Computational Molecular Bioscience
Vol.2 No.2(2012), Article ID:20466,16 pages DOI:10.4236/cmb.2012.22005
Molecular Features of Highly Pathogenic Avian and Human H5N1 Influenza A Viruses in Asia
Department of Computer Science, Houghton College, Houghton, USA
Email: wei.hu@houghton.edu
Received March 25, 2012; revised April 26, 2012; accepted May 3, 2012
Keywords: Entropy; H5N1; Hemagglutinin; Host Marker; Influenza; Informational Spectrum Method; Mutation; Mutual Information; Random Forest; Receptor Binding Specificity
ABSTRACT
The highly pathogenic avian H5N1 influenza virus could infect humans with high mortality rate, even though it has not yet become efficiently transmissible among humans. This proteomic study investigated the molecular basis of interspecies transmission and host range of this lethal virus in Asia, due to its potential pandemic threat. Although there are host markers located in previous research between general avian and human influenza viruses, the novelty of our work was to uncover host markers between highly pathogenic avian and human H5N1 viruses in Asia. Many host markers we found were not present in the previous general markers, thus expanding the current repertoire of host markers with these strain-specific host markers. Ranked by their order of importance, the top 10 host markers discovered in this report were PB2_627, HA_325, NS1_205, PB2_524, HA_86, NA_201, NP_373, NS1_7, HA_156, NA_74, confirming our current knowledge that PB2_627 is the most critical site for distinguishing avian and human H5N1. We also identified several naturally-occurred mutations in the HA protein that might shift the receptor binding preference of Asian avian H5N1, since early detection of mutations that might lead to emergence of a new pandemic virus is of prime importance. Finally, we analyzed the distinctive interaction patterns within and between proteins of avian and human H5N1 in Asia at protein level and individual residue level. From multiple viewpoints, our findings reinforced the experimental observation that multiple genes of Asian avian H5N1 are involved in its gradual adaptation to human hosts.
1. Introduction
The highly pathogenic avian H5N1 influenza virus was first discovered in Guangdong China in 1996, originated from its low pathogenic counterpart that is present in many wild bird species. The first case of human infection with highly pathogenic avian H5N1 occurred in Hong Kong in 1997 [1]. Since then, the number of confirmed cases of H5N1 infection has increased with high mortality rate and therefore poses a serious threat to public health with pandemic potential. Patients infected by this virus typically have severe pneumonia that often progresses rapidly to the acute respiratory distress syndrome. In general, influenza viruses from one host do not normally transmit or cause disease in other hosts. As such, avian influenza viruses including the highly pathogenic ones do not replicate efficiently or cause disease in humans. The concern is that H5N1 might acquire necessary mutations to increase its capability for avian-to-human infection and human-to-human transmission. Therefore, finding the molecular indicators of H5N1 that promote its transmission and replication in humans is relevant to the prevention and treatment of this disease.
The genome of influenza viruses is composed of eight gene segments, which code for 11 known proteins. The HA protein, the principal antigen on the viral surface, plays a key role in host range and cellular tropism of influenza through its preference for different receptors on host cells. A switch in receptor specificity of HA is a necessary adaptation step for influenza with avian HAs that like binding the receptors of avian type to gain the ability to bind the receptors of human type. The avianlike receptors typically are abundant in the intestinal tract of birds, whereas human-like receptors in the respiratory tract of humans. Additionally, avian and human receptors are shown to have a different structural conformation. It is obvious that an influenza virus with high affinity to both avian and human receptors could be disastrous, as the HA proteins from the H1N1 1918 pandemic virus bind efficiently to both avian and human receptors. The spread of avian H5N1 to human population has been limited so far. This lack of efficient avian H5N1 transmission and replication in humans might be in part caused by their avian-like receptor preference. Several mutations that could shift receptor preference of the HA proteins have been identified, some of which are located within or near the receptor binding domain while some are not [2- 4]. Interestingly, mutations that cause a receptor binding shift on the H1 and H3 proteins do not necessarily cause an equivalent shift on the H5 proteins [3].
Analysis on isolates of avian influenza H5N1 from patients in 1997 revealed that virulence factors included the highly cleavable HA protein, a specific substitution in the PB2 protein (E627K) that enhances replication and a substitution in the NS1 protein (D92E) [5]. The experiments in vivo or vitro are usually limited to the study of single mutations, while computational approaches such as those in [6,7] allow systematic screening of all possible mutations. Furthermore, current knowledge of the influenza host range and transmission suggests that multiple molecular factors from various viral genes are involved, thus making experimental determination of these molecular determinants challenging. An effective approach would be applying computational methods to discover candidate factors that are critical for host range, and then verify them experimentally.
Characteristic sites in the internal proteins of avian and human influenza viruses were uncovered with help of seven feature selection algorithms [8]. The validity of these signature sites was justified by their performance to differentiate between avian and human influenza strains using four machine learning classifiers. It further indicated that, in these selected sites, human influenza tended to mutate more than avian influenza each year. However, the correlation and connectivity between the avian sites was stronger than that of the human sites, and the percentage of sites with high connectivity was also greater in avian influenza.
Using several computational techniques, Asian and North American avian H5N1 were studied as examples of highly and low pathogenic avian viruses respectively [9]. Distinctive features were discovered between North American and Asian avian H5N1 based on HA receptor preference, HA cleavage site, Hamming distance, GC content of PB1-F2, NA second active site, and interaction patterns of HA and NA, and NP, PA, PB1, and PB2.
The avian H5N1 virus has a high mortality rate, but fortunately it has not exhibited sustained transmission and replication in humans. However, the pandemic 2009 H1N1 influenza A virus, a triple reassortant of avian, swine, and human viruses, displays an opposite trait. It is a mild human disease, but highly transmissible. One fear is that these two viruses might reassort into a new strain that is highly transmissible and pathogenic. The studies in [10,11] uncovered the characteristics of this new virus including new host markers that are not known in avian or swine viruses. A recent report showed a reassortant 2009 pandemic H1N1 influenza virus containing an H5N1 HA gene increased replication and enhanced pathogenicity in mice [12]. Viruses containing a HA gene from highly pathogenic avian H5N1 virus were found to be transmissible in ferrets, the animal model that most closely resembles humans in flu studies [13].
To date avian H5N1 has not developed the ability of human-to-human transmission, the possibility of this deadly virus becoming fully adapted to humans remains a serious global health concern. Despite intense research on H5N1, further understanding of the molecular mechanisms for avian influenza virus to cross species boundaries to infect humans is of major interest. Using the computational methodology developed in [14-24], the current proteomic study aimed to discover molecular characteristics in the proteins of avian and human H5N1 viruses in Asia and to identify viral traits including HA binding features that might promote the transmission of H5N1 from birds to humans.
2. Materials and Methods
2.1. Sequence Data
Protein sequences of avian and human H5N1 influenza viruses in Asia were retrieved from the EpiFlu Database (http://platform.gisaid.org) of GISAID and the Influenza Virus Resource (http://www.ncbi/nlm.nih.giv/genomes/FLU/FLU.html) of the National Center for Biotechnology Information (NCBI). Only the full length and unique sequences were selected. All sequences used in this study were aligned with MAFFT [25].
2.2. Informational Spectrum Method
The informational spectrum method (ISM) is a bioinformatics technique that can be used to analyze protein sequences [26]. The idea is to translate the protein sequences into numerical sequences based on electron-ion interaction potential (EIIP) of each amino acid. Then the Discrete Fourier Transform (DFT) can be applied to these numerical sequences, and the resulting DFT coefficients are used to produce the energy density spectrum. The informational spectrum (IS) comprises the frequencies and the amplitudes of this energy density spectrum. According to the ISM theory, the peak frequencies of IS of a protein sequence reflect its biological or biochemical functions. The ISM was successfully applied to quantify the effects of HA mutations on the receptor binding preference in [17,27,28]. Table 1 shows several common IS frequencies identified in [27,28].
It was observed in [20] that some of the strains of 2009 H1N1 displayed dual HA receptor binding preference. Consequently, in this study we used top two IS frequencies, one primary and one secondary, to describe the HA receptor selection.
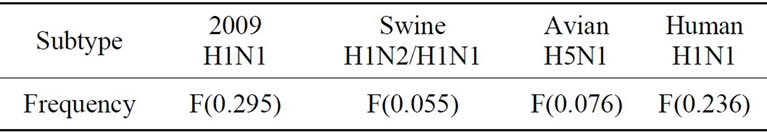
Table 1. Characteristic IS frequencies of HA proteins in 2009 H1N1, swine H1N1/H1N2, avian H5N1, and human H1N1.
2.3. Entropy and Mutual Information
In information theory [29,30], entropy is a measure of the uncertainty associated with a random variable. Let x be a discrete random variable that has a set of possible values  with probabilities
with probabilities  where
where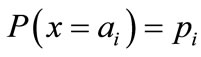 . The entropy H of x is
. The entropy H of x is
The mutual information of two random variables is a quantity that measures the mutual dependence of the two variables or the average amount of information that x conveys about y, which can defined as

where  is the entropy of x, and
is the entropy of x, and  is the joint entropy of x and y.
is the joint entropy of x and y.  if and only if x and y are independent random variables.
if and only if x and y are independent random variables.
In the current study, each of the n columns in a multiple sequence alignment of a set of influenza protein sequences of length N is considered as a discrete random variable xi (1 ≤ i ≤ N) that takes on one of the 20 (n = 20) amino acid types with some probability.  has its minimum value 0 if all the amino acids at position i are the same, and achieves its maximum if all the 20 amino acid types appear with equal probability at position i, which can be verified by the Lagrange multiplier technique. A position of high entropy means that the amino acids are often varied at this position. While
has its minimum value 0 if all the amino acids at position i are the same, and achieves its maximum if all the 20 amino acid types appear with equal probability at position i, which can be verified by the Lagrange multiplier technique. A position of high entropy means that the amino acids are often varied at this position. While  measures the genetic diversity at position i in our current study,
measures the genetic diversity at position i in our current study,  measures the correlation between amino acid substitutions at positions i and j.
measures the correlation between amino acid substitutions at positions i and j.
3. Results and Discussion
While much research has focused on the evolution of certain genes of influenza viruses, comprehensive analysis of full genome sequences is still in great need. Using different computational methods including entropy, mutual information, and ISM, our proteomic study provided elucidation of the protein sequence similarity and distinction between avian and human H5N1 viruses in Asia. Additionally, the variations of and the interactions among proteins of avian and human H5N1 were uncovered at protein level and individual residue level. The findings of this section supported with fine detail the experimental evidence [31] that multiple genes of highly pathogenic avian H5N1 virus are involved in its gradual adaptation to human hosts.
3.1. Protein Sequenced Entropy of Avian and Human H5N1 Viruses in Asia
To render a global picture of protein sequence variation of avian and human H5N1 viruses in Asia, their entropy was calculated (Figure 1).


Figure 1. Entropy of proteins of Asian avian and human H5N1.
In general, the average entropy of avian H5N1 proteins was higher than human H5N1 (Figure 2), in contrast to MI values of these proteins (Figure 4). The average of entropy of all proteins was 0.054 for avian and 0.046 for human viruses. Both entropies of avian and human proteins HA, NA, M1, NS1, and NS2 were above the average and the others were below the average. It was expected to see the high entropy of HA and NA, but it was intriguing to observe the same for M2, NS1, and NS2.
Interestingly, our entropy values for M1 and M2 in Figure 2 validated the notation that selective pressure on M2 is stronger than that on M1. In addition, selective pressure on M1 and M2 for human is stronger than that for avian H5N1 [32], which is the opposite of the other proteins as seen in Figure 2. The M2 proteins indicated much higher entropy than M1 proteins, implying a greater response to host-immune selective pressure or structural constraints on M2.
In addition to the protein entropy of avian and human H5N1 in Asia (Figures 1 and 2), the top entropy sites in proteins of these two viruses are displayed in Figure 3. There are 18 sites shared between avian and human viruses with HA and NA having the most: PB2_108, PB2_147, PB2_339, PA_101, PA_400, HA_94, HA_124, HA_138, HA_140, HA_155, HA_189, NA_44, NA_46, NA_70, NA_95, NA_340, M2_66, and NS2_52. Only M1 was missing in the high entropy sites in human H5N1, while M1 and NP were missing in avian because PB1, NP and M1 were the three proteins of the lowest entropy (Figure 2).
Figure 2. Average entropy of proteins of Asian avian and human H5N1.


Figure 3. Top 50 entropy sites in proteins of Asian avian and human H5N1.
The HA protein is the primary target of host immune responses. The three-dimensional location and structure of H3 HA antigenic epitopes have been characterized and five antigenic sites are identified (A, B, C, D, E). The antigenic sites of H1, H2, and H5 HAs are characterized and mapped on the H3 HA three-dimensional structure [33]. The amino acid sequences at these sites are extremely variable thanks to selection pressure. The primary purpose of adaptive mutations is to improve viral replicative fitness.
Two sites HA_140 and HA_189 had high entropy in both avian and human H5N1, and they were on the antigenic sites of highly pathogenic avian H5 HA [33]. In a study of highly pathogenic avian H5N1 viruses in Indonesia during 2007-2008 [34], amino acid differences were identified at six sites (83, 86, 124, 138, 140 and 141) within regions homologous to antigenic sites A, B, and E of the H3 HA protein. It was also noteworthy that HA_325 in human H5N1 was at the cleavage site of HA.
3.2. Mutual Information of Protein Sequences of Avian and Human H5N1 Viruses in Asia
Entropy measures the sequence variation at individual sites, but mutual information (MI) reveals the correlated variation of two sites. The sum of the MI values of various two sites represents this correlation within a protein or between two proteins. Our purpose of this section was to show the correlations among four groups of related proteins, PB1, PB2, PA, and NP, HA and NA, M1 and M2, and NS1 and NS2 (Figure 4).
Within the four proteins PB1, PB2, PA, and NP, the highest correlated pair of proteins was PB2 and PA, and highest correlated individual protein was PA. NS1 is a short sequence, but its correlation with itself and NS2 was high. As expected, the correlations within and between HA and NA were both very high. Surprisingly, the correlation of avian NS1 and NS2 was higher than human H5N1, in contrast to the other proteins (Figure 4). For most proteins, the interactions of inter-proteins were stronger than intra-proteins with one exception NS1.
In general, human H5N1 proteins had higher MI than avian H5N1 (Figure 4), while avian H5N1 had higher entropy than human H5N1 (Figure 2). The opposite patterns of entropy and mutual information of avian and human H5N1 suggested that the amino acid variation at a single site is a different molecular feature from amino acid correlation at two sites.
3.3. Host Specificity Markers between Avian and Human H5N1 Viruses in Asia
Identification of biological determinants for host adaptation is important. Different from the approaches in [10, 11] where each protein was evaluated individually, here we identified the major amino acid host markers between avian and human H5N1 in Asia from the concatenation of all protein sequences. Our method, in a sense, allowed these proteins to compete with each other in their contribution to the power of distinguishing between these two viruses. The benefits of this approach became apparent when we reported the top 50 important host markers ranked by Random Forests [35,36] that could significantly differentiate these two viruses (Figure 5). It detailed the involvement of particular residues, and demonstrated that the proteins PB2, PA and NS1 were the most contributing elements when compared to other internal proteins. As expected, HA and NA were the leading contributors among all proteins. The actual number of sites from each contributing protein is: PB1: 1, PB2: 6, PA: 6, NP: 2, HA: 13, NA: 9, M1: 1, M2: 2, NS1: 7, and NS2: 3. Interestingly, all the proteins considered contributed at least one site.
In the order of importance ranked by Random Forests, the top 10 amino acid host markers were PB2_627, HA_325, NS1_205, PB2_524, HA_86, NA_201, NP_ 373, NS1_7, HA_156, NA_74 (Figure 5), reaffirming our current knowledge that PB2_627 is the most critical site for distinguishing avian and human H5N1. Beyond PB2_627, our computational results suggested other sites MI sum of site pairs within and between avian and human H5N1 proteins in Asia
Figure 4. MI sum of site pairs within and between proteins of Asian avian and human H5N1.

Figure 5. Top 50 important sites ranked by Random Forests that could differentiate avian from human H5N1 proteins.
worthy of experimental verification. A recent report [37] discovered that PB2_627E and PB2_701N, besides being host markers as suggested in this study, could contribute to the virulence of influenza. Furthermore, HA_156 was on the antigenic sites of highly pathogenic avian H5 HA.
The work in [7,10,11] identified host adaption markers in the proteins of avian and human influenza, and proved that these markers are reproducible. The novelty of our work here was to unravel the host markers in avian and human H5N1 in Asia. In light of our results, it appeared that there was substantial difference in the host markers found in general human and avian viruses and those in highly pathogenic avian and human H5N1 in Asia, highlighting the need for and value of strain-specific host markers.
There were 13 host markers identified in the current work that were part of the general host markers found in previous research [10,11]: PB2_108, PB2_292, PB2_524, PB_627, NP_373, HA_94, HA_138, NA_340, M2_20, M2_50, NS1_171, NS1_79, NS2_14. Many host markers displayed in Figure 5 did not show high entropy when compared to the top entropy sites reported in Figure 3. But those that did were worthy of further attention. The host markers in Figure 5 that had high entropy in human H5N1 were PB2_108, PB2_292, PB2_524, PA_394, PA_520, NP_373, NP_400, HA_86, HA_94, HA_124, HA_138, HA_140, HA_162, HA_325, HA_482, HA_488, HA_533, NA_46, NA_48, NA_74, NA_95, NA_105, NA_338, NA_340, M2_20, NS1_7, NS1_205, NS1_206, NS1_214, NS2_7, and NS2_52. At the same time, the host markers that had high entropy in avian H5N1 were PB2_108, HA_94, HA_124, HA_138, HA_140, NA_95, NA_340, NS1_171, NS2_14, and NS2_52. All these sites also had high entropy in human H5N1 except NS1_171 and NS2_14.
The active site of NA is made of eight functional residues (R118, D151, R152, R224, E276, R292, R371, and Y406) and 11 framework residues (E119, R156, W178, S179, D198, I222, E227, H274, E277, N294, and E425) (N2 numbering) [38]. In contrast to HA, none of these important sites were part of the host markers from NA.
3.4. Correlated Sites in Protein Sequences of Avian and Human H5N1 Viruses in Asia
The results of MI analysis in section 3.2 exhibited the global correlation within and between proteins; here we zoomed in on individual sites in proteins to find those sites that were highly correlated. Due to functional and structural constrains, a mutation at one site in a protein tends to induce mutations at other sites. Recent experiments demonstrated that for H5 HA to bind human airway epithelium, multiple simultaneous mutations are required [39]. In view of this finding, detecting sites in a protein that incline to co-mutate is warranted. Our goal in this section was to unravel the different interface patterns of avian and human H5N1 in Asia at the level of individual sites.
3.4.1. Correlated Site Pairs in All Proteins of Avian and Human H5N1 in Asia
This subsection reported top 50 highly correlated site pairs in all proteins of avian and human H5N1 (Figure 6). To visualize the connectivity of these corrleated pairs, we connected in Figure 6 each pair with a stright line. There were two clusters of connected sites in human H5N1 and one such cluster in avian H5N1. Many sites in human H5N1 were highly connected, and as a result they belonged to either one of the two clusters with one pair NS1_7 and NS2_7 as an exception. On the contrary, there were more isolated pairs in avian than in human H5N1. At the same time, it appeared that the interaction of HA and NA with the viral polymerase complex was stronger in human than in avian H5N1. The prominent clustered PA sites in avian H5N1 seemed a feature of this virus.
Pathogenesis of influenza is a polygenic trait and HA, PB1, PB2, PA, NA and NS1 are involved in host range and pathogenicity. The non-structural protein NS1 is a virulence factor, known to interact with many cellular proteins to promote virus replication. The selected sites in Figure 6 provided fine details on their interactions at residue level. M1 and M2 are highly conservative, so they were not present as significant contributors to intermolecular correlations.
The top 50 highly correlated site pairs in Figure 6 were made of 24 sites in human H5N1 and 43 sites in avian H5N1, implying the selected sites of human H5N1 were more mutually correlated than avian H5N1.
3.4.2. Correlated Site Pairs in PB1, PB2, PA, and NP Proteins of Avian and Human H5N1 in Asia
The viral ribonucleoproteins (vRNP) PB1, PB2, PA, and NP play a critical role in viral replication through complex interactions of protein-protein and protein-RNA. The eight gene segments of influenza genome are wrapped around the NP protein to form vRNP complexes. To illustrate their interface, we reported top 50 highly correlated site pairs in these four proteins (Figure 7).
In human H5N1, there were two groups of connected sites, one made of PA sites only and one made of mixture of PB1, PA, and NP sites. In avian H5N1, there was one such group made of PA sites alone (Figure 7) and the top 50 pairs were mainly from PA, implying the interface of PA sites selected were much stronger in avian than in human H5N1 as seen in Figure 6 already. Sites PA_58, PA_101, PA_129, PA_204, PA_400, PA_712 were highly correlated to other PA sites in both avian and human H5N1. Of the highly correlated sites in Figure 7,

Figure 6. Top 50 highly correlated site pairs within and between all proteins of human (on left) and avian (on right) H5N1.

Figure 7. Top 50 highly correlated site pairs within and between PB1, PB2, PA, and NP proteins of human (on left) and avian (on right) H5N1.
PB2_108, PB2_292, PB2_524, PA_394, PA_520, NP_373, NP_400 were also host markers and had high entropy in human H5N1, and PB2_108 was a host marker and had high entropy in avian H5N1. The individual sites presented in Figure 7 confirmed the global interface between these four proteins of H5N1, i.e., the highest correlated pair of proteins was PB2 and PA, and highest correlated protein was PA (Figure 4).
3.4.3. Correlated Site Pairs in PB2 Protein of Avian and Human H5N1 in Asia
The RNA polymerase, made of PA, PB1, and PB2, is implicated in host range, replication, and pathogenicity. In particular, amino acid residue 627 in PB2 plays a key role. One of the mutations in PB2 commonly associated with adaptation of avian H5N1 to mammalian species is E627K, and the mutation D701N in PB2 is also frequently present. Both sites were recognized as significant host markers in this study and as major contributors to virulence of influenza [37]. However, mutations that enhance the polymerase activity of avian viruses in mammalian hosts are not fully characterized.
Here we reported top 50 highly correlated site pairs in PB2 (Figure 8) owing to the significance of this protein. There was a hub site PB2_524 among all the highly correlated sites in human H5N1, and in avian H5N1 a similar site like this was PB2_64, and the difference between the two sites was that the former directly connected to many sites whereas the latter indirectly connected to many sites. However, the same PB2_64 was either directly or indirectly connected to PB2_309, PB2_355, PB2_483, PB2_495 in both avian and human H5N1.
Site PB2_627 in avian H5N1 was connected to PB2_ 649 and PB2_451 that belonged to a group of mutually connected sites, thereby revealing several potential sites that might impact the critical site 627. Sites PB2_292, PB2_526, PB2_524 in Figure 8 were highly correlated to sites in PB1, PA, and NP of human H5N1 (Figure 7). Importantly, here PB2_524 actually was a hub site that connected many sites in PB2 of human H5N1, including site PB2_590, one of the two sites of SR polymorphism discovered in [40] that could enhance polymerase activity in human cells. The connection between PB2_590 and PB2_524 in human H5N1 suggested that the change at site 590 might be associated with the change at site 524, as well as other sites connected by site 524, which enlarged the number of potential sites that might influence site 590. Sites PB2_108, PB2_147, PB2_368, PB2_390 were contained in a group of sites that were mutually connected in both avian and human H5N1. Site PB2_108 was also a host marker with high entropy.
3.4.4. Correlated Site Pairs in HA and NA Proteins of Avian and Human H5N1 in Asia
The ability of avian H5N1 to break species barriers to infect humans depends mainly on modification of the HA and NA proteins. The efficient entry of virus into and release of virus from host cells requires a good coordination between the receptor-binding and receptor-destroying properties of HA and NA, respectively. In line with this requirement, our analysis highlighted specific sites in these two proteins that were considerably correlated (Figure 9).
The sites that were mutually connected tended to mutate together. One such example was the amino acid difference observed at six sites 83, 86, 124, 138, 140, 141 in the HA protein of avian H5N1 in Indonesia during 2007-2008 [34]. These six sites were under positive selection pressure among the H5N1 viruses from southern China and Southeast Asia during 2002-2005 [41].

Figure 8. Top 50 highly correlated site pairs in PB2 protein of human (on left) and avian (on right) H5N1.
Site HA_140 was a hub site for a group of connected sites in both avian and human H5N1. Site HA_86 was directly connected to many sites in human H5N1, but was indirectly connected to many sites in avian H5N1. However, the connectivity of NA_95 with other sites was reversed, indirect in human and direct in avian H5N1, making NA_95 a second hub site in avian H5N1.
The sites HA_94, HA_124, HA_140, HA_141 in human and HA_140, HA_156 in avian H5N1 in Figure 9 play an important role in HA receptor binding, and will be further analyzed in section 3.5. The high connectivity of HA_140 with other sites in both avian and human H5N1 suggested that its variation might be associated with the changes at other sites. Also HA_325 in human H5N1 was at the HA cleavage site, and HA_322 in avian H5N1 was near the HA cleavage site. Sites HA_94, HA_124, HA_138, HA_140, NA_95, and NA_340 were host markers and had high entropy in both avian and human H5N1.
3.4.5. Correlated Site Pairs in M1 and M2 Proteins of Avian and Human H5N1 in Asia
The M gene segment encodes the M1 matrix and the M2 ion channel proteins, both highly conserved. The positions 239-252 of M1 and positions 10-23 of M2 overlap in different reading frames. M1 forms a shell-like structure beneath the viral envelope and interacts with viral ribonucleoprotein (vRNP) complex, so it is critical in the assembly and budding process. As a transmembrane protein, M2 interacts with both the internal proteins and the host immune system. Experimental data showed that the interaction of M1 and M2 is essential for complete virus assembly [42].
On account of its high conservativity, the M2 protein is the target of the adamantane class of antiviral drugs, which confine the viral proliferation in the infected host cells. Two mutations on this protein (L26I and S31N) lead to drug resistance [43]. Sites M1_206, M2_26, M2_31, M2_64 were mutually connected in both avian and human H5N1, which implied that the variation at sites M2_26 and M2_31 might be connected to other sites including M2_64 and M1_206 (Figure 10).
There was one cluster of mutually connected sites in both human and avian H5N1 with M2_18 as a common site in both. Site M2_66 was indirectly connected to many sites in human, but directly in avian H5N1. The connectivity of M2_82 was the opposite of M2_66, direct in human and indirect in avian H5N1. Site M2_20 was a host marker and had high entropy in human H5N1. In the overlapping region, M2_13 is connected to M1_242 in avian, M2_20 to M1_249 in human H5N1 (Figure 10).
3.4.6. Correlated Site Pairs in NS1 and NS2 Proteins of Avian and Human H5N1 in Asia
The NS gene segment encodes the nonstructural protein NS1 and the nuclear export protein NS2. The positions 168-230 of NS1 and positions 11-73 of NS2 overlap in different reading frames. The non-structural protein NS1 is multifunctional, which regulates influenza virulence by modulating the host immune responses as well as other important aspects of viral replication [44]. Initially NS2 was considered as a non-structural protein, but later it was thought to be the adaptor between the cellular nuclear export machinery Crm1 and the newly amplified viral genomic segments (vRNPs). NS2 was then renamed nuclear export protein (NEP) [45].
Figure 9. Top 50 highly correlated site pairs within and between HA and NA proteins of human (on left) and avian (on right) H5N1.
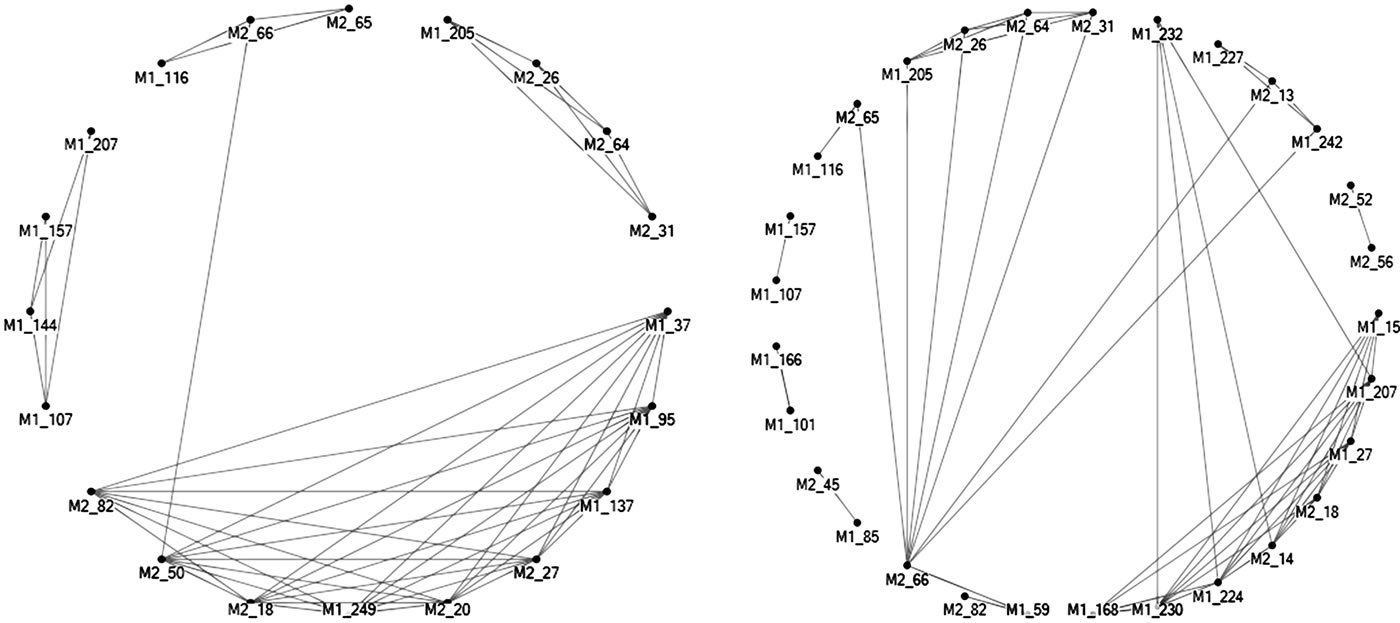
Figure 10. Top 50 highly correlated site pairs within and between M1 and M2 proteins of human (on left) and avian (on right) H5N1.

Figure 11. Top 50 highly correlated site pairs within and between NS1 and NS2 proteins of human (on left) and avian (on right) H5N1.
There are functional motifs at the C terminus of NS1. They include nuclear localization signal motif containing amino acids K219, R220, R224, K229, R231, and R232, the SH3-interacting motif formed by P212, P215, and K217, the PDZ-binding motif at positions 227 to 230 [46]. The highly correlated sites such as NS1_212, NS1_215, NS1_217, NS1_229 identified at the variable C terminus of NS1 (Figure 11) could be valuable when dealing with mutations in the region and their impact on other sites in NS1 and NS2. The amino acid at one site might compensate the loss of a particular amino acid at another site when mutated [46]. A deletion in residues 80 to 84 in NS1 has been associated with increased virulence, which appeared in many avian and human NS1 proteins used in this study.
In the overlapping region, NS2_14, NS2_36, N2S_49, NS2_55 were connected to their corresponding overlapping sites in NS1, NS1_171, NS1_193, NS1_206, and NS1_212 respectively in avian H5N1. Similarly, NS2_14, NS2_44, NS2_52, NS2_55, and NS2_58 were connected to NS1_171, NS1_201, NS1_209, NS1_212, and NS1_ 215 in human H5N1 (Figure 11). This phenomenon makes sense since these overlapping positions share two nucleotides within one codon, as a result making them easy to correlate.
Compared to other proteins, it was unusual to see that many sites including NS2_44, NS2_58, NS1_201, NS1_215 and NS2_52, NS1_209, NS1_210 and NS2_14, NS2_55, NS1_112, NS1_127, NS1_171, NS1_212, NS1_217 were all well-connected in both avian and human H5N1. Therefore, proteins NS1 and NS2 of avian and human H5N1 shared similar correlation pattern at these top 50 sites. Sites NS1_7, NS1_205, NS1_206, NS1_214, NS2_7, NS2_52 were host markers and had high entropy in human H5N1, and NS1_171, NS2_14, NS2_52 in avian H5N1.
3.5. Mutations of HA That Could Alter the Receptor Specificity of Avian H5N1 in Asia
Occasionally, the highly pathogenic avian H5N1 virus can infect humans and cause severe disease. So far, there has been no sign of sustained presence of this virus in human population. The HA protein initiates influenza virus infection by binding to receptors on host cells. Typically, avian H5N1 virus binds to avian-type receptors, and does not bind to human-type receptors. This receptor binding property of avian H5N1 is thought to be the main obstacle for the virus to transmit from person to person. However, the receptor binding preference of avian H5N1 could be changed by only a few amino acid substitutions in HA.
The receptor binding site of HA comprises three structural elements, a 190-helix (residues 188 to 190), a 130-loop (residues 134 to 138), and a 220-loop (residues 221 to 228) (H3 numbering). The most variable part of these three elements is the 220 loop. Several previous studies have showed that mutations, such as L129V, A134V, N182K, Q192R, Q222L, S223N, G224S, could enhance the binding of avian H5N1 virus to human-type receptors [3,4,47-52]. Moreover, Q222L, and G224S are also well-known adaptive mutations for H2 and H3 [4]. Experimental data suggest that mutations within the receptor binding domain can shift the binding specificity, but those at other sites can as well.
The main task of this section was to verify these mutations with ISM and, at the same time, to identify new mutations that could switch the HA receptor binding preference of Asian avian H5N1, in particular, those that could enhance the binding to human-type receptors. The new mutations identified in this study were all naturally occurred according to the amino acid changes observed in our dataset. Sometimes, artificially generated mutations were employed in other research.
In [20], the dual binding of HA proteins to avianand human-type receptors were quantified with the primary and secondary IS frequencies. Here we used the same approach to measure the change of binding preference conferred by various mutations through the amplitude variation of IS at the primary and secondary frequencies. We used the consensus HA sequence of Asian avian H5N1 as a baseline, and applied several mutations as seen in Table 3 to the consensus to detect the change of IS at these two frequencies. As observed in [17], the amount of IS changes caused by various mutations are additive.
Mutations at the sites 129, 133, 134, 156, 182, 188, 192, 222, 223, 224 were analyzed in [2], as they might cause the alteration of receptor binding preference. The amino acid makeup at these positions in the HA sequences of avian and human H5N1 in Asia was listed in Table 2, and some mutations presented in Table 2 were selected for receptor binding preference test as seen in Table 3.

Table 2. Amino acids in HA proteins of Asian avian and human H5N1 at 10 critical sites that might influence the HA binding specificity [2] (The number inside the parentheses represents the frequency of a particular amino acid in our dataset).
Table 3. The known and new mutations in the consensus HA protein sequence of Asian avian H5N1 that could modulate the HA binding specificity.
Table 3 contained 42 known or new mutations analyzed with ISM, which also presented the amplitude of IS at the primary and secondary frequencies caused by each mutation. Our findings confirmed that several known mutations A134V, N182K, Q192R, Q222L, and S223N increased avian binding and decreased human binding. However, we didn’t see the increase of human binding from mutation G224S. The reason was that the HA protein used in [3] was from A/Vietnam/1203/200, a human strain in Asia, while we used the consensus of avian HA protein sequences in Asia. Also we didn’t try mutation L129V, since the consensus amino acid of Asian avian H5N1 at site 129 was S. But we did test mutations S129L and S129V, and they enhanced both avian and human binding.
Beyond the known mutations found by past investigations, new mutations in HA at various sites were also considered (Table 3). One mutation N154D even reversed the primary frequency from F(0.076) to F(0.236), which demonstrated its significant power in switching the binding specificity. R323G is at the cleavage site of HA, and some strains contained this mutation. It turned out the this mutation increased the IS at F(0.076) and decreased the IS at F(0.236) with large amount, implying this mutation could increase the binding to avian-type receptors.
Of the mutations analyzed in Table 3, there were several host markers including HA 94, 124, 134, 138, 140, 156 (Figure 5), highlighting their multiple roles in Asian avian H5N1. They also had high entropy indicating their propensity for mutation (Figure 3) and were highly correlated to other sites in HA and NA (Figure 9), thus further suggesting their prime importance.
The results in Table 3 indicated that some mutations increased both avian and human binding, some decreased both, and some increased one while decreased the other. If a mutation changed the IS at one frequency then it always altered the other one as well.
4. Conclusions
Some of the H5N1 viruses isolated from humans cannot spread efficiently among humans. Hence, amino acid mutations in viral proteins other than HA might be required for transmission of avian H5N1 among humans [31]. This study sought to employ several computational techniques to search the molecular characteristics in all viral proteins giving rise to a human-adapted avian H5N1 virus in Asia. The high mortality for patients with H5N1 virus infection underlines the importance of understanding the biological traits of interspecies transmission of H5N1 from birds to humans.
The pathogenicity of H5N1 influenza virus is polygenic, including at least HA, NA, PB2, PB1, PA, NP and NS genes. Our analysis of highly correlated sites in subsection 3.4.1 detailed the interface between these various proteins at residue level. The viral polymerase complex comprising PA, PB1, and PB2 is involved in determining host range, replication, and pathogenicity, and the interactions of these three proteins plus NP were considered in subsections 3.4.2 and 3.4.3. The HA and NA proteins are main determinant of the host range of influenza. A balanced interplay between HA and NA is crucial for efficient viral replication and transmission, which could be seen from the correlated sites in subsection 3.4.4. Finally the M and NS genes encode two proteins, M1 and M2, and NS1 and NS2, respectively, and their intrinsic interface was revealed in subsections 3.4.5 and 3.4.6.
In addition to the study of the intraand inter-protein correlation at individual residue level, the interactions of all proteins were also considered at protein level using entropy and mutual information. It appeared that the selective pressure on M2 of Asian avian and human H5N1 was stronger than that on M1. Furthermore, the selective pressure on M1 and M2 for human is stronger than that for avian H5N1 [32], which is opposite of the other proteins as seen in Figure 2.
Our computational approach disclosed top 50 host specificity markers between Asian avian and human H5N1. Among all proteins, the top two contributors of these sites were HA and NA as expected, and PB2, PA, and NS1 contributed the most among the internal proteins.
We also discovered several naturally-occurred mutations in the HA protein that might shift the receptor binding preference of Asian avian H5N1, and demonstrated that ISM technique could be employed to detect such mutations efficiently and identify new H5N1 strains with pandemic potential. Early in the adaptation of avian virus to humans, it must acquire the ability to bind human-type receptors as this is the predominant type in the human airway. However, the presence of lesser amounts of avian-type receptors perhaps aids the initial crossover from avian species [53]. The ISM technique provides a numerical measurement of binding affinity change for avianand human-type receptors conferred by different amino acid substitutions, thereby ideal for monitoring the progressive nature of influenza evolution caused by such mutations.
In summary, the goal of the present proteomic study was to identify the molecular elements of Asian avian H5N1 virus that promote its transmission and replication in humans. In comparison with Asian human H5N1, our findings revealed these features in the proteins of this virus at protein level as well at individual residue level, thus providing comprehensive molecular details about its gradual adaptation to human hosts.
5. Acknowledgements
We thank Houghton College for its financial support.
REFERENCES
- K. Subbarao, A. Klimov, J. Katz, H. Regnery, W. Lim, H. Hall, M. Perdue, D. Swayne, C. Bender, J. Huang, M. Hemphill, T. Rowe, M. Shaw, X. Xu, K. Fukuda and N. Cox, “Characterization of an Avian Influenza A (H5N1) Virus Isolated from a Child with a Fatal Respiratory Illness,” Science, Vol. 279, No. 5349, 1998, pp. 393-396. doi:10.1126/science.279.5349.393
- Y. Gao, et al., “Identification of Amino Acids in HA and PB2 Critical for the Transmission of H5N1 Avian Influenza Viruses in a Mammalian Host,” PLoS Pathogens, Vol. 5, No. 12, 2009, Article ID: 1000709. doi:10.1371/journal.ppat.1000709
- J. Stevens, et al., “Structure and Receptor Specificity of the Hemagglutinin from an H5N1 Influenza Virus,” Science, Vol. 312, No. 5772, 2006, pp. 404-410. doi:10.1126/science.1124513
- P. Auewarakul, et al., “An Avian Influenza H5N1 Virus That Binds to a Human-Type Receptor,” Journal of Virology, Vol. 81, No. 18, 2007, pp. 9950-9955. doi:10.1128/JVI.00468-07
- J. H. Beigel, et al., “Avian influenza A (H5N1) infection in humans,” The New Journal of Medicine, Vol. 353, No. 13, 2005, pp. 1374-1385. doi:10.1056/NEJMra052211
- O. Miotto, et al., “Identification of Human-to-Human Transmissibility Factors in PB2 Proteins of Influenza A by Large-Scale Mutual Information Analysis,” BMC Bioinformatics, Vol. 9, Suppl. 1, 2008, p. 18.
- O. Miotto, et al., “Complete-Proteome Mapping of Human Influenza A Adaptive Mutations: Implications for Human Transmissibility of Zoonotic Strains,” PLoS One, Vol. 5, No. 2, 2010, p. 9025. doi:10.1371/journal.pone.0009025
- D. King, Z. Miller, W. Jones and W. Hu, “Characteristic Sites in the Internal Proteins of Avian and Human Influenza Viruses,” Journal of Biomedical Science and Engineering, Vol. 3, No. 10, 2010, pp. 943-955. doi:10.4236/jbise.2010.310125
- W. Hu, “Characterization of Asian and North American Avian H5N1,” American Journal of Molecular Biology, Vol. 1, No. 2, 2011, pp. 52-61. doi:10.4236/ajmb.2011.12007
- W. Hu, “Novel Host Markers in the 2009 Pandemic H1N1 Influenza A Virus,” Journal of Biomedical Science and Engineering, Vol. 3, No. 6, 2010, pp. 584-601. doi:10.4236/jbise.2010.36081
- W. Hu, “Nucleotide Host Markers in the Influenza A Viruses,” Journal of Biomedical Science and Engineering, Vol. 3, No. 7, 2010, pp. 684-699. doi:10.4236/jbise.2010.37093
- T. D. Cline, et al., “Increased Pathogenicity of a Reassortant 2009 Pandemic H1N1 Influenza Virus Containing an H5N1 Hemagglutinin,” Journal of Virology, Vol. 85, No. 23, 2011, pp. 12262-12270. doi:10.1128/JVI.05582-11
- Y. Kawaoka, “H5N1: Flu Transmission Work is Urgent,” Nature, Vol. 482, No. 7384, 2012, p. 155.
- W. Hu, “Analysis of Correlated Mutations, Stalk Motifs, and Phylogenetic Relationship of the 2009 Influenza A Virus Neuraminidase Sequences,” Journal of Biomedical Science and Engineering, Vol. 2, No. 7, 2009, pp. 550- 558. doi:10.4236/jbise.2009.27080
- W. Hu, “The Interaction between the 2009 H1N1 Influenza A Hemagglutinin and Neuraminidase: Mutations, Co-mutations, and the NA Stalk Motifs,” Journal of Biomedical Science and Engineering, Vol. 3, 2010, pp. 1-12. doi:10.4236/jbise.2010.31001
- W. Hu, “Identification of Highly Conserved Domains in Hemagglutinin Associated with the Receptor Binding Specificity of Influenza Viruses: 2009 H1N1, Avian H5N1, and Swine H1N2,” Journal of Biomedical Science and Engineering, Vol. 3, 2010, pp. 114-123. doi:10.4236/jbise.2010.32017
- W. Hu, “Quantifying the Effects of Mutations on Receptor Binding Specificity of Influenza Viruses,” Journal of Biomedical Science and Engineering, Vol. 3, 2010, pp. 227-240. doi:10.4236/jbise.2010.33031
- W. Hu, “Subtle Differences in Receptor Binding Specificity and Gene Sequences of the 2009 Pandemic H1N1 Influenza Virus,” Advances in Bioscience and Biotechnology, Vol. 1, 2010, pp. 305-314. doi:10.4236/abb.2010.14040
- W. Hu, “Correlated Mutations in the Four Influenza Proteins Essential for Viral RNA Synthesis, Host Adaptation, and Virulence: NP, PA, PB1, and PB2,” Natural Science, Vol. 2, No. 10, 2010, pp. 1138-1147. doi:10.4236/ns.2010.210141
- W. Hu, “Highly Conserved Domains in Hemagglutinin of Influenza Viruses Characterizing Dual Receptor Binding,” Natural Science, Vol. 2, No. 9, 2010, pp. 1005-1014. doi:10.4236/ns.2009.29123
- W. Hu, “Host Markers and Correlated Mutations in the Overlapping Genes of Influenza Viruses: M1, M2; NS1, NS2; and PB1, PB1-F2,” Natural Science, Vol. 2, No. 11, 2010, pp. 1225-1246. doi:10.4236/ns.2010.211150
- W. Hu, “Receptor Binding Specificity and Origin of 2009 H1N1 Pandemic Influenza Virus,” Natural Science, Vol. 3, No. 3, 2011, pp. 234-248. doi:10.4236/ns.2011.33030
- W. Hu, “New Mutational Trends in the HA Protein of 2009 H1N1 Pandemic Influenza Virus from May 2010 to February 2011,” Natural Science, Vol. 3, No. 5, 2011, pp. 379-387. doi:10.4236/ns.2011.35051
- W. Hu, “Molecular Determinants for Receptor Specificity in Hemagglutinin Protein of 2009 Pandemic H1N1,” Proceedings of the 6th International Conference on Bioinformatics and Biomedical Engineering, Shanghai, 17- 20 May 2012, pp. 629-632.
- K. Katoh, K. Kuma, H. Toh and T. Miyata, “MAFFT version 5: Improvement in Accuracy of Multiple Sequence Alignment,” Nucleic Acids Research, Vol. 33, No. 2, 2005, pp. 511-518. doi:10.1093/nar/gki198
- I. Cosic, “The Resonant Recognition Model of Macromolecular Bioreactivity, Theory and Application,” Birkhauser Verlag, Berlin, 1997. doi:10.1007/978-3-0348-7475-5
- V. Veljkovic, H. L. Niman, S. Glisic, N. Veljkovic, V. Perovic and C. P. Muller, “Identification of Hemagglutinin Structural Domain and Polymorphisms Which May Modulate Swine H1N1 Interactions with Human Receptor,” BMC Structural Biology, Vol. 9, 2009, p. 62. doi:10.1186/1472-6807-9-62
- V. Veljkovic, N. Veljkovic, C. P. Muller, S. Müller, S. Glisic, V. Perovic and H. Köhler, “Characterization of Conserved Properties of Hemagglutinin of H5N1 and Human Influenza Viruses: Possible Consequences for Therapy and Infection Control,” BMC Structural Biology, Vol. 7, 2009, pp. 9-21.
- T. A. Cover and J. A. Thomas, “Elements of Information Theory,” John Wiley and Sons, New York, 1991. doi:10.1002/0471200611
- D. MacKay, “Information Theory, Inference, and Learning Algorithms,” Cambridge University Press, Cambridge, 2003.
- M. Hatta, et al., “Growth of H5N1 Influenza A Viruses in the Upper Respiratory Tracts of Mice,” PLoS Pathogens, Vol. 3, No. 10, 2007, p. 133. doi:10.1371/journal.ppat.0030133
- Y. Furuse, A. Suzuki, T. Kamigaki, H. Oshitani, “Evolution of the M Gene of the Influenza A Virus in Different Host Species: Large-Scale Sequence Analysis,” Virology Journal, Vol. 6, 2009, p. 67. doi:10.1186/1743-422X-6-67
- N. V. Kaverin, et al., “Epitope Mapping of the Hemagglutinin Molecule of a Highly Pathogenic H5N1 Influenza Virus by Using Monoclonal Antibodies,” Virology Journal, Vol. 81, No. 23, 2007, pp. 12911-12917. doi:10.1128/JVI.01522-07
- H. Wibawa, et al., “A Molecular and Antigenic Survey of H5N1 Highly Pathogenic Avian Influenza Virus Isolates from Smallholder Duck Farms in Central Java, Indonesia during 2007-2008,” Virology Journal, Vol. 8, 2011, p. 425. doi:10.1186/1743-422X-8-425
- L. Breiman, “Random Forests,” Machine Learning, Vol. 45, No.1, 2001, pp. 5-32. doi:10.1023/A:1010933404324
- R. Díaz-Uriarte and S. A. de Andrés, “Gene Selection and Classification of Microarray Data Using Random Forest,” BMC Bioinformatics, Vol. 7, 2006, p. 3. doi:10.1186/1471-2105-7-3
- Y. G. Li, et al., “Characterization of H5N1 Influenza Viruses Isolated from Humans in Vitro,” Virology Journal, Vol. 7, 2010, p. 112. doi:10.1186/1743-422X-7-112
- N. A. Ilyushina, et al., “Effect of Neuraminidase Inhibitor-Resistant Mutations on Pathogenicity of Clade 2.2 A/Turkey/15/06 (H5N1) Influenza Virus in Ferrets,” PLoS Pathogens, Vol. 6, No. 5, 2010, Article ID: 1000933. doi:10.1371/journal.ppat.1000933
- G. Ayora-Talavera, et al., “Mutations in H5N1 Influenza Virus Hemagglutinin that Confer Binding to Human Tracheal Airway Epithelium,” PLoS One, Vol. 4, No. 11, 2009, p. 7836. doi:10.1371/journal.pone.0007836
- A. Mehle and J. A. Doudna, “Adaptive Strategies of the Influenza Virus Polymerase for Replication in Humans,” Proceedings of the National Academy of Sciences, Vol. 106, No. 50, 2009, pp. 21312-21316. doi:10.1073/pnas.0911915106
- G. J. Smith, et al., “Evolution and Adaptation of H5N1 Influenza Virus in Avian and Human Hosts in Indonesia and Vietnam,” Virology, Vol. 350, No. 2, 2006, pp. 258- 268. doi:10.1016/j.virol.2006.03.048
- B. J. Chen, et al., “The Influenza Virus M2 Protein Cytoplasmic Tail Interacts with the M1 Protein and Influences Virus Assembly at the Site of Virus Budding,” Journal of Virology, Vol. 82, No. 20, 2008, pp. 10059-10070. doi:10.1128/JVI.01184-08
- A. C. Hurt, et al., “Susceptibility of Highly Pathogenic A (H5N1) Avian Influenza Viruses to the Neuraminidase Inhibitors and Adamantanes,” Antiviral Research, Vol. 73, 2007, pp. 228-231.
- B. G. Hale, et al., “The Multifunctional NS1 Protein of Influenza A Viruses,” Journal of General Virology, Vol. 89, 2008, pp. 2359-2376. doi:10.1099/vir.0.2008/004606-0
- H. Akarsu, K. Iwatsuki-Horimoto, T. Noda, E. Kawakami, H. Katsura, F. Baudin, T. Horimoto and Y. Kawaoka, “Structure-Based Design of NS2 Mutants for Attenuated Influenza A Virus Vaccines,” Virus Research, Vol. 155, No. 1, 2011, pp. 240-248. doi:10.1016/j.virusres.2010.10.014
- K. Xu, et al., “Modification of Nonstructural Protein 1 of Influenza A Virus by Sumo1,” Journal of Virology, Vol. 85, No. 2, 2011, pp. 1086-1098. doi:10.1128/JVI.00877-10
- Q. Zhu, et al., “A Naturally Occurring Deletion in Its NS Gene Contributes to the Attenuation of an H5N1 Swine Influenza Virus in Chickens,” Journal of Virology, Vol. 82, 2008, pp. 220-228. doi:10.1128/JVI.00978-07
- A. S. Abdel-Moneim, et al., “Isolation and Characterization of Highly Pathogenic Avian Influenza Virus Subtype H5N1 from Donkeys,” Journal of Biomedical Science, Vol. 17, 2010, p. 25. doi:10.1186/1423-0127-17-25
- Y. Ha, et al., “X-Ray Structures of H5 Avian and H9 Swine Influenza Virus Hemagglutinins Bound to Avian and Human Receptor Analogs,” Proceedings of the National Academy of Sciences, Vol. 98, 2001, pp. 11181- 11186. doi:10.1073/pnas.201401198
- R. Harvey, et al., “Restrictions to the Adaptation of Influenza A Virus H5 Hemagglutinin to the Human Host,” Journal of Virology, Vol. 78, 2004, pp. 502-507. doi:10.1128/JVI.78.1.502-507.2004
- S. Yamada, et al., “Haemagglutinin Mutations Responsible for the Binding of H5N1 Influenza A Viruses to Human-Type Receptors,” Nature, Vol. 444, 2006, pp. 378- 382. doi:10.1038/nature05264
- W.-M. Jiang, et al., “Molecular Epidemiological Surveys of H5 subtype Highly Pathogenic Avian Influenza Viruses in Poultry in China during 2007-2009,” Journal of General Virology, Vol. 91, 2010, pp. 2491-2496.
- C. I. Thompson, et al., “Infection of Human Airway Epithelium by Human and Avian Strains of Influenza A Virus,” Journal of Virology, Vol. 80, No. 16, 2006, pp. 8060-8068. doi:10.1128/JVI.00384-06