Pharmacology & Pharmacy
Vol.3 No.2(2012), Article ID:18704,5 pages DOI:10.4236/pp.2012.32019
Topiramate: An in Vitro and in Vivo Comparison between the Pharmacokinetic Properties of a Generic (Sincronil) and the Reference (Topamax) Formulation
![]()
1Fidia Farmaceutici, Abano Terme, Italy; 2Pharmanet, Montreal, Canada; 3Department of Pharmacology, University of Firenze, Florence, Italy.
Email: flavio.moroni@unifi.it
Received January 15th, 2012; revised February 19th, 2012; accepted March 5th, 2012
Keywords: Topiramate; Migraine; Pharmacokinetics
ABSTRACT
The use of generic formulation of therapeutic agents may allow a significant reduction of costs for patients and the National Health Services. This is particularly true for drugs used in prolonged therapies such as topiramate which is effective in the treatment of epilepsy, migraine, alcohol abuse and psychiatric conditions. The purposes of this study were: 1) evaluate Topiramate (50 mg) release in vitro from a generic (Sincronil) and the reference formulation (Topamax); 2) compare the above mentioned generic and reference formulations in bioavailability studies in healthy volunteers. Dissolution tests in vitro showed that more than 95% of the active principle was released within 15 minutes both from the reference and the generic formulation. No difference in release kinetics was found between the two topiramate preparations. In vivo pharmacokinetic data were obtained by administering 1 tablet containing 50 mg of topiramate of each of the two formulations to 28 healthy volunteers under fasting conditions, using a randomized, single-dose, open-label, 2-way crossover design. The treatment phases were separated by a washout period of 21 days. The maximum concentration reached in plasma (Cmax) for the reference and the generic formulation, were 946 ± 308 and 849 ± 247 (ng/mL) and the area under the curve (AUC0-t) were 35,900 ± 7800 and 34,300 ± 8100 (ng·h/mL) respectively. The data indicate that the rate and extent of absorption of the reference or generic 50 mg topiramate formulation are not significantly different and suggest that the therapeutic effects of the two preparations do not significantly differ.
1. Introduction
The use of generic drugs significantly increased in the last ten years both in Europe and the United States [1,2] and, reasonably, this trend will continue because, in the next few years, patent protection of widely used therapeutic agents will expire. The lower cost of generics in comparison of the original (innovator) products is expected to reduce therapeutic expenses for patients and the National Health Services. In order to have safe and low cost generic drugs, increasing attention is devoted to the methods that should be applied to test the bioequivalence of these agents with the originator product [3]. Official regulations regarding generics require tests to establish whether the pharmacokinetic parameters are within established limits. A single dose cross over study with appropriate sample size and the evaluation of the most relevant pharmacokinetic parameters, such as the maximum concentration reached in plasma (Cmax) and the area under the curve (AUC) contrasting plasma concentration with time after the administration of either the test or the reference compound is now considered sufficient to establish bioequivalence of small molecules. Topiramate is a small molecule widely used for the treatment of epilepsy, migraine, bipolar disorders and alcoholism [4,5] and its availability as a generic formulation could significantly reduce the national health service costs since the total sales of the reference product in Europe reach approximately 3 bilion Euro. Recent publications, however, by focusing on anecdotal reports, attempt to question the pharmacokinetic and therapeutic bioequivalence of reference and generic products [6-8]. Most of the medical literature, however, does not consider a problem the substitution of the originator with a generic drug even when the therapeutic index is narrow [8,9]. One of the parameter to be considered in bioequivalence evaluation is the dose to be tested and since most of the studies so far performed to obtain generic formulations of topiramate compared preparations containing 200 mg [9] we report here bioequivalence studies performed with a significantly lower dose (50 mg). In fact, while epileptic patients, often assume preparations of 200 mg, the prescription of topiramate for other medical conditions such as migraine [10] or alcoholism [5] may suggest the use of lower doses.
2. Material and Methods
2.1. Dissolution Test in Vitro
Dissolution test was performed using a paddle apparatus at 75 rpm and 500 ml of HCL 0.01 N. The measurement of released topiramate at different time intervals (5, 10, 15, 20, 25 and 30 min) was performed for 50 mg filmcoated tablets. The reference we used was Topamax (Jansen-Cilag Ltd. batch 03HS042 and the generic was Sincronil (Pharmathen S.A. batch 053024). Each batch was tested 6 times using an HPLC Liquid Chromatographer Shimadzu LC10AD with RID-10A detector. The Column was Inertsil ODS-2, 250 mm lenght, 4.6 mm internal diameter; the mobile phase was a mixture (50% each) of acetonitrile and water and the flow rate was set at 1.0 ml/min. Column temperature was 30˚C while for RID detector the cell temperature was 50˚C.
2.2. In Vivo Study
This was a single-center, randomized, single-dose, openlabel, 2-way crossover bioequivalence study to compare the rate and extent of absorption of 50 mg film coated tablets of Sincronil versus Topamax, under fasting conditions. The study was conducted at Anapharm Inc. (now known as Pharmanet, Sainte-Foy, Quebec, Canada). The Clinical Study Protocol was approved in the Ethics Review Committee (Approval Form dated May 25, 2004) and a letter of non-objection from the Canadian heath authorities was obtained. All clinical work was conducted in compliance with Good Clinical Practice Rules (GCP) as referenced in the ICH guidelines (ICH E6), local regulatory requirements, and the principles enunciated in the Declaration of Helsinki. Healthy adult subjects, aged 18 - 55 years, body mass indices below 30.0 kg/m2 were judged eligible for enrolment in this study, on the basis of medical and medication histories, demographic data (including sex, age, race, body weight [kg], height [cm] and BMI [kg/m2]), vital signs, 12-lead ECG, physical examination, a urine drug screen, urine pregnancy tests (female subjects), clinical laboratory tests (hematology, biochemistry, urinalysis, HIV and hepatitis C [HCV] antibodies, and hepatitis B antigen [HBSAg]).
A total of 35 healthy subjects signed the study-specific Informed Consent Form; of these subjects, 28 (16 males and 12 females) were dosed and were enrolled in the study; 27 of these enrolled subjects completed the study. Prior to study commencement, subjects were randomly assigned to a treatment in accordance with the randomization scheme generated by Anapharm Inc. Subjects were confined to the Anapharm Clinical Research Facility from at least 10 hours prior to drug administration until after the 24.0 hour post-dose blood draw, in each of the two periods. Single oral doses were separated by a washout periods of 21 days. Treatment was a single-dose of 50 mg filmcoated tablet per subject in accordance with the randomization scheme, either of the test preparation (Pharmathen S.A., Greece, batch 053024) or the reference formulation (Topamax, Janssen-Cilag Ltd., UK, batch 03KS24H).
2.3. Blood Sampling and GC/MS Assay Method
Blood samples were collected prior to drug administration and 0.333, 0.667, 1.00, 1.25, 1.50, 1.75, 2.00, 2.33, 2.67, 3.00, 3.50, 4.00, 5.00, 6.00, 8.00, 12.0, 24.0, 48.0, 72.0, 96.0, 144, 192, and 240 hours post-dose (50 mg) in each period. Plasma concentrations of Topiramate were then evaluated using Gas-Chromatographic separation with Mass Spectrometry Detection (GC/MS). Briefly, a known amount of 1,2:5,6-Di-O-isopropylidene-3-O- (methylsulfoyl)-α-D-glucofuranose (Aldrich; Oakville, Canada) used as the internal standard and 0.5 ml of phosphate buffer (0.5 M pH 7) were added to 0.250 ml of plasma and the mixture was placed on a activated extraction cartridge (C8, 1 ml/100 mg; Varian, Mississauga, Canada) and centrifuged at 400 rpm for 5 min. The cartridge was washed with 1 ml water and ten with 0.3 ml hexanes. Finally, topiramate was eluted with 0.75 ml ethyl acetate, dried under N2, re-suspended in pure methanol (0.1 ml) and allowed to react with 0.2 ml 2 M (trimethylsilyl) diazomethane in hexanes (Aldrich, Oakville, Canada) at 40˚ for 45 min. The reaction mixture was evaporated under N2 in a Turbo Vap, reconstituted with isooctane (0.075 ml) and placed in capped vials fitting the automatic sampler. The GC/MS apparatus consisted in the automatic sampler HP7683 (Hewlett Packard, Palo Alto, CA, USA), a gas chromatograph Hewlett Packard 6890, an analytical column HP1 15 m × 0.25 mm × 0.25 µm (Agilent technology; Mississauga, Canada), a mass selective detector Hewlett Packard 5973 and a data integration computer Chemstation G1701 BA (Hewlett Packard). The chromatographic conditions were: Injection temperature 250˚C; Oven 190˚C; Transfer line 280˚C and Source 230˚C. Carrier gas was Helium at a flow rate of 1.3 mL/min. Detection was performed at m/z 323 for the internal standard and at m/z 352 for topiramate. Another m/z routinely monitored for topiramate was 338. The retention time for the internal standard was approximately 2.16 and for topiramate 3.70 min. Quantitation was based on the ratio of the peak areas of topiramate and the internal standard on the basis of a calibration curve ranging from 9.97 (lower limit of detection) to 1495 ng/ml. Variation between-run was 2.74%. All the determinations were carried out at Anapharm Laboratories (Canada) using a validate procedure (ANI 8347.02).
2.4. Statistical Analysis
Plasma topiramate concentrations were determined in each subject in order to directly obtain the maximum plasma concentration (Cmax) and the time to maximum plasma concentration (Tmax). The other pharmacokinetic parameters including the half-time of drug elimination during the terminal phase (t1/2), the area under the plasmaconcentration-time curve from 0 to the last measurable time (AUC0-t) or from 0 to infinity (AUC0-inf) were estimated using a non-compartmental model method and the computer program Bioequiv (version 3.40). ANOVA was performed and a non-parametric test (Wilcoxon’s Signed-Rank test) was carried out to compare the Tmax between treatments. Ratios of least-squares means and 90% geometric confidence interval were calculated for ln-transformed AUC0-t, AUC0-inf and Cmax. The safety data were evaluated descriptively and no statistical analysis was performed.
2.5. Tolerability Assessment
The following clinical laboratory tests were performed for each subject at the time of the screening and poststudy procedures: Hematology: Complete blood cell count, hemoglobin, hematocrit; Biochemistry: sodium, potassium, chloride, phosphate and calcium ions, glucose, total bilirubin, creatinine, urea nitrogen, total protein, albuminalkaline phosphatase, alanine aminotransferase (ALT/ SGPT), aspartate aminotransferase (AST/SGOT). Urinalysis: pH, specific gravity, protein, glucose, ketones, bilirubin, occult blood and cells, nitrite, urobilinogen, leukocytes microscopic examination. Physical examinations (blood pressure, heart rate, respiratory rate, and oral temperature) and electrocardiogram were performed at the time of screening. Throughout the study, subjects were monitored for adverse events. At the admission, subjects were asked a standard probe question concerning the onset of a new illness since their last visit and at the time of discharge, each subject was asked how he/she was feeling. All adverse events included those reported within 21 days following last drug administration, were recorded onto appropriate data sheets.
3. Results
3.1. In Vitro Data
The release of the active principle was measured in vitro under standard conditions from the test 50 mg tablet formulation (Sincronil) and from the reference product (Topamax). The data reported in Table 1 show that the two products almost completely released topiramate in 15 min, with a large percentage of the process taking place in the first 5 minutes. The variability between tests was rather low and the results strongly suggest that the rate and extent of the solubility process of the two formulations is not significantly different.
3.2. In Vivo Data
We evaluated 28 adult healthy subjects (16 males and 12
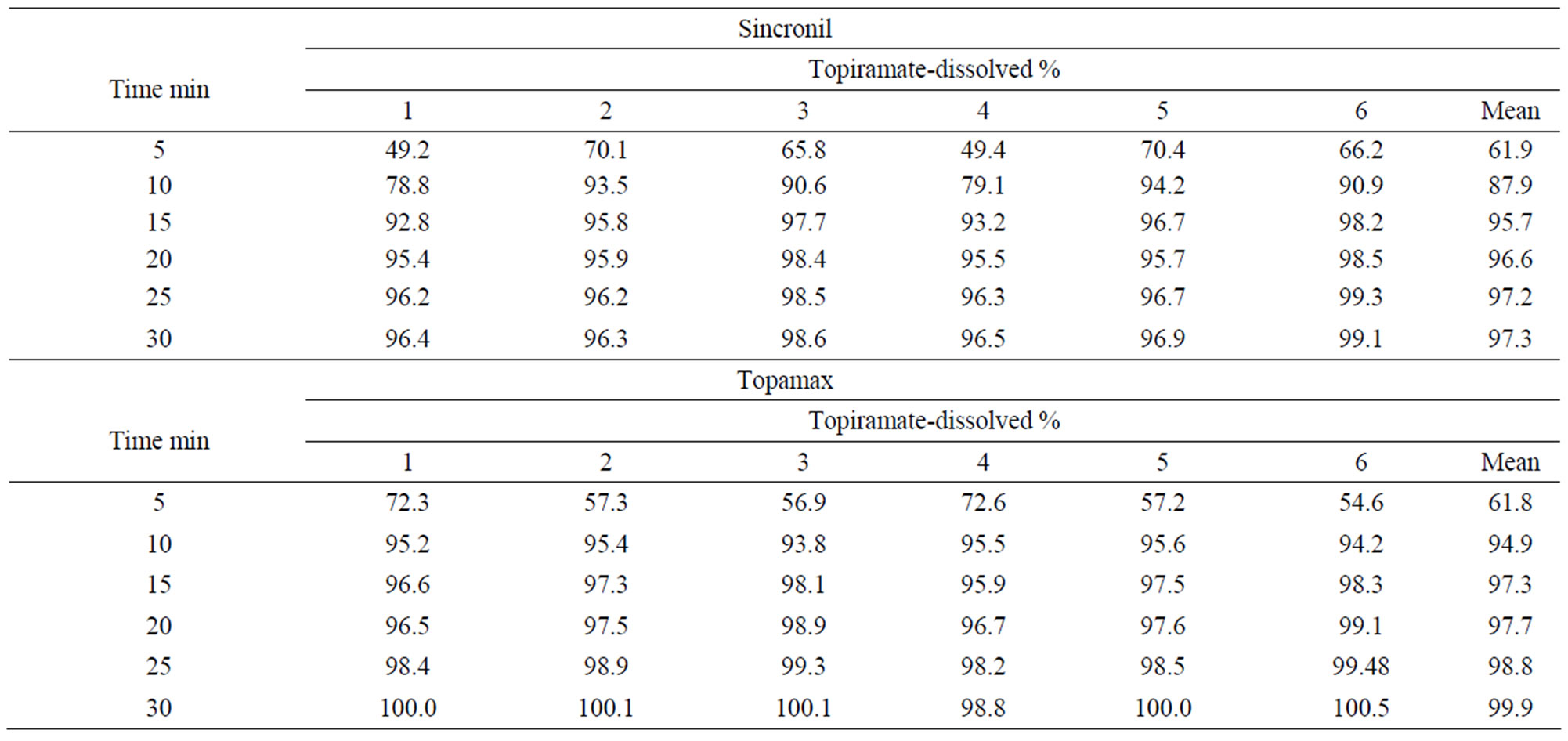
Table 1. Solubility of a 50 mg Sincronil film coated tablet and the tablet Topamax 50 mg (Batch 03HS042).
females). In accordance with the study protocol, the results obtained in the first 24 subjects who completed the study, were used for pharmacokinetic and statistical analysis. None of the subject treated with 50 mg tablets had serious or significant adverse events. However, 35 post-dose complaints were mentioned and recorded and their relationship with drug treatment was judged as “probable” for 2, “possible” for 17, “remote”, for 6 and “unrelated” for 10. These events were either mild (24) or moderate (9) with “headache”, reported in 10 occasions, being the most frequent complaint. In 2 cases it was impossible to have data on the onset of the events and it was therefore impossible to ascribe them to either the reference or the test compound. For the remaining 33 events recorded, 20 occurred following the administration of the test (Sincronil) and 13 following administration of the reference compound (Topamax). Topiramate was detectable in all the subjects after administration while it was undetectable before the second administration, with one exception. A subject treated with the test formulation had low but detectable plasma levels of topiramate (18 ng/ml, a value close to the detection limit) before administration of the reference formulation. Figure 1 shows the time course of the mean plasma topiramate levels after either the reference or the test preparation. Table 2, shows that the AUC0-t was 35,943 ± 7835 for the reference and 34,323 ± 8157 (ng·h/mL) for the test formulation indicating that both products behave quite similarly in terms of total absorption. Furthermore, the Cmax was 946 ± 307 and 849 ± 247 ng/mL (means ± SD) respectively, again indicating very similar behavior in terms of the rate of drug absorption. Finally, as reported in Table 3, the ratio of least-squares means of the AUC0-inf calculated for Sincronil and Topamax was 94% and was well within the 90% geometric confidence interval required to accept the bioequivalence of the products. In a similar manner, the least-squares means for the AUC0-t and those for Cmax were well within the 90% geometric confidence interval.
4. Discussion
Current regulations recommend performing solubility evaluation and a randomized, two-period, two-sequence single dose crossover design study in order to evaluate the bioequivalence of two preparations of the same drug. In this work we report that two formulations, Topamax (the reference) and Sincronil (the generic), both containing 50 mg of topiramate, had a comparable rate and extent of solubility process in vitro and, when tested in a group of healthy volunteers, their pharmacokinetic profiles were well within the bioequivalence required limits and none of them caused significant side effects. To the best of our knowledge, this is the first time that pharma-

Figure 1. Time course of plasma topiramate concentrations in healthy volunteers treated with Topomax (open squares) or with Sincronil (cruciform lines) 50 mg film coated tablets. Each value is the mean of the values obtained in 24 subjects.
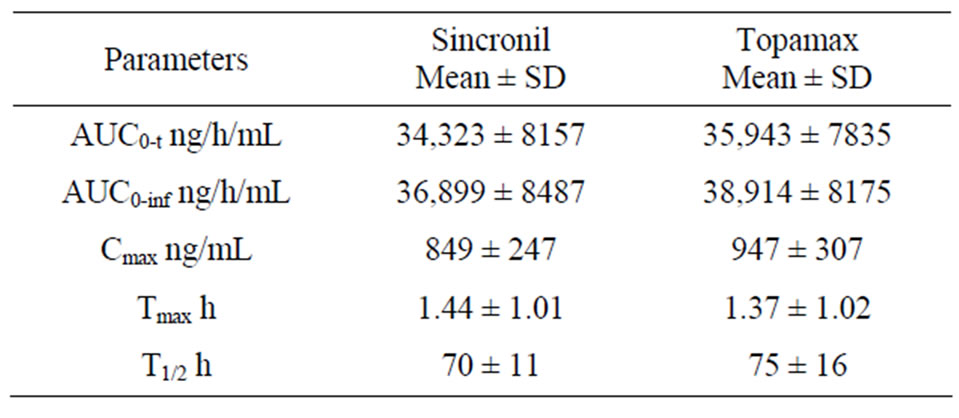
Table 2. Pharmacokinetic parameters of Sincronil and Topamax.
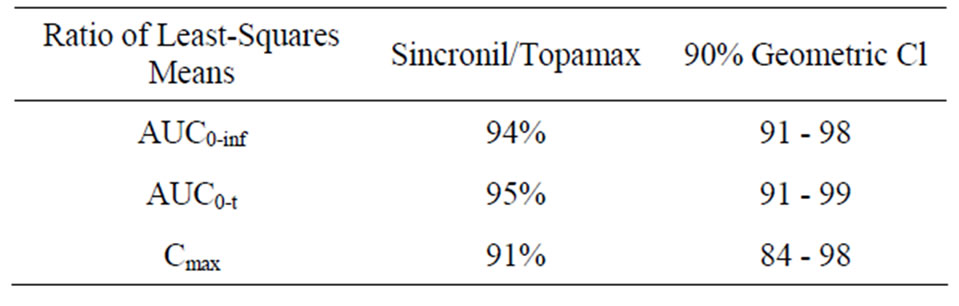
Table 3. Bioequivalence of Topamax and Sincronil as evaluated with the ratio of the least squares means and the 90% geometric confidence interval (CI) of the AUC0-inf AUC0-t and Cmax.
cokinetic data for preparations containing 50 mg topira mate are reported, since in previous studies in healthy subjects, the dose of topiramate studied were 100 - 200 mg, possibly because the doses of the drug used for the treatment of epilepsy are larger than those used in migraine prevention [11-14]. Considering published data on topiramate pharmacokinetic after the administration of 100 - 200 mg, our results extend previous observation on a linear decrease/increases in plasma Cmax proportional to the dose administered. As previously reported, topiramate half-life was relatively long (see Table 2) and for this reason the wash out period for the present study was set at 21 days, a period long enough to allow the clearance of the drug after each dosing. Finally, it is important to mention that topiramate absorption should not change after food intake, thus the data obtained in the fasting condition reasonably apply to the fed condition.
Topiramate efficacy in epilepsy and migraine is included in several treatment guidelines [5,15-17] and excellent clinical results have been obtained for other pathological conditions such as alcoholism [5]. The overall risk/benefit ratio of its standard use is considered favorable and a recent report dealing with migraine prevention, states that topiramate inclusion in the drug armamentarium used in these patients is one of the outstanding advances of the last ten years in this field [10].
REFERENCES
- C. A. Jackevicius, M. M. Chou, J. S. Ross, N. D. Shah, and H. M. Krumholz, “Generic Atorvastatin and Health Care Costs,” New England Journal of Medicine, Vol. 366, No. 3, 2011, pp. 201-204. doi:10.1056/NEJMp1113112
- C. Chu, E. Rudant, M. Bonvalet, H. Agostini, P. Cavalie, L. Bonhomme-Faivre, J. Frenkiel, J. Taillandier, A. Boissonnas, D. Vittecoq and B. Wyplosz, “Generic Drug Prescriptions Following Hospital Discharge: A Prospective Study in France,” European Journal of Internal Medicine, Vol. 22, No. 5, 2011, pp. e45-e49. doi:10.1016/j.ejim.2011.01.016
- M. Yamada and T. E. Welty, “Generic Substitution of Antiepileptic Drugs: A Systematic Review of Prospective and Retrospective Studies,” The Annals of Pharmacotherapy, Vol. 45, No. 11, 2011, pp. 1406-1415. doi:10.1345/aph.1Q349
- R. P. Shank and B. E. Maryanoff, “Molecular Pharmacodynamics, Clinical Therapeutics, and Pharmacokinetics of Topiramate,” CNS Neuroscience & Therapeutics, Vol. 14, No. 2, 2008, pp. 120-142. doi:10.1111/j.1527-3458.2008.00041.x
- B. A. Johnson, N. Rosenthal, J. A. Capece, F. Wiegand, L. Mao, K. Beyers, A. McKay, N. Ait-Daoud, R. F. Anton, D. A. Ciraulo, H. R. Kranzler, K. Mann, S. S. O’Malley, and R. M. Swift, “Topiramate for Treating Alcohol Dependence: A Randomized Controlled Trial,” JAMA, Vol. 298, 2007, pp. 1641-1651. doi:10.1001/jama.298.14.1641
- P. Crawford, M. Feely, A. Guberman and G. Kramer, “Are There Potential Problems with Generic Substitution of Antiepileptic Drugs? A Review of Issues,” Seizure, Vol. 15, No. 3, 2006, pp. 165-176. doi:10.1016/j.seizure.2005.12.010
- J. E. Desmarais, L. Beauclair and H. C. Margolese, “Switching from Brand-Name to Generic Psychotropic Medications: A Literature Review,” CNS Neuroscience & Therapeutics, Vol. 17, No. 6, 2011, pp.750-760. doi:10.1111/j.1755-5949.2010.00210.x
- A. S. Kesselheim, M. R. Stedman, E. J. Bubrick, J. J. Gagne, A. S. Misono, J. L. Lee, M. A. Brookhart, J. Avorn and W. H. Shrank, “Seizure Outcomes Following the Use of Generic versus Brand-Name Antiepileptic Drugs: A Systematic Review and Meta-Analysis,” Drugs, Vol. 70, No. 6, 2010, pp. 605-621. doi:10.2165/10898530-000000000-00000
- M. Maliepaard, N. Banishki, C. C. Gispen-de Wied, S. Teerenstra and A. J. Elferink, “Interchangeability of Generic Anti-Epileptic Drugs: A Quantitative Analysis of Topiramate and Gabapentin,” European Journal of Clinical Pharmacology, Vol. 67, No. 10, 2011, pp. 1007- 1016. doi:10.1007/s00228-011-1041-4
- L. Edvinsson and M. Linde, “New Drugs in Migraine Treatment and Prophylaxis: Telcagepant and Topiramate,” Lancet, Vol. 376, No. 9741, 2010, pp. 645-655. doi:10.1016/S0140-6736(10)60323-6
- W. E. Rosenfeld, “Topiramate: A Review of Preclinical, Pharmacokinetic, and Clinical Data,” Clinical Therapeutics, Vol. 19, No. 6, 1997, pp. 1294-1308. doi:10.1016/S0149-2918(97)80006-9
- J. H. Park, Y. S. Park, M. H. Lee, S. Y. Rhim, J. C. Song, S. J. Lee, J. M. Kim, L. M. Shaw, and J. S. Kang, “Determination of Plasma Topiramate Concentration Using LC-MS/MS for Pharmacokinetic and Bioequivalence Studies in Healthy Korean Volunteers,” Biomedical Chromatography, Vol. 22, No. 8, 2008, pp. 822-829. doi:10.1002/bmc.995
- A. Pineyro-Lopez, E. Pineyro-Garza, M. Gomez-Silva, R. Reyes-Araiza, M. A. Flores-Diego, S. Borrego-Alvarado, M. E. Gamino-Pena, R. Vargas-Zapata and M. E. Salazar-Leal, “Bioequivalence of Single 100-mg Doses of Two Oral Formulations of Topiramate: An Open-Label, Randomized-Sequence, Two-Period Crossover Study in Healthy Adult Male Mexican Volunteers,” Clinical Therapeutics, Vol. 31, 2009, pp. 411-417. doi:10.1016/j.clinthera.2009.02.001
- I. Saavedra, E. Tamayo, A. Gamboa, J. Sasso, N. Varela, I. Moreno, D. Marchant, D. Caceres and L. Quinones, “Relative Bioavailability Study with Two Oral Formulations of Topiramate Using a Validated UPLC-MS/MS Method,” International Journal of Clinical Pharmacology, Therapy, & Toxicology, Vol. 48, 2010, pp. 342-348.
- E. Beghi, “Efficacy and Tolerability of the New Antiepileptic Drugs: Comparison of Two Recent Guidelines,” Lancet Neurology, Vol. 3, No. 10, 2004, pp. 618-621. doi:10.1016/S1474-4422(04)00882-8
- A. May, M. Leone, J. Afra, M. Linde, P. S. Sandor, S. Evers and P. J. Goadsby, “EFNS Guidelines on the Treatment of Cluster Headache and Other TrigeminalAutonomic Cephalalgias,” European Journal of Neurology, Vol. 13, 2006, pp. 1066-1077. doi:10.1111/j.1468-1331.2006.01566.x
- S. Evers, J. Afra, A. Frese, P. J. Goadsby, M. Linde, A. May and P. S. Sandor, “EFNS Guideline on the Drug Treatment of Migraine—Revised Report of an EFNS Task Force,” European Journal of Neurology, Vol. 16, 2009, pp. 968-981.doi:10.1111/j.1468-1331.2009.02748.x