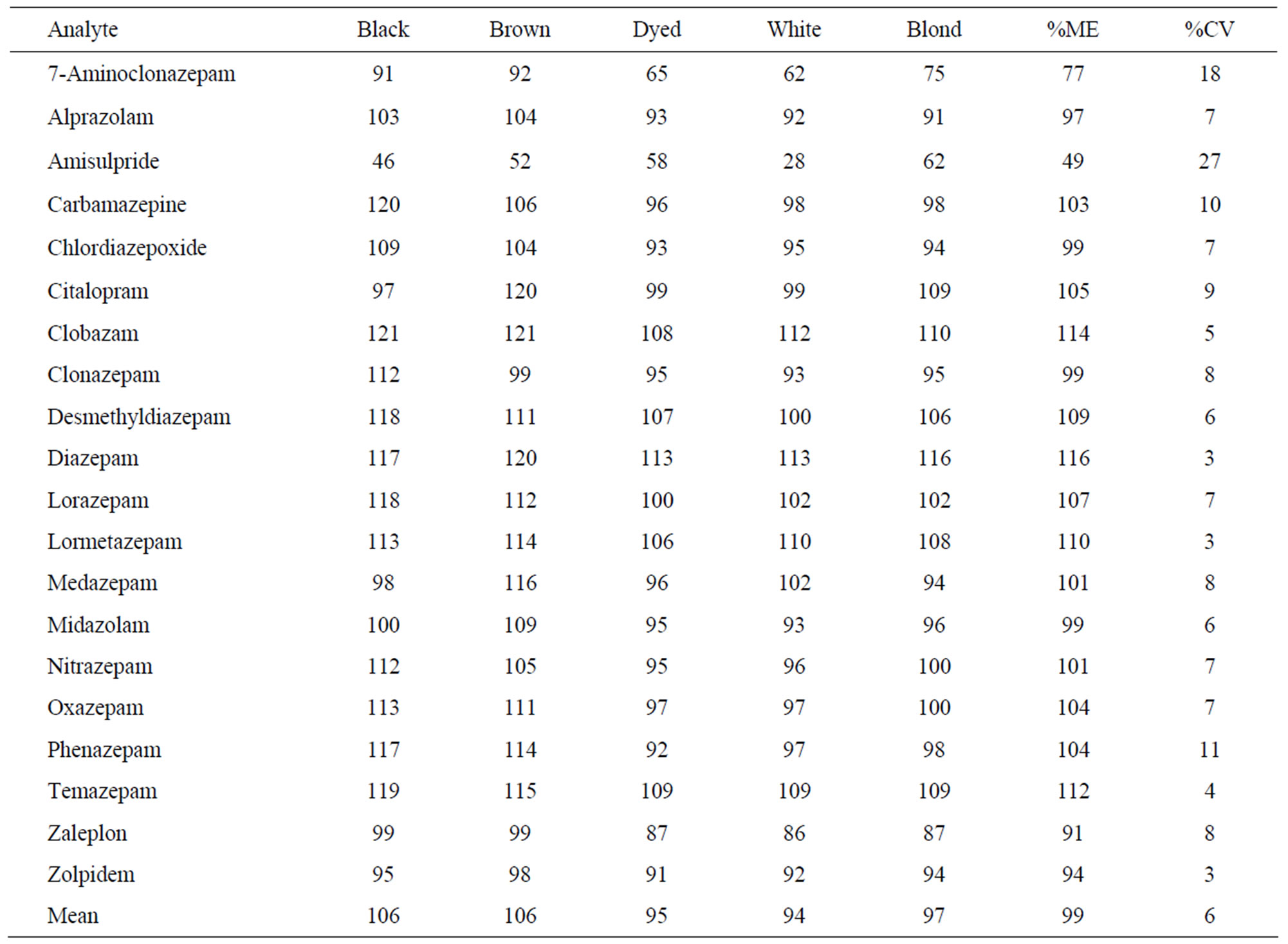
Table 4. Matrix effect (ME) results for 0.2 ng/mg hair samples spiked with the analytes (n = 5).
with the mean peak area ratio of product ion/internal standard of unextracted/unincubated standards prepared in 25% MeOH at the same concentration (n = 3).
3.4.9. Stability/Recovery with Alkaline Digestion
Extraction of hair with various solvents following alkaline digestion has been widely mentioned in many publications [11,19-21]. In order to investigate whether the proposed method has any advantages over the alkaline digestion method, the first step was to check how stable the analytes would be during the incubation/extraction with NaOH/MTBE. Twenty five milligrams of blank/ washed hair was incubated for 3 hours at 50˚C with the analytes at a concentration of 1 ng/mg and mixed with 0.5 mL 1 M NaOH. Extraction was with 2 mL of MTBE on an orbital shaker for 30 minutes followed with back extraction with 150 µL of 1% formic acid. Twenty microlitres were injected into the LCMSMS system.
The stability results shown in Table 5 indicate that almost all the analytes are stable under the extraction/incubation conditions of the methanolic method. The least stable analyte was oxazepam with 56.1% recovery. The average recovery was 89.6% with coefficient of variation of 4.2%. While stability of analytes under the alkaline condition was remarkably lower with an average of 51.7%. The results are also demonstrated in Figure 1.
3.5. Real Samples (Methanolic vs. Alkaline Extraction)
The methanolic method was applied to 10 cases, one of them postmortem. Hair samples were taken from the posterior vertex region of the scalp. Nine of these cases were patients taking doses of different classes of basic drugs. Seven of the nine were addicts under rehabilitation.
About 0.6 cm segment from the root end (proximal) of each sample was decontaminated and extracted by the methanolic method. However, there was an additional step added to the analysis of those cases. After full incubation and extraction steps, hair sample remained in the tubes were not discarded. Instead they were re-extracted with the alkaline digestion method described earlier. Results are shown in Table 6 and Figure 2 and discussed in the following section. Some of the real samples chromatograms are shown in Figure 3.
4. Discussion and Conclusion
The development of the proposed basic drugs extraction method was based on previous studies showing that methanolic and aqueous solution is capable of extracting drugs from hair samples [2]. However, a comparison between this extraction solution and the digestion with
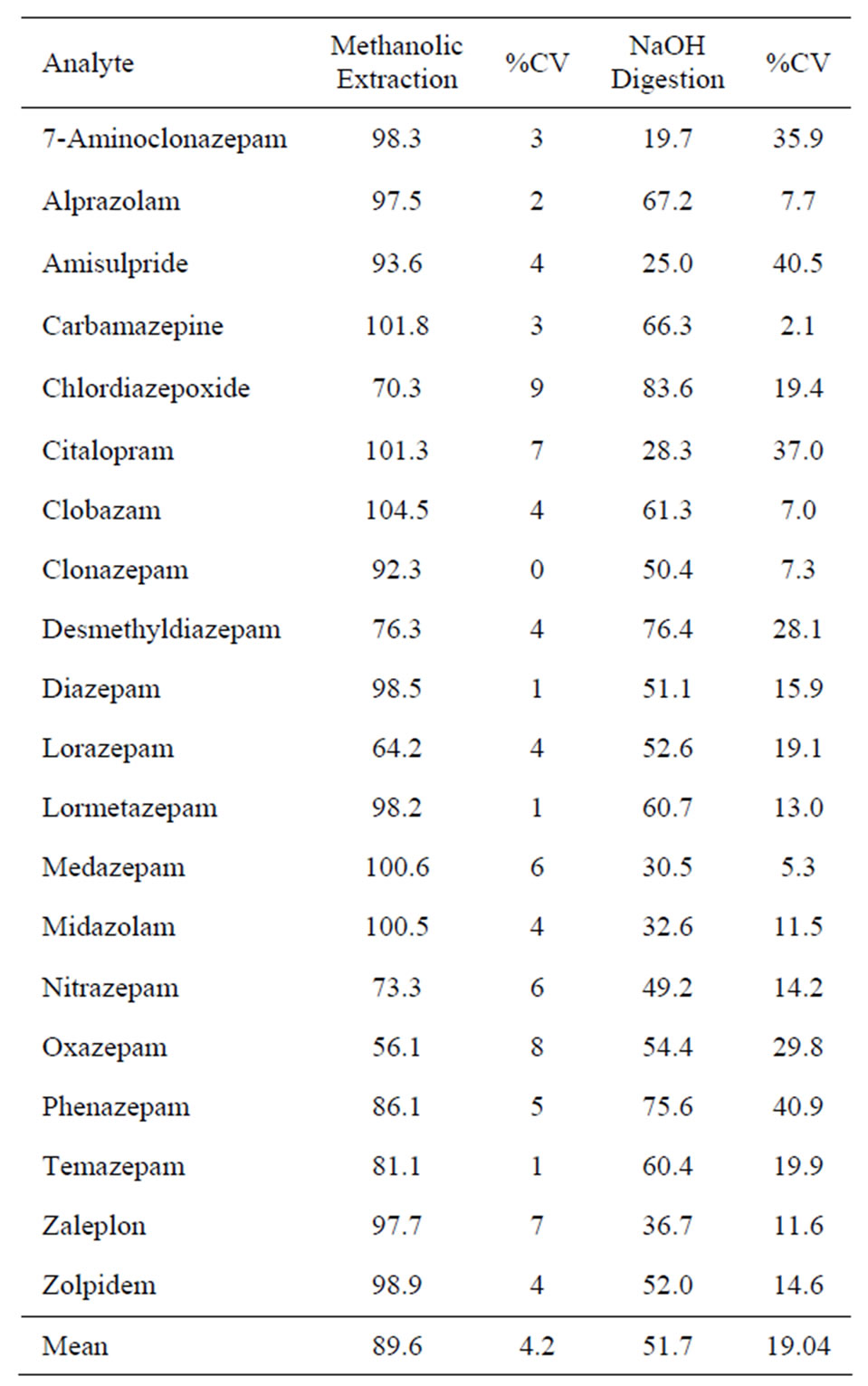
Table 5. Stability (%) of the analytes after sonication/incubation at 50˚C/18h (n = 3) and after extraction and digestion with 1 M NaOH at 50˚C/3h (n = 2).
NaOH was not probably suggested in this specified wayparticularly with general basic drugs screening. Therefore, the mixture of methanol, formic acid and acetonetrile (methanolic solution) was proposed to extract basic drugs. Methanol and 0.1% formic acid are protic solvents and capable of swelling the hair and attracting/ionizing basic drugs entrapped inside the hair shaft, at least theoretically. Acetonitrile was added in a small amount (10%) as it was recommended by some articles [15,22].
The first early recovery experiments with the methanolic method produced extracts that were turbid in colour which brought about the necessity of adding a filtration step to the method and checking the effect of these filters on the drugs’ recovery. Mini-UniPrep filters were chosen as they are syringeless, simple and fast. As these filters come in different materials, a comparison study was carried out and showed that PTFE based filters gave the highest recoveries for the analytes.
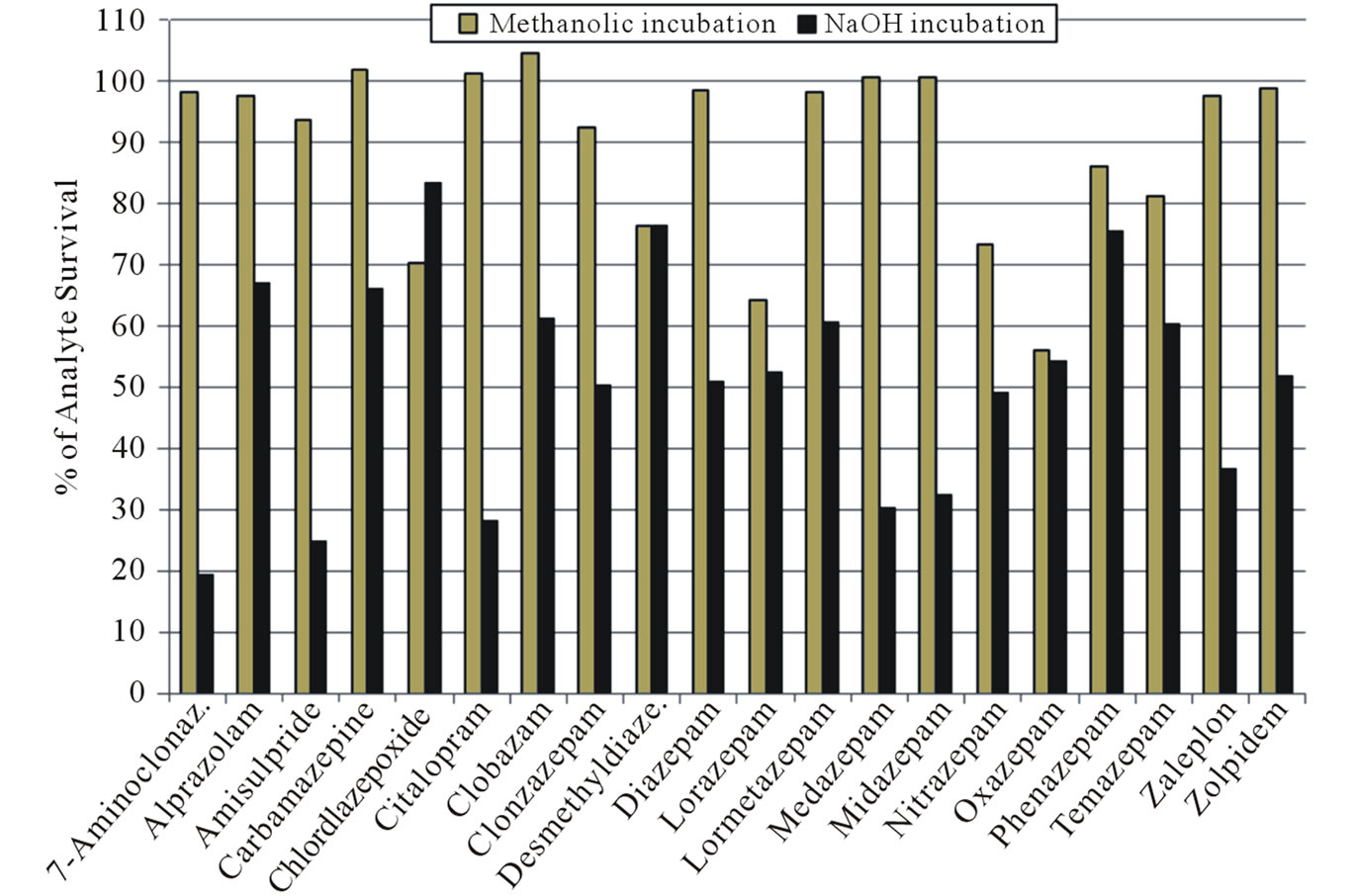
Figure 1. Stability of analytes during methanolic and alkaline conditions.
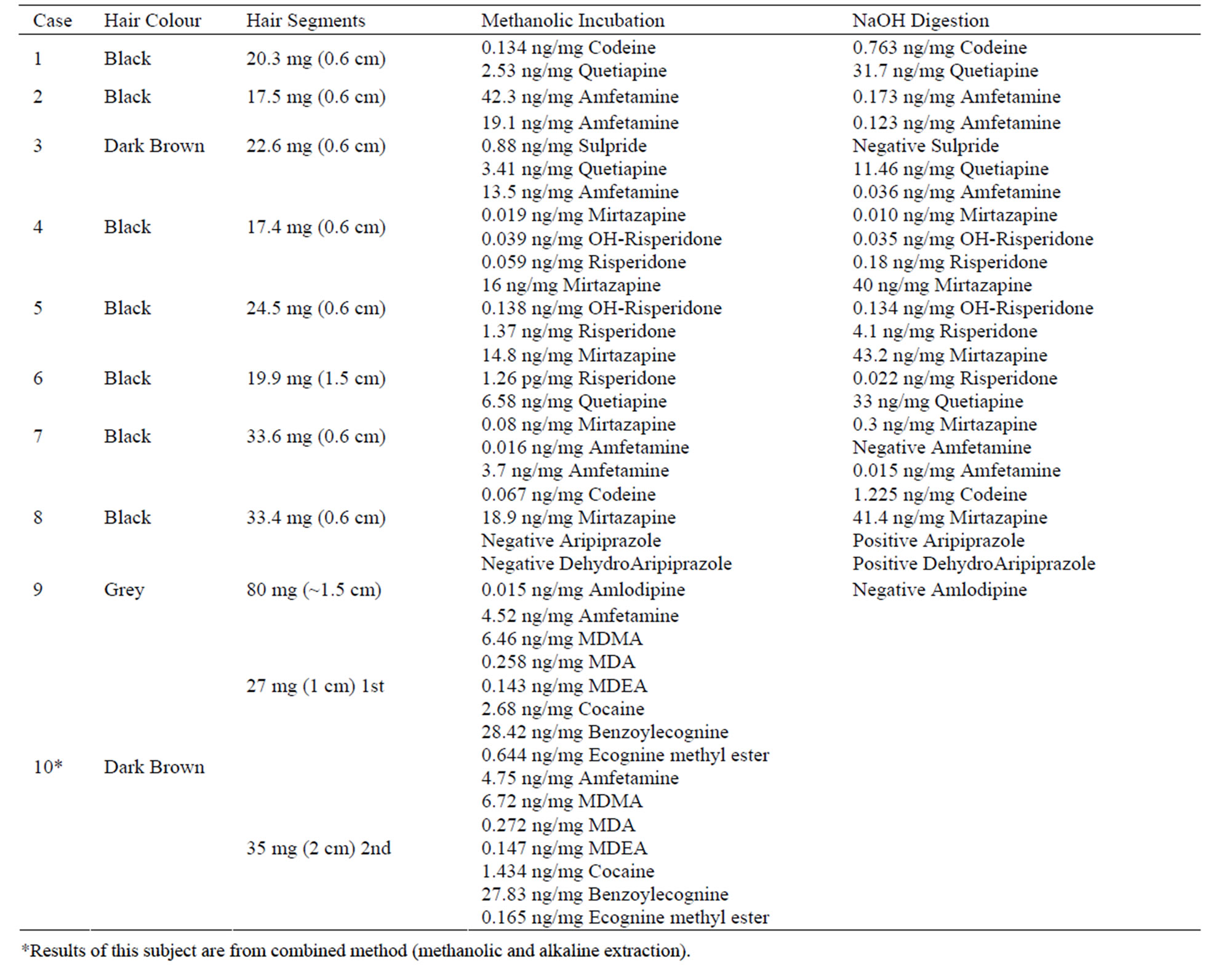
Table 6. Real samples: comparison of results.
*Results of this subject are from combined method (methanolic and alkaline extraction).
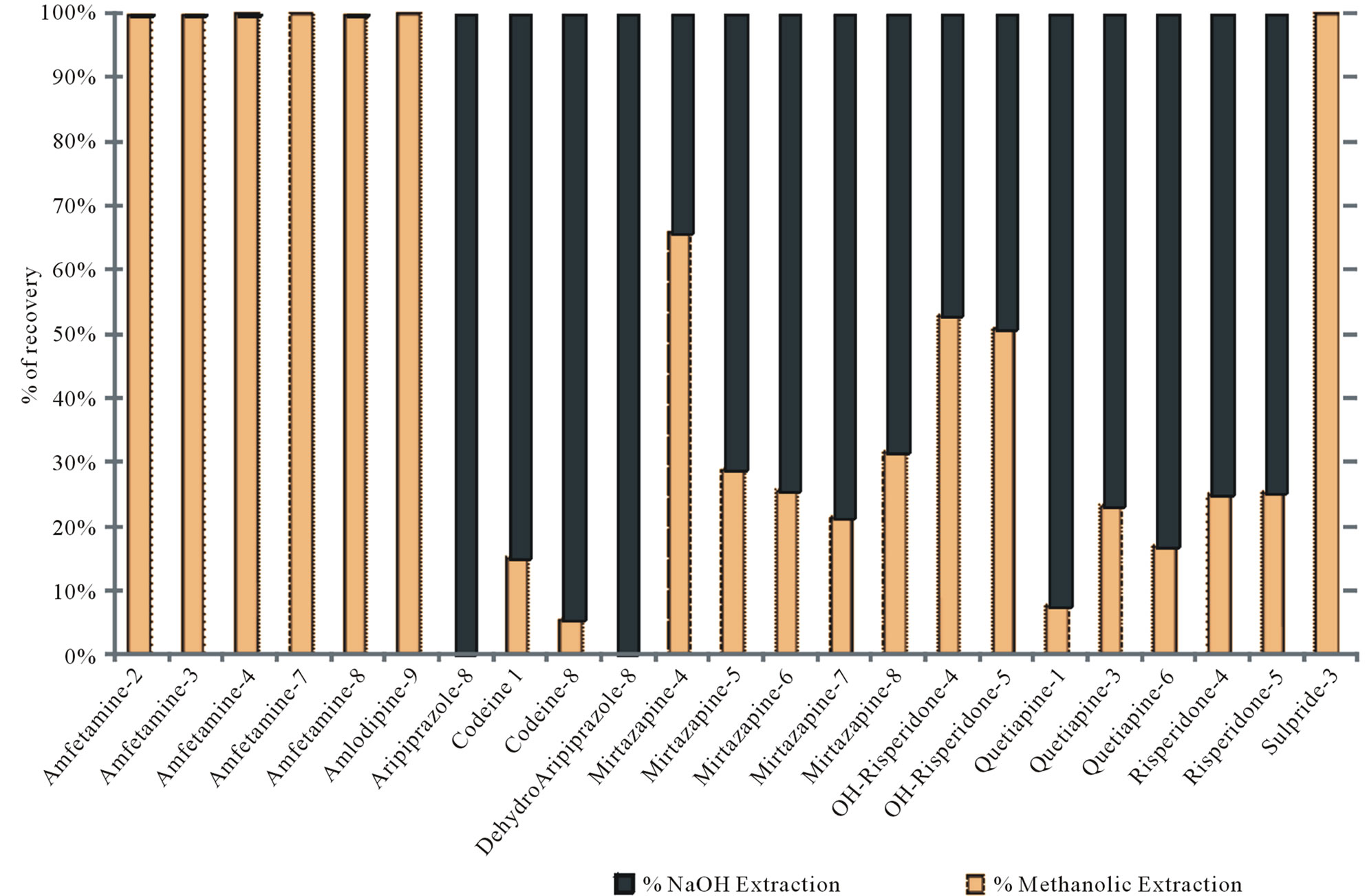
Figure 2. Real samples: comparison of results for methanolic and alkaline extraction (Drug-Case No.).
It was seen from the stability results that digestion with NaOH reduces the stability of most of the analytes whereas methanolic incubation has no such effect on stability as the great majority of drugs showed >90% stability with or without blank hair.
Some method validation steps have been carried out on the methanolic method, such as recovery, LOD/LOQ, matrix effect and selectivity. Absolute recovery experiments showed that all the analytes have a recovery ranging between 58.4% and 119.3% with a CV from 0.2 to 13.4% after excluding the results of two drugs. Amisulpride and citalopram gave low recoveries (12.9% - 29.8%, CV 2.5% - 34.7%). During the matrix effect study five different sources of hair have been incorporated (different colours and ethnicities). Results showed that all the drugs have ME ranging between 91.4 and 115.6% (CV 3.2% - 11.1%) except 7-aminoclonazepam and amisulpride (ME 77% and 49.1%, CV 18.4% and 26.8%, respectively). The selectivity study also showed no major problem affecting the method selectivity as no analyte eluted in the same retention time and window of the other analytes and no false positive from blank hair.
Last but not least, analysis of authentic hair samples from a group of subjects taking basic or weak basic drugs revealed the usefulness of the proposed method at least for the detection of basic drugs in hair. To further examine the extent of extraction the same samples were digested with 1 M NaOH after the methanolic extraction to compare the extractability of both methods on the administered drugs in a way by using the exact same samples. Results revealed that the methanolic method was able to extract most of the investigated drugs from hair samples, but the downside about it is the fact that it only extracts part of the incorporated drug in hair. In addition, this part or this percentage of the incorporated drug varied between the investigated compounds. Results showed that 99% to 100% of detected amfetamine from authentic hair samples was extracted by the methanolic method as shown in Figure 3, while less than 1% was revealed by the alkaline digestion. Amlodipine and sulpiride were only detected by the methanolic extraction method. On the contrary, aripiprazole and its active metabolite (dehydroaripiprazole) were revealed only with the alkaline digestion. Codeine showed a weak response with the methanolic method as only 5% - 15% of the incorporated drug revealed, while the 85% - 95% of it was detected with the alkaline method. Mirtazapine was also recovered better with the alkaline digestion (69% - 79%) except in one sample (35%). Similarly, risperidone was recovered better with the alkaline digestion (75%) while
Figure 3. Some of the real sample chromatograms, between brackets are precursor ion/product ion.
its metabolite showed a 50/50 detection in both methods. Quetiapine as well showed like pattern to risperidone and mirtazapine with a 77% - 93% found with the alkaline digestion method.
These results suggest that the incorporated drugs in hair need either complete digestion of the hair samples or maybe powdering the samples to ensure complete release of the entrapped compounds. Although some drugs have been extracted or revealed only by methanolic method without digestion, like sulpiride and amlodipine, it does not mean that the percentage of recovery was really 100%. It is likely that these drugs were not stable under NaOH digestion which may have led to their degradation and disappearance. High pH possibly has caused hydrolysis to both amlodipine’s ester moiety and sulpride’s amide type link.
In conclusion, the methanolic method is capable of extracting most basic drugs in hair samples but only part of the total incorporated drug. As all the drug in the real samples were detected by the methanolic method except one drug, aripiprazole. Therefore, these results suggest that a combination of both methods (methanolic and alkaline extractions) in hair sample processing for the detection of basic and weak basic drugs seems to provide better extraction yield than either method alone. However, not all basic drugs are suitable for alkaline digestion. A further study with more real samples is needed and preferably in comparison with other hair extraction techniques, such as powdering the hair and other digestion methods.
4.1. Limitations
Ideally, the method should be applied to more samples especially from subjects taking the same drugs as the drugs used in the method development. Unfortunately, only those patients who were studied agreed to participate. Also, the access time on the LCMSMS was very limited and optimization of every variable was not feasible. Moreover, the method is intended to analyse basic drugs in general and basic drugs are very common so it is almost impossible to include all of them. Alkaline digestion has limited application to only analytes stable under basic conditions, while methanolic extraction showed partial extraction ability even after 18 hours of incubation.
4.2. Ethical Notes
Collection of hair samples from patients was performed after receiving the written approval of the health research ethics committee in ministry of health, Kuwait and informed consent was also signed by patients after explanation of the study and its aim.
REFERENCES
- I. M. Kempson and E. Lombi, “Hair Analysis as a Biomonitor for Toxicology, Disease and Health Status,” Chemical Society Reviews, Vol. 40, No. 7, 2011, pp. 3915- 3940. doi:10.1039/c1cs15021a
- P. Kintz, “Analytical And Practical Aspects of Drug Testing in Hair,” CRC Press, New York, 2007.
- F. Pragst and M. A. Balikova, “State of the Art in Hair Analysis for Detection of Drug and Alcohol Abuse,” Clinica Chimica Acta, Vol. 370, No. 1-2, 2006, pp. 17-49. doi:10.1016/j.cca.2006.02.019
- R. Smolders, K. W. Schramm, M. Nickmilder and G. Schoeters, “Applicability of Non-Invasively Collected Matrices for Human Biomonitoring,” Environmental Health, Vol. 8, No. 1, 2009, p. 8. doi:10.1186/1476-069X-8-8
- F. Musshoff and B. Madea, “New Trends in Hair Analysis and Scientific Demands on Validation and Technical Notes,” Forensic Science International, Vol. 165, No. 2-3, 2007, pp. 204-215. doi:10.1016/j.forsciint.2006.05.024
- M. Villain, M. V. C. Concheiro and P. Kintz, “Screening Method for Benzodiazepines and Hypnotics in Hair at pg/mg Level by Liquid Chromatography-Mass Spectrometry/Mass Spectrometry,” Journal of Chromatography B, Vol. 825, No. 1, 2005, pp. 72-78. doi:10.1016/j.jchromb.2004.12.036
- M. Laloup, M. Ramirez Fernandez Mdel, G. De Boeck, M. Wood, V. Maes and N. Samyn, “Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method for the Simultaneous Determination of 26 Benzodiazepines and Metabolites, Zolpidem and Zopiclone, in Blood, Urine, and Hair,” Journal of Analytical Toxicology, Vol. 29, No. 7, 2005, pp. 616-26.
- F. Musshoff and B. Madea, “Analytical Pitfalls in Hair Testing,” Analytical and Bioanalytical Chemistry, Vol. 388, No. 7, 2007, pp. 1475-1494. doi:10.1007/s00216-007-1288-x
- F. J. Couper, I. M. McIntyre and O. H. Drummer, “Extraction of Psychotropic Drugs from Human Scalp Hair,” Journal of the Forensic Science, Vol. 40, No. 1, 1995, pp. 83-86.
- M. Josefsson, R. Kronstrand, J. Andersson and M. Roman, “Evaluation of Electrospray Ionisation Liquid Chromatography-Tandem Mass Spectrometry for Rational Determination of a Number of Neuroleptics and Their Major Metabolites in Human Body Fluids and Tissues,” Journal of Chromatography B, Vol. 789, No. 1, 2003, pp. 151- 167. doi:10.1016/S1570-0232(03)00207-1
- J. Villamor, A. Bermejo, P. Fernandez and M. Tabernero, “TECHNICAL NOTE: A New GC-MS Method for the Determination of Five Amphetamines in Human Hair,” Journal of Analytical Toxicology, Vol. 29, No. 2, 2005, pp. 135-139.
- R. Gottardo, F. Bortolotti, G. De Paoli, J. P. Pascali, I. Miksík and F. Tagliaro, “Hair Analysis for Illicit Drugs by Using Capillary Zone Electrophoresis-Electrospray Ionization-Ion Trap Mass Spectrometry,” Journal of Chromatography A, Vol. 1159, No. 1-2, 2007, pp. 185- 189. doi:10.1016/j.chroma.2007.01.0118
- S. Broecker, S. Herre and F. Pragst, “General Unknown Screening in Hair by Liquid Chromatography-Hybrid Quadrupole Time-of-Flight Mass Spectrometry (LCQTOF-MS),” Forensic Science International, Vol. 218, No. 1, 2012, pp. 68-81.
- M. K. K. Nielsen, S. S. Johansen, P. W. Dalsgaard and K. Linnet, “Simultaneous Screening and Quantification of 52 Common Pharmaceuticals and Drugs of Abuse in Hair Using UPLC-TOF-MS,” Forensic Science International, Vol. 196, No. 1-3, 2010, pp. 85-92. doi:10.1016/j.forsciint.2009.12.027
- S. Hegstad, H. Z. Khiabani, L. Kristoffersen, N. Kunøe, P. P. Lobmaier and A. S. Christophersen, “Drug Screening of Hair by Liquid Chromatography-Tandem Mass Spectrometry,” Journal of Analytical Toxicology, Vol. 32, No. 5, 2008, pp. 364-372.
- C. Hartmann, J. Smeyers-Verbeke, D. Massart and R. McDowall, “Validation of Bioanalytical Chromatographic Methods,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 17, No. 2, 1998, pp. 193-218. doi:10.1016/S0731-7085(97)00198-2
- F. T. Peters, O. H. Drummer and F. Musshoff, “Validation of New Methods,” Forensic Science International, Vol. 165, No. 2-3, 2007, pp. 216-224. doi:10.1016/j.forsciint.2006.05.021
- B. K. Matuszewski, M. L. Constanzer and C. M. ChavezEng, “Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLCMS/MS,” Analytical Chemistry, Vol. 75, No. 13, 2003, pp. 3019-3030. doi:10.1021/ac020361s
- A. Pelander, J. Ristimaa, I. Rasanen, E. Vuori and I. Ojanperä, “Screening for Basic Drugs in Hair of Drug Addicts by Liquid Chromatography/Time-of-Flight Mass Spectrometry,” Therapeutic Drug Monitoring, Vol. 30, No. 6, 2008, pp. 717-724. doi:10.1097/FTD.0b013e3181897cfa
- M. Villain, “Determination of Bromazepam, Clonazepam and Metabolites after a Single Intake in Urine and Hair by LC-MS/MS: Application to Forensic Cases of Drug Facilitated Crimes,” Forensic Science International, Vol. 145, No. 2-3, 2004, pp. 123-130.
- R. Stanaszek and W. Piekoszewski, “Simultaneous Determination of Eight Underivatized Amphetamines in Hair by High-Performance Liquid Chromatography-Atmospheric Pressure Chemical Ionization Mass Spectrometry (HPLC-APCI-MS),” Journal of Analytical Toxicology, Vol. 28, No. 2, 2004, pp. 77-85.
- R. Kronstrand, I. Nyström, J. Strandberg and H. Druid, “Screening for Drugs of Abuse in Hair with Ion Spray LC-MS-MS,” Forensic Science International, Vol. 145, No. 2-3, 2004, pp. 183-190. doi:10.1016/j.forsciint.2004.04.034