Modeling and Numerical Simulation of Material Science
Vol. 2 No. 4 (2012) , Article ID: 23504 , 9 pages DOI:10.4236/mnsms.2012.24009
A First Principles Investigation of the Mechanical Properties of g-TlN
Department of Mechanical, Aerospace and Nuclear Engineering,
Rensselaer Polytechnic Institute, Troy, USA
Email: *qpeng.org@gmail.com
Received September 3, 2012; revised October 5, 2012; accepted October 16, 2012
Keywords: g-TlN; Mechanical Properties; High Order Elastic Constants; Density Functional Theory; 2D Materials
ABSTRACT
We investigate the structure and mechanical properties of proposed graphene-like hexagonal thallium nitride monolayer (g-TlN) using first-principles calculations based on density-functional theory. Compared to graphene-like hexagonal boron nitride monolayer (g-BN), g-TlN is much softer, with 12% in-plane stiffness, 25%, 22%, and 20% ultimate strengths in armchair, zigzag, and biaxial strains respectively. However, g-TlN has a larger Poisson’s ratio, 0.69, about 3.1 times that of g-BN. It was found that the g-TlN also sustains much smaller strains before rupture. We obtained the second, third, fourth, and fifth order elastic constants for a rigorous continuum description of the elastic response of g-TlN. The second order elastic constants, including in-plane stiffness, are predicted to monotonically increase with pressure while the Poisson’s ratio monotonically decreases with increasing pressure.
1. Introduction
Fruitful studies and applications of graphene triggered the new era of the two-dimensional (2D) nanomaterials [1-10]. Graphene analogues of BN (g-BN), an insulating material that serves as an excellent dielectric substrate for graphene electronics, was exfoliated recently and subjected to extensive studies with promising applications in electronics and energy storage [11-17]. Besides the nanosheet, tremendous researches were carried on other nanostructures of g-BN, such as nanotubes [18-24], nanoshells [25], antidotes [26], bilayer of graphene and g-BN [10,27-29], quantum dots and nanorods of graphene embedded in g-BN [30], hybrid graphene/g-BN monolayer [30-33], and graphyne BN analog [34]. As a consequence, other III-nitrides have raised a lot of attention [35-39].
Hexagonal thallium nitride monolayer (g-TlN) is a proposed graphene-like 2D material, which is only monoatomically thick (Figure 1). Although g-TlN has not been fabricated currently, the theoretical study of the g-TlN could expand the range of possible applications of III-nitrides, and open new perspectives for miniaturization in engineering functional nano-devices and interconnects by a chemical modification. The bulk thallium nitride (TlN) was predicted to have a small energy gap, indicating a semi-metallic character [40,41]. The combi-
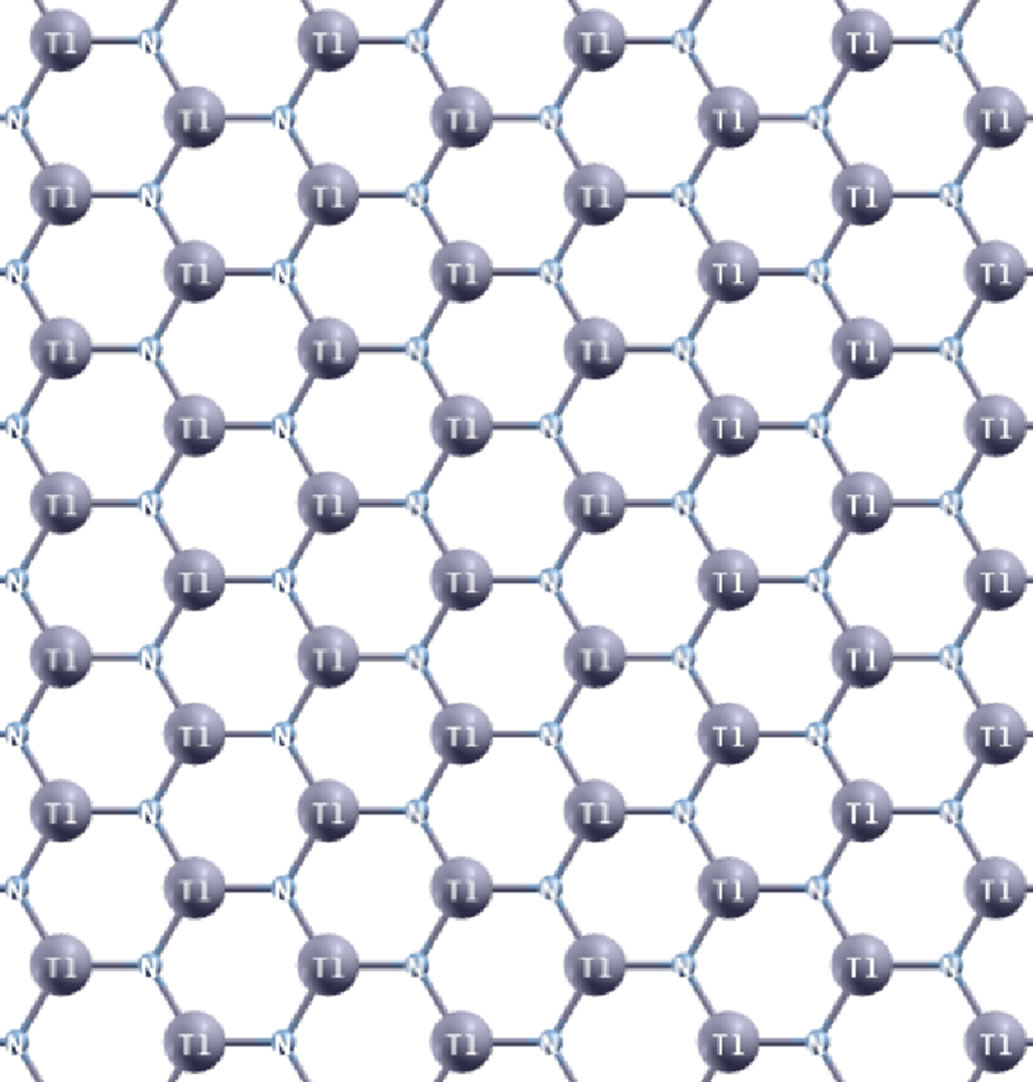
Figure 1. g-TlN: graphene-like hexagonal thallium nitride monolayer.
nation of thallium with other group III atoms which has wide gap in III-nitrides yields promising semiconductors for optical communication systems (laser diodes, detectors) with small band gap, down to the infrared energy region [42-47]. Structural stability [48], defects [49]phonon and elastic instabilities under pressure [50] were well studied in bulk TlN. The structural and reactivity parameters of novel N12X12H12 (X = B, Al, Ga, In, Tl) nitrides, in their coronene-like (C24H12) structure was investigated [51], where Tl-N bond length is reported as 2.15 - 2.20 Å.
Mechanical properties are critical in designing parts or structures with g-TlN regarding the practical applications. Strain engineering is a common and important approach to tailor the functional and structural properties of the nanomaterials [52]. One can expect that the properties of g-TlN will be affected by applied strain too. In addition, g-TlN is vulnerable to be strained with or without intent because of the monoatomic thickness. For example, there are strains because of the mismatch of lattices constants or surface corrugation with substrate [53,54]. Therefore, the knowledge of mechanical properties of g-TlN is highly desired.
Depending on the loading, the mechanical properties are divided into four strain domains: linear elastic, nonlinear elastic, plastic, and fracture. Materials in first two strain domains are reversible, i.e., they can restore to equilibrium status after the release of the loads. On the contrary, the last two domains are non-reversible. Defects are nucleated and accumulated with the increase of the strain, until rupture. As in graphene, the nonlinear mechanical properties are prominent since it remained elastic until the intrinsic strength was reached [55,56]. Thus it is of great interest to examine the nonlinear elastic properties of g-TlN, which is necessary to understand the strength and reliability of structures and devices made of g-TlN.
Several previous studies have shown that 2D monolayers present a large nonlinear elastic deformation during the tensile strain up to the ultimate strength of the material, followed by a strain softening until fracture [33,56-59]. We expect that the g-TlN behaves in a similar manner. Under large deformation, the strain energy density needs to be expanded as a function of strain in a Taylor series to include quadratic and higher order terms. The higher order terms account for both nonlinearity and strain softening of the elastic deformation. They can also express other anharmonic properties of 2D nanostructures including phenomena such as thermal expansion, phonon-phonon interaction, etc. [55].
The goal of this paper is to study the mechanical behaviors of g-TlN at large strains and find an accurate continuum description of the elastic properties from abinitio density functional theory calculations. The total energies of the system, forces on each atom, and stresses on the simulation boxes are directly obtained from DFT calculations. The response of g-TlN under the nonlinear deformation and fracture is studied, including ultimate strength and ultimate strain. The high order elastic constants are obtained by fitting the stress-strain curves to analytical stress-strain relationships that belong to the continuum formulation [58]. We compared this proposed new material with the well known 2D materials such as g-BN, graphene, and graphyne. Based on our result of the high order elastic constants, the pressure dependence properties, such as sound velocities and the second order elastic constants, including the in-plane stiffness, are predicted. Our results for the continuum formulation could also be useful in finite element modeling of the multiscale calculations for mechanical properties of g-TlN at the continuum level. The remainder of the paper is organized as follows. Section 2 presents the computational details of DFT calculations. The results and analysis are in Section 3, followed by conclusions in Section 4.
2. Density Functional Theory Calculations
We consider a conventional unit cell containing 6 atoms (3 thallium atoms and 3 nitrogen atoms) with periodic boundary conditions (Figure 2). The 6-atom conventional unit cell is chosen to capture the “soft mode”, which is a particular normal mode exhibiting an anomalous reduction in its characteristic frequency and leading to mechanical instability. This soft mode is a key factor in limiting the strength of monolayer materials can only be captured in unit cells with hexagonal rings [60].
The total energies of the system, forces on each atoms, stresses, and stress-strain relationships of g-TlN under the desired deformation configurations are characterized via first-principles calculations with density-functional theory (DFT). DFT calculations were carried out with the Vienna Ab-initio Simulation Package (VASP) [61-64] which is based on the Kohn-Sham Density Functional Theory (KS-DFT) [65,66] with the generalized gradient approximations as parameterized by Perdew, Burke, and Ernzerhof (PBE) for exchange-correlation functions [67]. The electrons explicitly included in the calculations are the 5d106s26p1 electrons for thallium atoms and the 1s22s22p1 electrons for nitrogen atoms. The core electrons of thallium are replaced by the projector augmented wave (PAW) and pseudo-potential approach [68,69]. A plane-wave cutoff of 600eV is used in all the calculations. The calculations are performed at zero temperature.
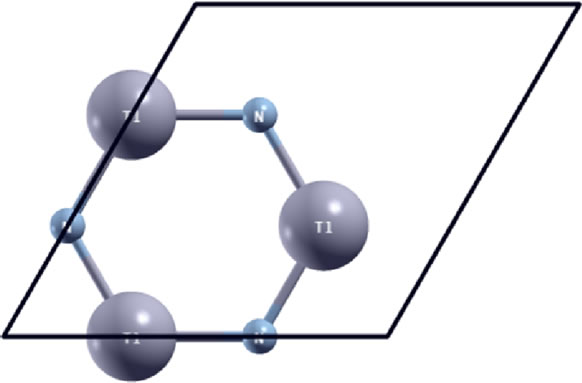
Figure 2. Atomic structure of g-TlN in the conventional unit cell (6 atoms) in the un-deformed reference configuration.
The criterion to stop the relaxation of the electronic degrees of freedom is set by total energy change to be smaller than 10–6 eV. The optimized atomic geometry was achieved through minimizing Hellmann-Feynman forces acting on each atom until the maximum forces on the ions were smaller than 0.001 eV/Å.
The atomic structures of both deformed and un-deformed configurations are obtained by fully relaxing a 6 atom-unit cell with all atoms placed in one plane. The simulation invokes periodic boundary conditions for the two in-plane directions while the displacement to out ofplane direction is forbidden.
The irreducible Brillouin Zone was sampled with a Gamma-centered 17 × 17 × 1 k-mesh. Such a large kmesh was used to reduce the numerical errors caused by the strain of the systems. The initial charge densities were taken as a superposition of atomic charge densities. There was a 15 thick vacuum region to reduce the inter-layer interaction to model the single layer system. To eliminate the artificial effect of the out-of-plane thickness of the simulation box on the stress, we used the second Piola-Kirchhoff stress [33] to express the 2D forces per length with units of N/m.
For a general deformation state, the number of independent components of the second, third, fourth and fifth order elastic tensors are 21, 56, 126, and 252 respectively. However, there are only fourteen independent elastic constants need to be explicitly considered due to the symmetries of the atomic lattice point group D6h which consists of a six-fold rotational axis and six mirror planes [56].
The fourteen independent elastic constants of g-TlN are determined by a least-squares fit to the stress-strain results from DFT based first-principles studies in two steps, detailed in our previous work [33]. In the first step, we use a least-squares fit of five stress-strain responses. Five relationships between stress and strain are necessary because there are five independent fifth-order elastic constants (FFOEC). We obtain the stress-strain relationships by simulating the following deformation states: uni-axial strain in the zigzag direction (zigzag); uni-axial strain in the armchair direction (armchair); and equibiaxial strain (biaxial). From the first step, the components of the second-order elastic constants (SOEC), the thirdorder elastic constants (TOEC), and the fourth order elastic constants (FOEC) are over-determined (i.e., the number of linearly independent variables are greater than the number of constrains), and the fifth-order elastic constants are well-determined (the number of linearly independent variables are equal to the number of constrains). Under such circumstances, the second step is needed: least-square solution to these overand well-determined linear Equations.
3. Results and Analysis
3.1. Atomic Structure
We first optimize the equilibrium lattice constant for g-TlN. The total energy as a function of lattice spacing is obtained by specifying nine lattice constants varying from 3.3 Å to 4.1 Å, with full relaxations of all the atoms. A least-square fit of the energies versus lattice constants with a fourth-order polynomial function yields the equilibrium lattice constant as a = 3.731 Å. The most energetically favorable structure is set as the strain-free structure in this study and the atomic structure, as well as the conventional cell is shown in Figure 2. Specifically, the bond length of Tl-N bond is 2.154 Å, which is 0.704 Å (or 49%) longer than the bond length of B-N bond in g-BN. The N-Tl-N and Tl-N-Tl angles are 120˚ and all atoms are within one plane. Our result of bond length is in good agreement with previous DFT calculations of N12Tl12H12.
3.2. Strain Energy
When the strains are applied, all the atoms are allowed full freedom of motion within their plane. A quasiNewton algorithm is used to relax all atoms into equilibrium positions within the deformed unit cell that yields the minimum total energy for the imposed strain state of the super cell.
Both compression and tension are considered with Lagrangian strains ranging from −0.1 to 0.3 with an increment of 0.01 in each step for all three deformation modes. We define strain energy per atom Es = (Etot − E0)/n, where Etot is the total energy of the strained system, E0 is the total energy of the strain-free system, and n = 6 is the number of atoms in the unit cell. This size-independent quantity is used for the comparison between different systems. Figure 3 shows the Es of g-TlN as a function of
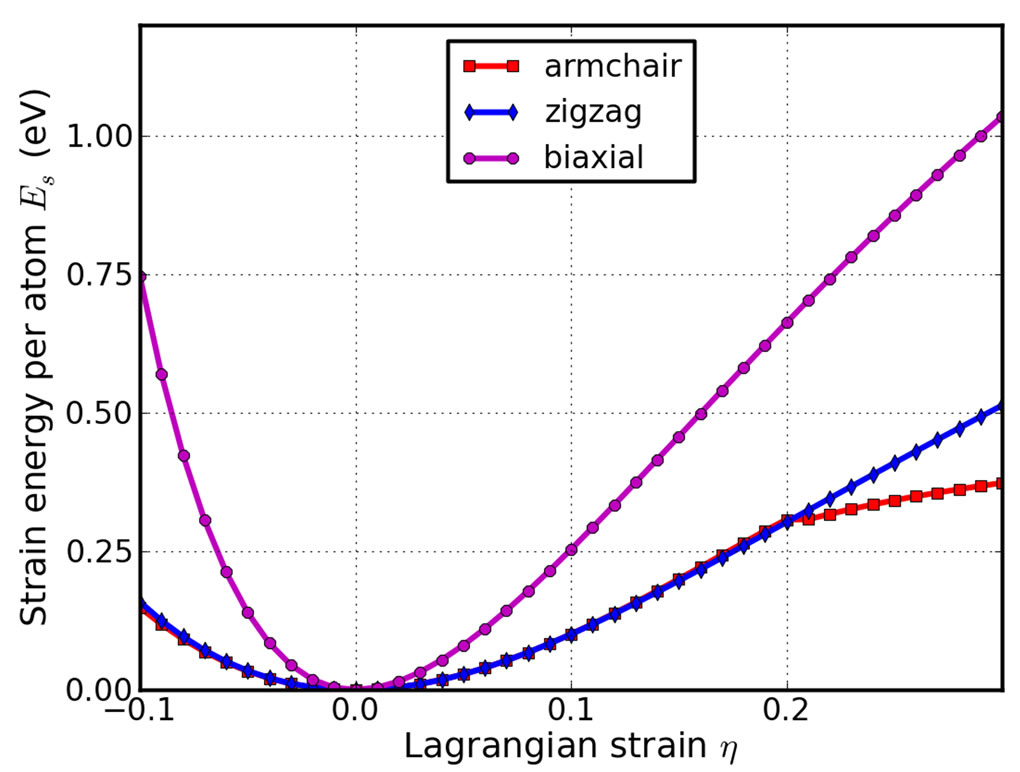
Figure 3. Energy-strain responses for uni-axial strain in armchair and zigzag directions, and biaxial strains.
strain in uni-axial armchair, uni-axial zigzag, and biaxial deformation. Es is seen to be anisotropic with strain direction. Es is non-symmetrical for compression (η < 0) and tension (η > 0) for all three modes. This non-symmetry indicates the anharmonicity of the g-TlN structures. The harmonic region where the Es is a quadratic function of applied strain can be taken between −0.02 < η < 0.02. The stresses, derivatives of the strain energies, are linearly increasing with the increase of the applied strains in the harmonic region. The anharmonic region is the range of strain where the linear stress-strain relationship is invalid and higher order terms are not negligible. With even larger loading of strains, the systems will undergo irreversible structural changes, and the systems are in plastic region where they may fail. The maximum strain in the anharmonic region is the critical strain. The critical strain is 0.2 under armchair deformation. However, for other two directions, the critical strains are not spotted. The ultimate strains are determined as the corresponding strain of the ultimate stress, which is the maxima of the stress-strain curve, as discussed in the following section.
3.3. Stress-Strain Curves
The second P-K stress versus Lagrangian strain relationship for uni-axial strains along the armchair and zigzag directions, as well as biaxial strains are shown in Figure 4. The stresses are the derivatives of the strain energies with respect to the strains. The ultimate strength is the maximum stress that a material can withstand while being stretched, and the corresponding strain is the ultimate strain. Under ideal conditions, the critical strain is larger than the ultimate strain. The systems of perfect g-TlN under strains beyond the ultimate strains are in a metastable state, which can be easily destroyed by long wavelength perturbations, vacancy defects, as well as high temperature effects [70]. The ultimate strain is determined by the intrinsic bonding strengths and acts as a lower limit of the critical strain. Thus it has a practical meaning in considering for its applications.
The ultimate strengths and strains corresponding to the different strain conditions are in Table 1, compared with that of g-BN, graphene, and graphyne. The material behaves in an asymmetric manner with respect to compressive and tensile strains. With increasing strains, the Tl-N bonds are stretched and eventually rupture. When the strain is applied in the armchair direction, the bonds of those parallel with this direction are more severely stretched than those in other directions. The ultimate strain in armchair deformation is 0.17, smaller than that of g-BN, graphene, and graphyne. The critical strain is 0.2 under armchair deformation, where there is big drop of the stresses (Figure 4 top panel), indicating the failure of the system. However, for other two directions, the
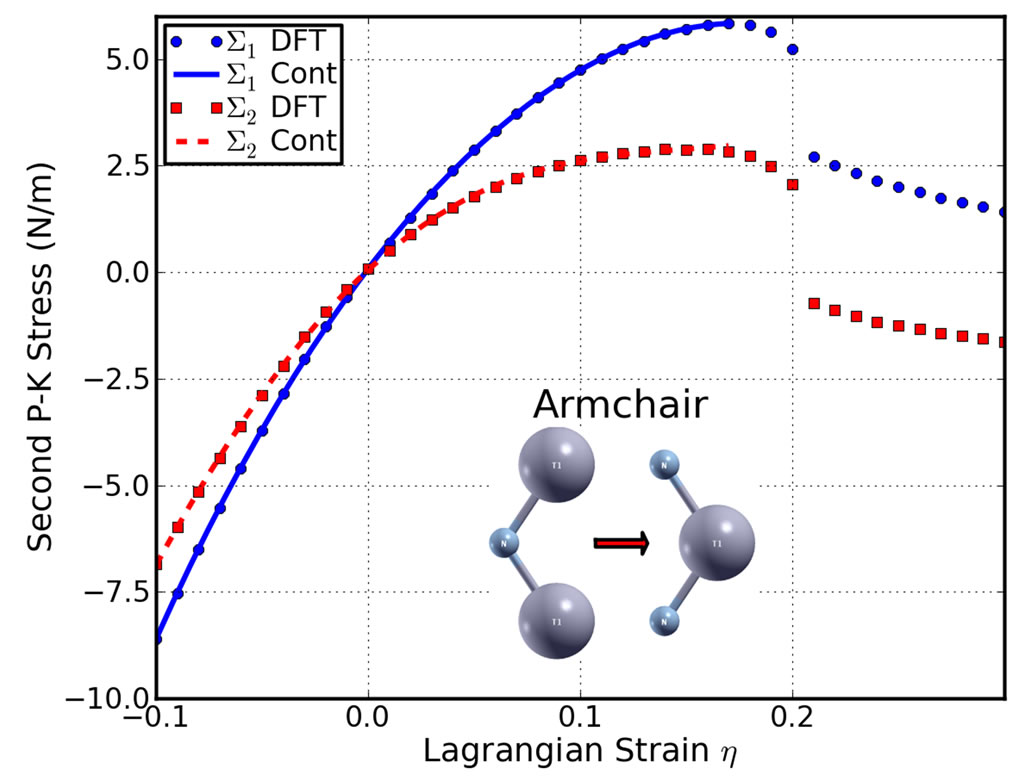

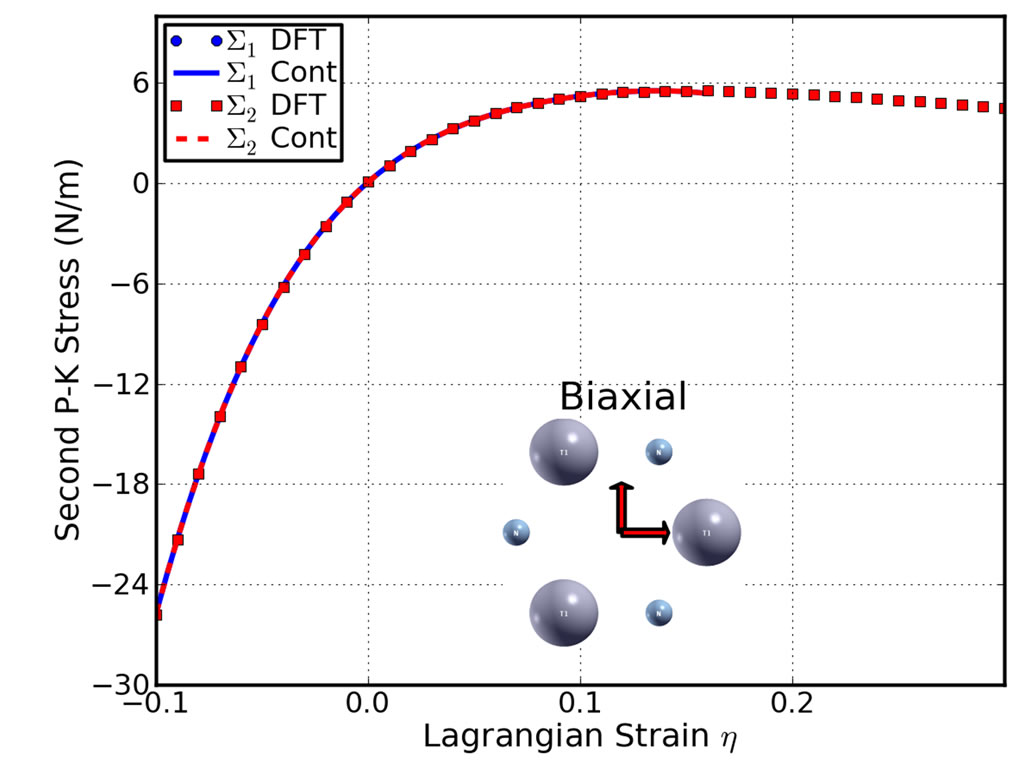
Figure 4. Stress-strain responses of g-TlN under the armchair (top), zigzag (middle), and biaxial (bottom) strains. ∑1 (∑2) denotes the x (y) component of stress. “Cont” stands for the fitting of DFT calculations (“DFT”) to continuum elastic theory.
critical strains are not spotted. Under the zigzag deformation, in which the strain is applied perpendicular to the armchair, there is no bond parallel to this direction. The bonds incline to the zigzag direction with an angle of 30˚ are more severely stretched than those in the armchair direction. The ultimate strain in this zigzag deformation is 0.21, smaller than that of g-BN and graphene, while the same as graphyne. At this ultimate strain, the bonds that are at an incline to the armchair direction appear to be ruptured (Figure 4 middle panel). Under the biaxial deformation, the ultimate strain is 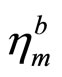 = 0.16, which is the smallest among those of g-BN, graphene, and graphene. As such ultimate strain applied, all the Tl-N bonds are observed to be ruptured (Figure 4 bottom panel).
= 0.16, which is the smallest among those of g-BN, graphene, and graphene. As such ultimate strain applied, all the Tl-N bonds are observed to be ruptured (Figure 4 bottom panel).
It should be noted that the softening of the perfect g-TlN under strains beyond the ultimate strains only occur for ideal conditions. The systems under this circumstance are in a meta-stable state, which can be easily destroyed by long wavelength perturbations and vacancy defects, as well as high temperature effects, and enter a plastic state [70]. Thus only the data within the ultimate strain has physical meaning and was used in determining the high order elastic constants in the following subsection.
3.4. Elastic Constants
The elastic constants are critical parameters in finite element analysis models for mechanical properties of materials. Our results of these elastic constants provide an accurate continuum description of the elastic properties of g-TlN from ab-initio density functional theory calculations. They are suitable for incorporation into numerical methods such as the finite element technique.
The second order elastic constants model the linear elastic response. The higher (>2) order elastic constants are important to characterize the nonlinear elastic response of g-TlN using a continuum description. These can be obtained using a least squares fit of the DFT data and are reported in Table 2. Corresponding values for graphene are also shown.
The in-plane Young’s modulus Ys and Poison’s ratio may be obtained from the following relationships: Ys = 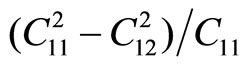 and v =
and v =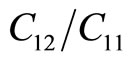 . We have Ys = 34.5 (N/m) and v = 0.689. The in-plane stiffness of g-TlN is
. We have Ys = 34.5 (N/m) and v = 0.689. The in-plane stiffness of g-TlN is

Table 1. Ultimate strengths ( ,
, 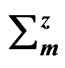 ,
,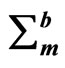 ) in units of N/m and ultimate strains (
) in units of N/m and ultimate strains (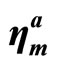 , ηmz, ηmb) under uni-axial strain (armchair and zigzag) and biaxial from DFT calculations, compared with g-BN, graphene, and graphyne.
, ηmz, ηmb) under uni-axial strain (armchair and zigzag) and biaxial from DFT calculations, compared with g-BN, graphene, and graphyne.
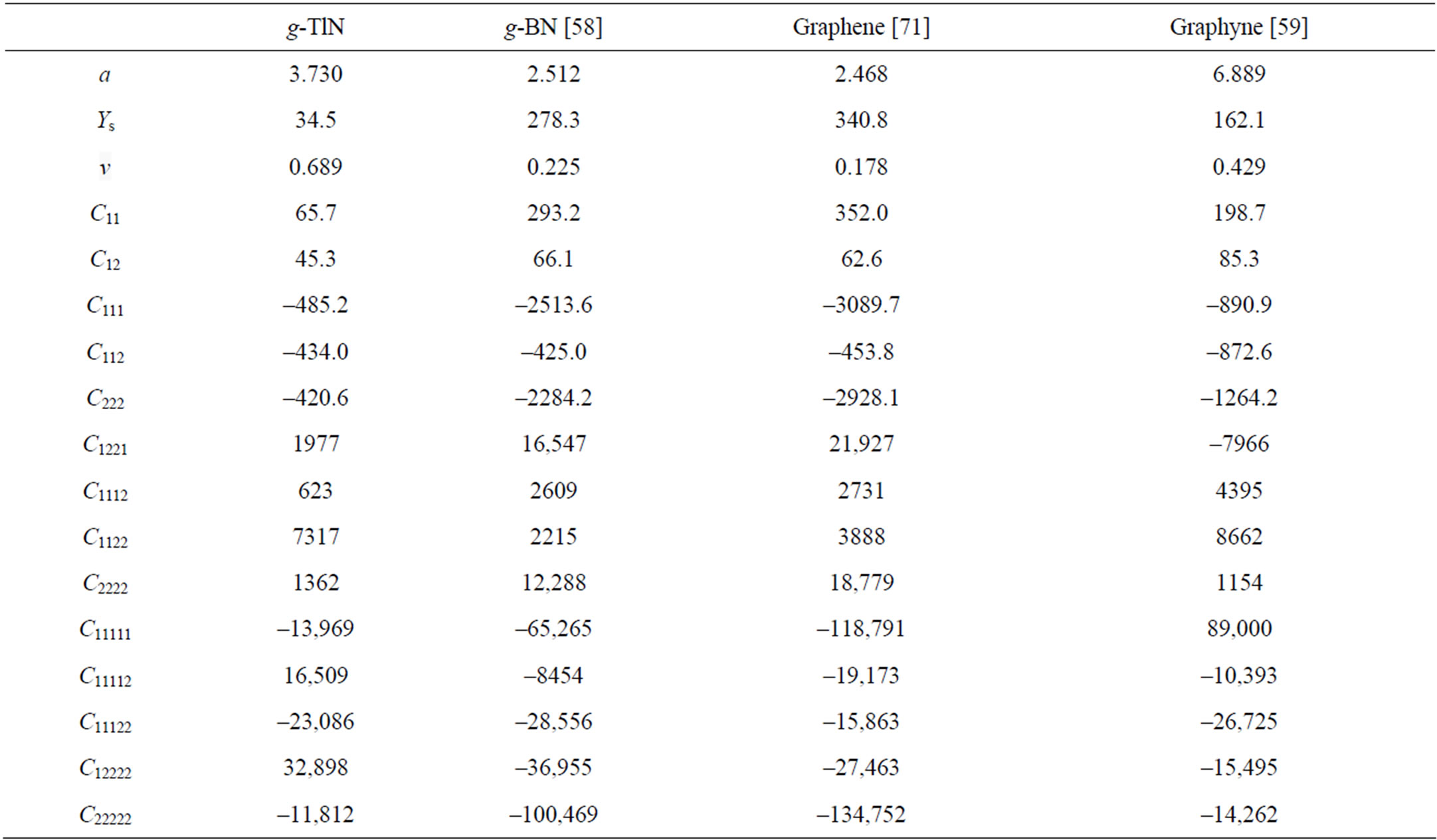
Table 2. Nonzero independent components for the SOEC, TOEC, FOEC, and FFOEC tensor components, Poisson’s ratio v and in-plane stiffness Ys of g-TlN from DFT calculations, compared with g-BN, graphene, and graphyne.
very small compared to g-BN (12%), graphene (10%), and graphyne (21%). The reduction of in-plane stiffness from g-BN to g-TlN is a result of the weakened bond of Tl-N compared to the B-N bond in g-BN. While all other things being equal, bond length is inversely related to bond strength and the bond dissociation energy, as a stronger bond will be shorter. Considering the bond length, in g-TlN the bond length of Tl-N is 2.154 Å, about 49 percents larger than B-N bond length in g-BN (1.45 Å). The bonds can be viewed as being stretched in prior by the introducing of thallium atoms, in reference to g-BN. These stretched bonds are weaker than those un-stretched, leading to a reduction of the mechanical strength.
Knowledge of higher order elastic constants is very useful in understanding the anharmonicity. Using the higher order elastic continuum description, one can calculate the stress and deformation state under uni-axial stress, rather than uni-axial strain [56]. Explicitly, when pressure is applied, the pressure dependent second-order elastic moduli can be obtained from the high order elastic continuum description [32,59,72,73]. The third-order elastic constants are important in understanding the nonlinear elasticity of materials such as changes in acoustic velocities due to finite strain. As a consequence, the nano devices such as nano surface acoustic wave sensors and nano waveguides could be synthesized by introducing local strain [33,59].
Stress-strain curves in the previous section show that they will soften when the strain is larger than the ultimate strain. From the view of electron bonding, this is due to the bond weakening and breaking. This softening behavior is determined by the TOECs and FFOECs in the continuum aspect. The negative values of TOECs and FFOECs ensure the softening of g-TlN monolayer under large strain.
The hydrostatic terms (C11, C22, C111, C222, and so on) of g-TlN monolayers are smaller than those of g-BN and graphene, consistent with the conclusion that the g-TlN is “softer”. The shear terms (C12, C112, C1122, etc.) in general are smaller than those of g-BN and graphene, which contributes to its high compressibility. Compared to graphene, graphyne, and g-BN, one can conclude that the mechanical behavior of g-TlN is similar to graphyne, and much softer than graphene and g-BN.
3.5. Pressure Effect on the Elastic Moduli
With third-order elastic moduli, we can study the effect of the second-order elastic moduli on the pressure P acting in the plane of g-TlN. Explicitly, C when pressure is applied, the pressure dependent second-order elastic moduli (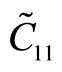 ,
, ,
,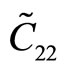 ) can be obtained C11, C12, C22, C111, C112, C22, Ys and v as:
) can be obtained C11, C12, C22, C111, C112, C22, Ys and v as:
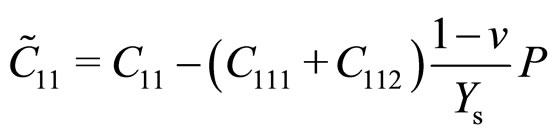 (1)
(1)
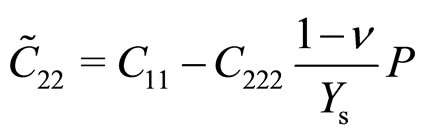 (2)
(2)
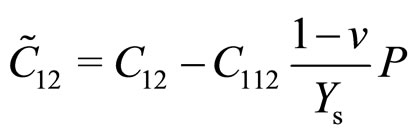 (3)
(3)
The second-order elastic moduli of g-TlN are seen to increase linearly with the applied pressure (Figure 5). However, Poisson’s ratio decreases monotonically with the increase of pressure. 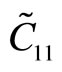 is asymmetrical to
is asymmetrical to 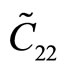 unlike the zero pressure case.
unlike the zero pressure case. 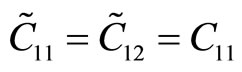 only occurs when the pressure is zero. This anisotropy could be the outcome of anharmonicity.
only occurs when the pressure is zero. This anisotropy could be the outcome of anharmonicity.
4. Conclusions
In summary, we studied the mechanical response of g-TlN under various strains using DFT based first principles calculations. It is observed that g-TlN exhibits a nonlinear elastic deformation up to an ultimate strain, which is 0.17, 0.21, and 0.16 for armchair, zigzag, and biaxial directions, respectively. The deformation and failure behavior and the ultimate strength are anisotropic. It has low in-plane stiffness (34.5 N/m) and large Poisson ratio compared to g-BN and graphene. Compared to gBN, g-TlN has 12% in-plane stiffness, 25%, 22%, and 20% ultimate strengths in armchair, zigzag, and biaxial strains respectively, and a 3.1 times of Poisson’s ratio. We also found that the g-TlN can sustain much smaller strains before the rupture.
The nonlinear elasticity of g-TlN was investigated. We found an accurate continuum description of the elastic properties of g-TlN by explicitly determining the fourteen independent components of high order (up to fifth order) elastic constants from the fitting of the stress-
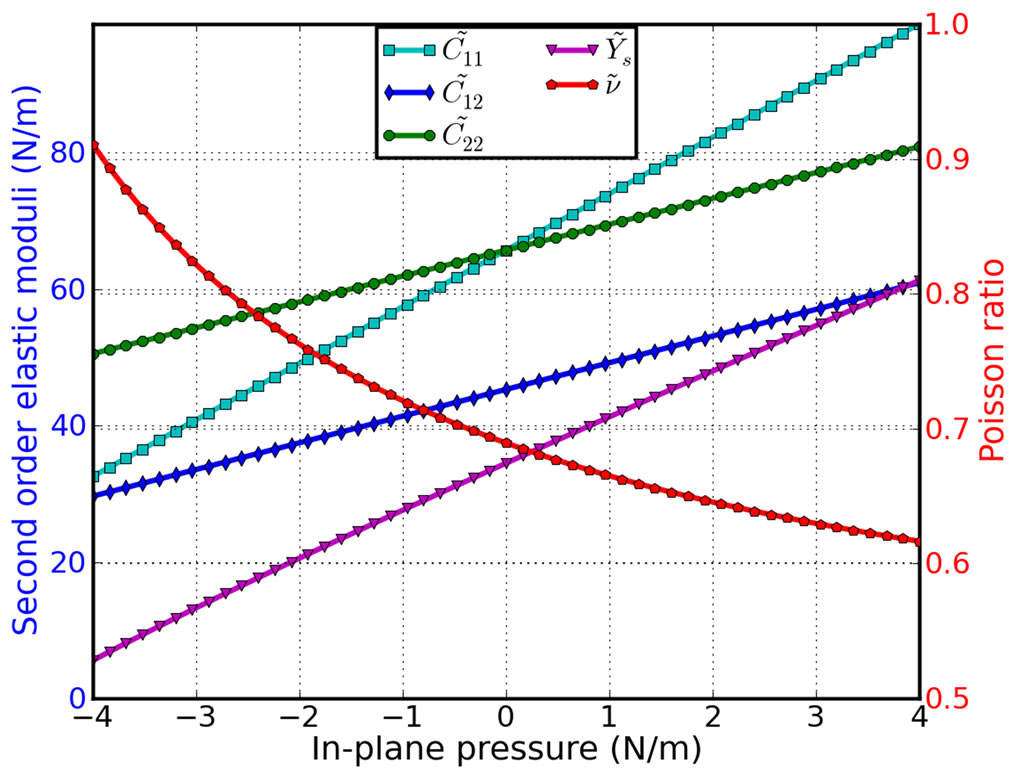
Figure 5. Second-order elastic moduli and Poisson ratio as a function of the pressure.
strain curves obtained from DFT calculations. This data is useful to develop a continuum description which is suitable for incorporation into a finite element analysis model for its applications in large scale. The second order elastic constants including in-plane stiffness are predicted to monotonically increase with pressure while Poisson’s ratio monotonically decreases with increasing pressure.
5. Acknowledgements
The authors would like to acknowledge the generous financial support from the Defense Threat Reduction Agency (DTRA) Grant #BRBAA08-C-2-0130, the U.S. Nuclear Regulatory Commission Faculty Development Program under contract #NRC-38-08-950, and U.S. Department of Energy (DOE) Nuclear Energy University Program (NEUP) Grant #DE-NE0000325.
REFERENCES
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, “Electric Field Effect in Atomically Thin Carbon Films,” Science Magazine, Vol. 306, No. 5696, 2004, pp. 666-669. doi:10.1126/science.1102896
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, “Two-Dimensional Atomic Crystals,” Proceedings of the National Academy of Sciences, USA, Vol. 102, No. 30, 2005, pp. 10451-10453. doi:10.1073/pnas.0502848102
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, “Two-Dimensional Gas of Massless Dirac Fermions in Graphene,” Nature, Vol. 438, No. 7065, 2005, pp. 197-200. doi:10.1038/nature04233
- Y.-M. Lin, K. A. Jenkins, A. Valdes-Garcia, J. P. Small, D. B. Farmer and P. Avouris, “Operation of Graphene Transistors at Gigahertz Frequencies,” Nano Letters, Vol. 9, No. 1, 2009, pp. 422-426. doi:10.1021/nl803316h
- Y.-M. Lin, C. Dimitrakopoulos, K. A. Jenkins, D. B. Farmer, H.-Y. Chiu, A. Grill and P. Avouris, “100-Ghz Transistors from Wafer-Scale Epitaxial Graphene,” Science Magazine, Vol. 327, No. 5966, 2010, p. 662. doi:10.1126/science.1184289
- L. Liao, Y.-C. Lin, M. Bao, R. Cheng, J. Bai, Y. Liu, Y. Qu, K. L. Wang, Y. Huang and X. Duan, “High-Speed Graphene Transistors with a Self-Aligned Nanowire Gate,” Nature, Vol. 467, No. 7313, 2010, pp. 305-308. doi:10.1038/nature09405
- Y. Ma, Y. Dai, M. Guo and B. Huang, “Graphene-Diamond Interface: Gap Opening and Electronic Spin Injection,” Physics Review B, Vol. 85, No. 23, 2012, pp. 235448-223452. doi:10.1103/PhysRevB.85.235448
- G. Brumfiel, “Graphene Gets Ready for the Big Time,” Nature, Vol. 458, No. 7237, 2009, pp. 390-391. doi:10.1038/458390a
- R. Mas-Balleste, C. Gomez-Herrero, J. Gómez-Herrero and F. Zamora, “2D Materials: To Graphene and Beyond,” Nanoscale, Vol. 3, No. 1, 2011, pp. 20-30.
- T. P. Kaloni, Y. C. Cheng and U. Schwingenschloegl, “Electronic Structure of Superlattices of Graphene and Hexagonal Boron Nitride,” Journal of Material Chemistry, Vol. 22, No. 3, 2012, pp. 919-922. doi:10.1039/c1jm14895h
- J. N. Coleman, M. Lotya, A. O’neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. Mccomb, P. D. Nellist and V. Nicolosi, “Two-Dimensional Nanosheets Produced by Liquid Exfoliation of Layered Materials,” Science Magazine, Vol. 331, No. 6017, 2011, pp. 568-571. doi:10.1126/science.1194975
- A. Nag, K. Raidongia, K. P. Hembram, R. Datta, U. V. Waghmare and C. N. Rao, “Graphene Analogues of Bn: Novel Synthesis and Properties,” ACS Nano, Vol. 4, No. 3, 2010, pp. 1539-1544. doi:10.1021/nn9018762
- C. Li, Y. Bando, C. Zhi, Y. Huang and D. Golberg, “Thickness-Dependent Bending Modulus of Hexagonal Boron Nitride Nanosheets,” Nanotechnology, Vol. 20, No. 38, 2009, pp. 385707-385712. doi:10.1088/0957-4484/20/38/385707
- L. Song, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, “Large Scale Growth and Characterization of Atomic Hexagonal Boron Nitride Layers,” Nano Letters, Vol. 10, No. 8, 2010, pp. 3209-3215. doi:10.1021/nl1022139
- M. Topsakal, E. Aktürk and S. Ciraci, “First-Principles Study of Twoand One-Dimensional Honeycomb Structures of Boron Nitride,” Physics Review B, Vol. 79, No. 11, 2009, pp. 115442-115452. doi:10.1103/PhysRevB.79.115442
- K. Watanabe, T. Taniguchi and H. Kanda, “Direct-Bandgap Properties and Evidence for Ultraviolet Lasing of Hexagonal Boron Nitride Single Crystal,” Nature Material, Vol. 3, No. 6, 2004, pp. 404-409. doi:10.1038/nmat1134
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, “Large-Scale Fabrication of Boron Nitride Nanosheets and Their Utilization in Polymeric Composites with Improved Thermal and Mechanical Properties,” Advanced Material, Vol. 21, No. 28-32, 2009, pp. 2889-2893.
- G. Y. Guo and J. C. Lin, “Systematic ab Initio Study of the Optical Properties of Bn Nanotubes,” Physics Review B, Vol. 71, No. 16, 2005, pp. 165402-165413. doi:10.1103/PhysRevB.71.165402
- W. Han, W. Mickelson, J. Cumings and A. Zettl, “Transformation of Bxcynz Nanotubes to Pure Bn Nanotubes,” Applied Physics Letters, Vol. 81, No. 6, 2002, pp. 1110-1112. doi:10.1063/1.1498494
- K. Suenaga, C. Colliex, N. Demoncy, A. Loiseau, H. Pascard and F. Willaime, “Synthesis of Nanoparticles and Nanotubes with Well-Separated Layers of Boron Nitride and Carbon,” Science Magazine, Vol. 278, No. 5338, 1997, pp. 653-655. doi:10.1126/science.278.5338.653
- A. P. Suryavanshi, M. F. Yu, J. G. Wen, C. C. Tang and Y. Bando, “Elastic Modulus and Resonance Behavior of Boron Nitride Nanotubes,” Applied Physics Letters, Vol. 84, No. 14, 2004, pp. 2527-2529. doi:10.1063/1.1691189
- X. Blase, A. Rubio, S. G. Louie and M. L. Cohen, “Stability and Band-Gap Constancy of Boron-Nitride Nanotubes,” Europhyics Letter, Vol. 28, No. 5, 1994, pp. 335-341. doi:10.1209/0295-5075/28/5/007
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. Tang and C. Zhi, “Boron Nitride Nanotubes and Nanosheets,” ACS Nano, Vol. 4, No. 6, 2010, pp. 2979-2993. doi:10.1021/nn1006495
- L. Liu, Y. P. Feng and Z. X. Shen, “Structural and Electronic Properties of H-Bn,” Physics Review B, Vol. 68, No. 10, 2003, pp. 104102-104109. doi:10.1103/PhysRevB.68.104102
- K. N. Kudin, G. E. Scuseria and B. I. Yakobson, “C2f, Bn and C Nanoshell Elasticity from ab Initio Computations,” Physics Review B, Vol. 64, No. 23, 2001, pp. 235406-235415. doi:10.1103/PhysRevB.64.235406
- A. Zhang, H. F. Teoh, Z. Dai, Y. P. Feng and C. Zhang, “Band Gap Engineering in Graphene and Hexagonal Bn Antidot Lattices: A First Principles Study,” Applied Physics Letters, Vol. 98, No. 2, 2011, pp. 023105-023107. doi:10.1063/1.3536517
- T. Kawasaki, T. Ichimura, H. Kishimoto, A. A. Akbar, T. Ogawa and C. Oshima, “Double Atomic Layers of Graphene/Monolayer H-Bn on Ni(111) Studied by Scanning Tunneling Microscopy and Scanning Tunneling Spectroscopy,” Surface Review and Letters, Vol. 9, No. 3-4, 2002, pp. 1459-1464. doi:10.1142/S0218625X02003883
- J. Slawinska, I. Zasada, P. Kosinski and Z. Klusek, “Reversible Modifications of Linear Dispersion: Graphene between Boron Nitride Monolayers,” Physics Review B, Vol. 82, No. 8, 2010, pp. 085431-085435. doi:10.1103/PhysRevB.82.085431
- G. Giovannetti, P. A. Khomyakov, G. Brocks, P. J. Kelly and J. Van Den Brink, “Substrate Induced Band Gap in Graphene on Hexagonal Boron Nitride: Ab Initio Density Functional Calculations,” Physics Review B, Vol. 76, No. 7, 2007, pp. 073103-073106. doi:10.1103/PhysRevB.76.073103
- S. Bhowrnick, A. K. Singh and B. I. Yakobson, “Quantum Dots and Nanoroads of Graphene Embedded in Hexagonal Boron Nitride,” The Journal of Physical Chemistry C, Vol. 115, No. 20, 2011, pp. 9889-9893. doi:10.1021/jp200671p
- L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li, A. Srivastava, Z. F. Wang, K. Storr, L. Balicas, F. Liu and P. M. Ajayan, “Atomic Layers of Hybridized Boron Nitride and Graphene Domains,” Nature Material, Vol. 9, No. 5, 2010, pp. 430-435. doi:10.1038/nmat2711
- Q. Peng and S. De, “Tunable Band Gaps of Mono-Layer Hexagonal BNC Heterostructures,” Physical E, Vol. 44, No. 7-8, 2012, pp. 1662-1666. doi:10.1016/j.physe.2012.04.011
- Q. Peng, A. R. Zamiri, W. Ji and S. De, “Elastic Properties of Hybrid Graphene/Boron Nitride Monolayer,” Acta Mechanica, in press, 2012. doi:10.1007/s00707-012-0714-0
- J. Zhou, K. Lv, Q. Wang, X. S. Chen, Q. Sun and P. Jena, “Electronic Structures and Bonding of Graphyne Sheet and Its Bn Analog,” Journal of Chemical Physics, Vol. 134, No. 17, 2011, pp. 174701-174706. doi:10.1063/1.3583476
- A. Schleife, F. Fuchs, C. Roedl, J. Furthmueller and F. Bechstedt, “Branch-Point Energies and Band Discontinuities of Iii-Nitrides and Iii-/Ii-Oxides from Quasiparticle Band-Structure Calculations,” Applied Physics Letter, Vol. 94, No. 1, 2009, pp. 012104-012106. doi:10.1063/1.3059569
- F. Bernardini, V. Fiorentini and D. Vanderbilt, “Spontaneous Polarization and Piezoelectric Constants of Iii-V Nitrides,” Physics Review B, Vol. 56, No. 16, 1997, pp. R10024-R10027. doi:10.1103/PhysRevB.56.R10024
- O. Ambacher, “Growth and Applications of Group Iii Nitrides,” Journal of Physics D, Vol. 31, No. 20, 1998, pp. 2653-2710. doi:10.1088/0022-3727/31/20/001
- M. Ferhat and A. Zaoui, “Do All Iii-V Compounds Have the Zinc-Blende or Wurtzite Ground State Structure,” Applied Physics Letter, Vol. 88, No. 16, 2006, pp. 161902-161904. doi:10.1063/1.2196050
- Y. Wang and S. Shi, “Structural and Electronic Properties of Monolayer Hydrogenated Honeycomb Iii-V Sheets from First-Principles,” Solid State Communications, Vol. 150, No. 31, 2010, pp. 1473-1478. doi:10.1016/j.ssc.2010.05.031
- N. S. Dantas, J. S. De Almeida, R. Ahuja, C. Persson and A. F. Da Silva, “Novel Semiconducting Materials for Optoelectronic Applications: Al1-Xtlxn Alloys,” Applied Physics Letter, Vol. 92, No. 12, 2008, pp. 121914-121916. doi:10.1063/1.2901146
- H. M. A. Mazouz, A. Belabbes, A. Zaoui and M. Ferhat, “First-Principles Study of Lattice Dynamics in ThalliumV Compounds,” Superlattices Microstructures, Vol. 48, No. 6, 2010, pp. 560-568. doi:10.1016/j.spmi.2010.09.012
- M. Vanschilfgaarde, A. Sher and A. B. Chen, “Intlsb—An Infrared Detector Material,” Applied Physics Letter, Vol. 62, No. 16, 1993, pp. 1857-1859. doi:10.1063/1.109523
- M. Vanschilfgaarde, A. B. Chen, S. Krishnamurthy and A. Sher, “Intip—A Proposed Infrared Detector Material,” Applied Physics Letter, Vol. 65, No. 21, 1994, pp. 2714-2716. doi:10.1063/1.112567
- K. Yamamoto, H. Asahi, M. Fushida, K. Iwata and S. Gonda, “Gas Source Molecular Beam Epitaxy Growth of Tlinp for New Infrared Optical Devices,” Journal of Applied Physics, Vol. 81, No. 4, 1997, pp. 1704-1707. doi:10.1063/1.364013
- R. Beneyton, G. Grenet, P. Regreny, M. Gendry, G. Hollinger, B. Canut and C. Priester, “Experimental and Theoretical Investigation into the Difficulties of Thallium Incorporation into Iii-V Semiconductors,” Physics Review B, Vol. 72, No. 12, 2005, pp. 125209. doi:10.1103/PhysRevB.72.125209
- L. Shi, Y. Duan, X. Yang, G. Tang, L. Qin and L. Qiu, “Structural, Electronic and Elastic Properties of WurtziteStructured Tlxal1-Xn Alloys from First Principles,” Materials Science in Semiconductor Processing, Vol. 15, No. 5, 2012, pp. 499-504. doi:10.1016/j.mssp.2012.03.013
- N. Saidi-Houat, A. Zaoui, A. Belabbes and M. Ferhat, “Ab Initio Study of the Fundamental Properties of Novel Iii-V Nitride Alloys Ga1-Xtlxn,” Materials Science and Engineering: B, Vol. 162, No. 1, 2009, pp. 26-31. doi:10.1016/j.mseb.2009.01.031
- N. Saidi-Houat, A. Zaoui and M. Ferhat, “Structural Stability of Thallium-V Compounds,” Journal of Physics: Condensed Matter, Vol. 19, No. 10, 2007, pp. 106221-106239. doi:10.1088/0953-8984/19/10/106221
- L. Shi, Y. Duan and L. Qin, “Structural Phase Transition, Electronic and Elastic Properties in Tlx (X = N, P, as) Compounds: Pressure-Induced Effects,” Computational Materials Science, Vol. 50, No. 1, 2010, pp. 203-210. doi:10.1016/j.commatsci.2010.07.027
- S. Li-Wei, D. Yi-Feng, Y. Xian-Qing and T. Gang, “Phonon and Elastic Instabilities in Zincblende Tln under Hydrostatic Pressure from First Principles Calculations,” Chinese Physics Letter, Vol. 28, No. 10, 2011, pp. 100503-100507. doi:10.1088/0256-307X/28/10/100503
- E. Chigo Anota, M. Salazar Villanueva and H. Hernandez Cocoletzi, “Electronic Properties of Group Iii: A Nitride Sheets by Molecular Simulation,” Physica Status Solidi (c), Vol. 7, No. 7-8, 2010, pp. 2252-2254. doi:10.1002/pssc.200983499
- F. Guinea, M. I. Katsnelson and A. K. Geim, “Energy Gaps and a Zero-Field Quantum Hall Effect in Graphene by Strain Engineering,” Nature Physics, Vol. 6, No. 1, 2010, pp. 30-33. doi:10.1038/nphys1420
- Y. Ma, Y. Dai, W. Wei, C. Niu, L. Yu and B. Huang, “First-Principles Study of the Graphene@Mose2 Heterobilayers,” The Journal of Physical Chemistry C, Vol. 115, No. 41, 2011, pp. 20237-20241. doi:10.1021/jp205799y
- Z. H. Aitken and R. Huang, “Effects of Mismatch Strain and Substrate Surface Corrugation on Morphology of Supported Monolayer Graphene,” Journal of Applied Physics, Vol. 107, No. 12, 2010, pp. 123531-123540. doi:10.1063/1.3437642
- C. Lee, X. Wei, J. W. Kysar and J. Hone, “Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene,” Science Magazine, Vol. 321, No. 5887, 2008, pp. 385-388. doi:10.1126/science.1157996
- X. Wei, B. Fragneaud, C. A. Marianetti and J. W. Kysar, “Nonlinear Elastic Behavior of Graphene: Ab Initio Calculations to Continuum Description,” Physics Review B, Vol. 80, No. 20, 2009, pp. 205407-205414. doi:10.1103/PhysRevB.80.205407
- Q. Peng, W. Ji and S. De, “Domain Size Effect on Mechanical Properties of the Hybrid Graphene/Boron Nitride Monolayer,” under review.
- Q. Peng, W. Ji and S. De, “Mechanical Properties of the Hexagonal Boron Nitride Monolayer: Ab Initio Study,” Computational Materials Science, Vol. 10, No. 8, 2012, pp. 11-17. doi:10.1016/j.commatsci.2011.12.029
- Q. Peng, W. Ji and S. De, “Mechanical Properties of Graphyne Monolayer: A First-Principles Study,” Physical Chemistry Chemical Physics, Vol. 14, 2012, pp. 13385-13391. doi:10.1039/c2cp42387a
- C. A. Marianetti and H. G. Yevick, “Failure Mechanisms of Graphene under Tension,” Physics Review Letter, Vol. 105, No. 24, 2010, pp. 245502-245505. doi:10.1103/PhysRevLett.105.245502
- G. Kresse and J. Hafner, “Ab Initio Molecular Dynamics for Liquid Metals,” Physics Review B, Vol. 47, No. 1, 1993, pp. 558-561. doi:10.1103/PhysRevB.47.558
- G. Kresse and J. Hafner, “Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semicon Ductor Transition in Germanium,” Physics Review B, Vol. 49, No. 20, 1994, pp. 14251-14269. doi:10.1103/PhysRevB.49.14251
- G. Kresse and J. Furthuller, “Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set,” Computational Materials Science, Vol. 6, No. 1, 1996, pp. 15-50. doi:10.1016/0927-0256(96)00008-0
- G. Kresse and J. Furthuller, “Efficient Iterative Schemes for ab Initio Total-Energy Calculations Using a PlaneWave Basis Set,” Physics Review B, Vol. 54, No. 16, 1996, pp. 11169-11186. doi:10.1103/PhysRevB.54.11169
- P. Hohenberg and W. Kohn, “Inhomogeneous Electron Gas,” Physical Review B, Vol. 136, No. 3B, 1964, pp. B864-B871. doi:10.1103/PhysRev.136.B864
- W. Kohn and L. J. Sham, “Self-Consistent Equations Including Exchange and Correlation Effects,” Physics Review, Vol. 140, No. 4A, 1965, pp. A1133-A1138. doi:10.1103/PhysRev.140.A1133
- J. Perdew, K. Burke and M. Ernzerhof, “Generalized Gradient Approximation Made Simple,” Physics Review Letter, Vol. 77, No. 18, 1996, pp. 3865-3868. doi:10.1103/PhysRevLett.77.3865
- P. E. Blochl, “Projector Augmented-Wave Method,” Physics Review B, Vol. 50, No. 24, 1994, pp. 17953-17979. doi:10.1103/PhysRevB.50.17953
- R. O. Jones and O. Gunnarsson, “The Density Functional Formalism, Its Applications and Prospects,” Reviews of Modern Physics, Vol. 61, No. 3, 1989, pp. 689-746. doi:10.1103/RevModPhys.61.689
- M. Topsakal, S. Cahangirov and S. Ciraci, “The Response of Mechanical and Electronic Properties of Graphane to the Elastic Strain,” Applied Physics Letter, Vol. 96, No. 9, 2010, pp. 091912-091914. doi:10.1063/1.3353968
- Q. Peng, C. Liang, W. Ji and S. De, “Change of Nonlinear Mechanical Properties by Hydrogenation: Graphane vs Graphene,” under review.
- J. F. Nye, “Physical Properties of Crystals,” Oxford Science Publications, Oxford, 1995.
- S. Y. Davydov, “Third Order Elastic Moduli of Single Layer Graphene,” Physics of the Solid State, Vol. 53, No. 3, 2011, pp. 665-668. doi:10.1134/S1063783411030073
NOTES
*Corresponding author.