Journal of Biosciences and Medicines
Vol.06 No.05(2018), Article ID:84732,26 pages
10.4236/jbm.2018.65009
Proinflammatory Signaling Cascades of Periodontopathic Oral Pathogen Porphyromonas gingivalis
Bronislaw L. Slomiany, Amalia Slomiany
Research Center, Rutgers School of Dental Medicine, Rutgers, The State University of New Jersey, Newark, USA
Copyright © 2018 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: April 20, 2018; Accepted: May 20, 2018; Published: May 23, 2018
ABSTRACT
Porphyromonas gingivalis, is the most prominent member of the bacteria flora associated with pathogenesis of periodontitis, a chronic inflammatory disease resulting in tooth loss. The extent of oral mucosal reaction to P. gingivalis invasion relays heavily on Toll-like receptors (TLRs) that recognize structurally common motifs of pathogens and initiate antibacterial responses. Among the virulence factors of P. gingivalis implicated in TLRs activation and triggering inflammatory responses leading to the development of periodontitis is the bacterium cell-wall lipopolysaccharide (LPS). The engagement by the LPS of oral mucosal TLR4 leads to initiation of signaling events characterized by the activation of mitogen-activated protein kinase (MAPK) and IκB-kinase complex (IKK) cascades, induction of phosphoinositide-specific phospholipase C (PLC)/protein kinase C (PKC)/PI3K pathway, up-regulation in TGF-α ectodomain shedding and EGFR transactivation, and the amplification of proinflammatory signals by spleen tyrosine kinase (Syk). These events, in turn, exert their control over transcription factors implicated in the induction of the expression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) genes that lead to up-regulation in the inflammatory mediators, PGE2 and NO. The systems involved in transcription factors activation, furthermore, remain under additional regulatory control through S-nitrosylation. Moreover, the LPS-induced TLR4 activation provides a docking site for Syk, the activation of which leads to amplification of the inflammatory signals by affecting transcription factors activation and their assembly to transcriptional complexes. Interestingly, the extent of oral mucosal inflammatory response to P. gingivalis remains under modulatory influence by two biologically active peptide hormones, leptin and ghrelin. Therefore, the presence of these multifunctional peptides in oral mucosa and saliva may be of significance in countering the destructive consequences of P. gingivalis―induced chronic mucosal inflammation that characterizes periodontitis.
Keywords:
P. gingivalis, Periodontal Disease, Oral Mucosa, Inflammatory Response, LPS, TLR4, Signaling Cascades

1. Introduction
Porphyromonas gingivalis is a non-motile, rod-shaped, Gram-negative anaerobic bacterium, which together with more than 700 bacterial species colonize the epithelial surfaces of oral mucosa, soft gingival tissue and tooth enamel [1] [2] [3] . Under normal physiological conditions, this diverse community of oral microbiota exists in commensal harmony with the host and this status is maintained primarily by saliva, a viscous secretion elaborated by the major and minor salivary glands located within the oral cavity [4] [5] . The proteins and glycoproteins of saliva not only participate in the formation of the tenacious protective coating covering tooth enamel and oral mucosa, but are also endowed by a large repertoire of biological functions that play a major role in bacterial aggregation and clearance from the oral cavity [4] [5] [6] [7] [8] . The disturbances in salivary glands secretory function and the ensuing decline in the protective potential of saliva heralds the weakening of oral mucosal defense perimeter and provides an opportunity for bacterial infection, and the development of chronic mucosal inflammation that leads to periodontal disease [5] [8] [9] [10] .
The bacterial species implicated as primary etiological agents of periodontal diseases are the members of so called “red bacterial complex”, and include Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia [11] [12] , while the role of facultative anaerobe, Actinobacillus actinomycetemcomitans, in etiology of periodontal disease is less apparent, although the bacterium is more frequently found in localized aggressive form of periodontitis [1] [13] [14] . Based on subgingival plaque analysis data, P. gingivalis is by far the most prominent member of the bacterial flora found in periodontal packets of people with gum disease [15] , and is generally recognized as a major culprit in the pathogenesis of periodontitis, a chronic inflammatory disease that affects about 15% of population and leads to progressive destruction of teeth-supporting tissue, and is the major cause of adult tooth loss [1] [12] [16] . Apparently, the first step in P. gingivalis invasion of gingival epithelium is the evasion of host defense mechanisms followed by the bacterium attachment to the epithelial surfaces through cell-surface specific receptors, and the unleash of a large panel of virulence factors that provoke the inflammatory response of the host tissue [2] [5] [12] . The factors implicated in the progressive destruction of periodontal tissues by P. gingivalis include its cell surface major (FimA) and minor (Mfa1) fimbriae proteins [17] [18] , elaborated arginine and lysine specific cysteine proteinases, referred to as gingipain R and gingipain K [16] [19] , collagenases, capable of extracellular matrix proteins degradation and activation of the host matrix metalloproteinases [2] [20] [21] [22] [23] , and the secreted sufatases directed towards cell membrane glycosphingolipids as well as proteoglycans of extracellular matrix [2] [24] .
A factor of particular significance to the pathogenic action of P. gingivalis leading to the development of periodontitis is its cell-wall lipopolysaccharide (LPS) [25] [26] [27] . Indeed, the oral mucosal responses to P. gingivalis LPS are manifested by a marked increase in epithelial cell apoptosis and proinflammatory cytokine expression, up-regulation in endothelin-1 (ET-1) and TNF-α release, stimulation in NF-κB nuclear translocation, excessive nitric oxide (NO) and prostaglandin (PGE2) generation, and the proinflammatory signal propagation associated with the activation of epidermal growth factor receptor (EGFR) and mitogen-activated protein kinase (MAPK) cascade [25] - [31] . Moreover, studies into the events underlying the inflammatory processes elicited by bacterial invaders revealed that these responses are mediated by Tool-like receptors (TLRs), a class of transmembrane pattern recognition receptors (PRRs) that recognize structurally common motifs of bacterial, fungal and protozoan origin [32] [33] [34] . Among the bacterial products implicated in TLRs activation and triggering vigorous inflammatory responses are lipoteichoic proteoglycans of Gram-positive bacteria and LPS of Gram-negative bacteria, including that of P. gingivalis [26] [28] [35] [36] [37] .
Therefore considering the preponderance of the evidence as to the primary role of P. gingivalis in the initiation and propagation of oral mucosal inflammatory responses leading to the development of periodontitis, in this article we review data on the signaling cascades triggered by the bacterium LPS activation of TLR4, and examine how these cascades are modulated by the endogenous peptide hormones, leptin and ghrelin.
2. Tool-Like Receptors
2.1. Structure and Activation
TLRs are a family of ten type I transmembrane glycoproteins, consisting of a highly N-glycosylated extracellular domain characterized by the presence of a leucine-rich repeat motifs (LRR) involved in ligand recognition, a single transmembrane spanning region, and an intracellular domain that contains a Toll-Il-1 receptor (TIR) region involved in the activation of signaling cascade [32] [38] . Majority of TLRs are located mainly in the cell membrane (TLR1, TLR2, TLR4, TLR5, and TLR6), with some (TLR7, and TLR9) being also present in the endosomes (34), and their TIR domains are not only found in the receptors but also in the adaptors involved in IL-1 signaling [39] .
In general, the stimulation of TLRs by the respective ligands triggers receptor dimerization followed by the recruitment of different adaptor molecules to the cytoplasmic domain (TIR) of the receptor, such as myeloid differentiation factor 88 (MyD88) or Toll/Interleukin-1 receptor (TIR) domain-containing adaptor-inducing interferon-(TRIF) [32] [39] . Depending on which of the adaptors are involved, these molecules trigger a different response and affect different downstream signaling cascades converging on NF-κB, interferon response factors (IRFs), and MAPKs [32] [34] . Indeed, with the exception of TLR3, all TLRs utilize MyD88 adaptor pathway. In this pathway, TLR binding leads to recruitment of MyD88, which in conjunction with another TIR-containing adaptor molecule, Mal (MyD88 adaptor-like), form complex with IRAK kinases, followed by polyubiquitination and activation of TAK1 [33] [34] . This results in activation of IκB-kinase complex (IKK) and MAPK pathways involving extracellular signal-regulated kinase (ERK), c-Jun terminal kinase (JNK), and p38 [32] [40] . Activation of IKK leads to phosphorylation of NF-κB inhibitory protein IκBα and translocation of NF-κB to the nucleus and induction of the expression of proinflammatory genes, while MAPKs are involved in the activation of activator protein-1 complex (AP-1) transcription program [33] [34] [41] .
Based on the ligand recognition and binding, TLR4 is now well-documented as a receptor that mediates inflammatory responses to bacterial LPS, including that of P. gingivalis [25] [26] [27] . Indeed, studies show that the exposure of salivary gland acinar cells to P. gingivalis LPS results in the TLR4 activation through autophosphorylation (Figure 1) on several critical Tyr residues that are essential for the initiation of downstream signaling events [42] .
2.2. LPS and Its Interaction with TLR4
The Lipopolysaccharide is an integral glycolipid component of the outer membrane of Gram-negative bacteria where it plays important role in the maintenance of cellular and structural integrity, folding and insertion of membrane proteins, and the entry of hydrophobic molecules [2] [32] [37] . The glycolipid consists of membrane anchored hydrophobic domain, referred to as lipid A, non-repeating core oligosaccharide region, and a distal polysaccharide structure referred to as O-antigen chain. The lipid A region is composed of phosphorylated β(1,6)-linked diglucosamine backbone with four to seven fatty acyl chains ranging from 10 to 16 carbon units attached to it. Of these, four acyl chains are directly linked to the glucosamine backbone, and the remaining being attached to the hydroxyl groups of the lipid chains [38] [39] . The non-repeating core oligosaccharide region is rich in heptose and KDO (keto-deoxyoctulosonate), and coupled to the O-specific highly heterogeneous set of carbohydrate chains extending from the cell surface [32] [38] [39] .
The endotoxin and the immunological properties of LPS reside mainly in the heterogeneous acylation character of lipid A domain, which in different bacterial species shows considerable variation in the acylation pattern and the number of phosphate groups [36] [39] [43] . The core and O-specific regions of LPS appear to have only minor role in recognition by host immune receptors, as the chemical removal of carbohydrate chains has only limited effect on the inflammatory

Figure 1. Structural elements of TLR4 signaling pathway activated by LPS binding. The LPS-induced dimerization of the TLR4/MD2 complex elicits phosphorylation of the intracellular TIR domain at Tyr, followed by its dimerization and the recruitment of the adaptor molecules myD88, Mal, and TAK1. This leads to the activation of IκB-kinase complex (IKK) and MAP kinases, and resulting in the induction and translocation to the nucleus of NF-κB and AP-1 transcription factors involved in the activation of proinflammatory genes. pY, phosphotyrosine.
potential of LPS [38] [39] [44] . It should be noted, however, that due to its amphipathic character, the extent of inflammatory response to LPS is controlled by the accessory protein molecules, LPS-binding protein (LBP), CD14, and myeloid differentiation-2 (MD-2). The LBP and CD14 molecules, apparently, determine the level of the enhancement in detection of LPS by TLR4: with LBP binding to LPS aggregates and delivering them to CD14, and the CD14 transferring the bound LPS to the TLR4/MD-2 aggregates on the plasma membrane [39] [41] .
Indeed, studies show that LBP binds tenaciously to the LPS oligomers release from bacterial membrane and transfers them to the binding factor, CD14, existing either as membrane-anchored through glycosylphosphatidylinositol tail or as soluble glycoprotein in the serum. The LPS aggregates are sorted on CD14 into monomeric forms and as such presented to the TLR4/MD2 complex [33] [41] . While MD2 has neither transmembrane or signaling domain, its hydrophobic cluster offers a seamless interface for binding with the hydrophilic lipid A region of the LPS, whereas the exposed oligosaccharide portion of LPS presents an ideal perimeter for the interaction with the second TLR4/MD2 complex [32] [39] . The LPS-induced and symmetrically arranged TLR4/MD2 dimerization on the cell surface is at the core of TLR4 activation that is then followed by TIR domain dimerization and induction of binding of the adaptor molecules. Indeed, after binding LPS, the TIR domain of membrane-located TLR4 interacts with the TIR domain-containing adaptor protein Mal (MyD88 adaptor-like), followed by the interaction with the TIR region of myeloid differentiation factor 88 (MyD88). Apparently, in this process, dimerization of the extracellular domains leads to juxtaposition of the intracellular domains and their dimerization, followed by the recruitment of signaling adaptors [39] . The My88 aggregation signal is then transmitted to IRAK kinases, ultimately leading to activation of signaling kinases involved in the activation through phosphorylation of two sets of kinases, IKK and MAPKs (Figure 1). Activation of IKK result in translocation of NF-κB transcription factors to the nucleus and induction of the expression of proinflammatory genes, while MAPKs are involved in the activation of AP-1 transcription complex-mediated signaling cascades [36] [39] [41] .
3. Endogenous Modulators of Oral Mucosal Inflammatory Responses to P. gingivalis
3.1. Leptin
Advances in identification of salivary constituents of significance to the maintenance of oral mucosal integrity have brought to focus the importance of salivary bioactive peptides in the protection of oral cavity against the hordes of invading bacteria. Historically, the classical example of biologically active peptide of salivary origin, identified nearly 70 years ago and recognized for its role in cellular proliferation, mucosal protection and wound healing, is epidermal growth factor [45] [46] . Equally important, although more recent development, was the identification in saliva and oral mucosal tissue of two biologically active peptides of a broad physiological relevance, leptin and ghrelin [6] [7] .
Leptin, a multifunctional16 kDa peptide hormone encoded by the obese (ob) gene and initially acknowledged for its role in the maintenance of body energy stores and weight homeostasis, has emerged as an important regulator of mucosal inflammatory responses to bacterial infection [28] [47] [48] . The biological activities of leptin are mediated through the interaction with the specific membrane receptor, OB-R, a member of the class I cytokine receptor superfamily, which exists in several variant forms sharing the same extracellular domain but differing in the length of transmembrane coding regions [48] . Aside of its hypothalamic site of action, leptin and leptin receptors have been identified along the alimentary tract, including oral mucosa and the acinar cells of salivary gland [6] [28] [47] . Indeed, leptin was found to interfere with the detrimental effects of P. gingivalis LPS on the synthesis of salivary mucins, increase in the level of leptin characterizes gastric mucosal inflammatory responses to H. pylori infection, and the exogenous leptin has been demonstrated to affect the extend of inflammatory responses to E. coli LPS [28] [49] [50] . Moreover, leptin released locally within mucosal tissue has been implicated in the interaction with proinflammatory cytokines to control local inflammations, and the regulation of proinflammatory mediators, NO and PGE2 production [51] [52] .
3.2. Ghrelin
Another peptide hormone, identified more recently as an important modulator of oral mucosal inflammatory responses to P. gingivalis is ghrelin [7] [29] [53] . This 28-amino acid peptide, found in saliva and oral mucosal tissue has gained recognition as principal modulator of the local inflammatory responses by affecting the cross-talk between nitric oxide synthase isozyme system (NOS), and the cyclooxygenase synthase (COX) enzymes. The signaling mechanism that underlies this regulatory of mode of ghrelin action involves the stimulation of growth-hormone secretagogue receptor type 1a (GHS-R1a), a seven-transmembrane G protein-coupled receptor (GPCR), that leads to activation of heterotrimeric G-protein-dependent network of protein kinases, including phospholipase C (PLC)/protein kinase C (PKC) implicated in phosphatidylinositol 3-kinase (PI3K)/Src and Akt signaling, as well as those involved in the regulation of NO and PGE2 production [54] [55] [56] [57] .
4. LPS and Proinflammatory Mediators, NO and PGE2, Production
4.1. Porphyromonas Gingivalis LPS-Induced NOS/COX Cross-Talk
The oral mucosal responses to P. gingivalis and its key virulence factor, LPS are characterized by a massive rise in epithelial cell apoptosis and proinflammatory cytokine expression, excessive NO generation, and a marked increase in PGE2 production [22] [28] [58] . A growing volume of literature, moreover, points towards the existence of functional and signaling relationship between NO, generated by the action of NOS, and the formation of PGE2 synthesized from arachidonic acid (AA) by the action of COX systems [30] [59] [60] . While NO and PGE2 generated by the constitutive nitric oxide synthase (cNOS) and cyclooxygenase-1 (COX-1) enzyme systems are deemed essential to maintaining normal housekeeping functions, the induction of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) expression in response to inflammatory stimulus, including that of P. gingivalis LPS, have been demonstrated to promote the proinflammatory events propagation [37] [59] [61] .
Indeed, the stimulation of NO production through iNOS induction has been shown to lead to the up-regulation in COX-2 activation, whereas the inhibition in iNOS activation results in a decrease in PGE2 formation [60] [61] [62] . Moreover, we have demonstrated that COX-2 activation and the ensuing increase in PGE2 generation is the result of iNOS-derived and NO-induced COX-2 enzyme protein S-nitrosylation [30] [60] . Hence, the LPS elicited induction in iNOS expression and the resulting COX-2 activation through S-nitrosylation leads the excessive PGE2 generation (Figure 2). PGE2 then acts

Figure 2. Signaling pathways and transcription factors involved in oral mucosal inflammatory responses to P. gingivalis LPS. Ligation of TLR4 by the LPS results in the activation of IKK complex and MAP kinases, ERK, p38 and JNK, and triggers the nuclear translocation of transcription factors involved in the induction of COX-2 (AP-1), CREB and C/EBP) and iNOS (NF-κB). This leads to iNOS-induced up-regulation in NO generation and COX-2 activation through S-nitrosylation that results in the excessive PGE2 production. pY, phosphotyrosine; P, phosphate.
locally through binding of one or more of its four eicosanoid receptors (ER) and triggers the persistence of inflammatory microenvironment by promoting local vasodilation, attraction and activation of neutrophils, macrophages, natural killer cells, and dendritic cells [63] [64] .
Furthermore, P. gingivalis-induced enhancement in oral mucosal iNOS activation and the excessive NO generation results in cNOS S-nitrosylation that interferes with its activation through Akt-mediated phosphorylation on Ser1179 [29] . The induction in iNOS expression by P. gingivalis LPS also leads to the increase in salivary gland acinar cell apoptosis as well as Akt kinase inactivation through S-nitrosylation, that is manifested by a decrease in the kinase phosphorylation on Ser473 [29] [56] [65] [66] . There are also data suggesting that P. gingivalis LPS-induced up-regulation in the acinar cell iNOS leads to disturbances in cNOS phosphorylation that exerts the detrimental effect on the processes of cSrc kinase activation through cNOS-mediated S-nitrosylation (57). S-nitrosylation has been also convincingly linked to the regulation of IκB-kinase complex (IKK) activity responsible for NF-κB nuclear translocation, and the activation processes of cytosolic phospholipase A2 (cPLA2) involved in AA release [37] [67] [68] . Remarkably, peptide hormone, ghrelin, exerts the modulatory impact on all of these proinflammatory effects of P. gingivalis LPS by influencing the extent of Src/Akt-dependent up-regulation in cNOS activation [29] [30] [56] [57] [65] [66] [68] .
4.2. Transcriptional Factors in LPS-Induced iNOS and Cox-2 Activation
The events underlying the proinflammatory signal regulation in oral mucosa in response to P. gingivalis challenge relay heavily on the bacterial LPS engagement of TLR4, the ligation of which results in the receptor dimerization followed phosphorylation of its intracellular domain at the several critical Tyr residues [43] . This leads to recruitment to the cytoplasmic domain of the TLR4 of several different adaptor molecules involved in the propagation of signaling events, including Mal and MY88, and triggers the activation of downstream signaling cascades converging on two sets of kinases, IKK and MAPK [32] [36] [39] . Activation of MAPK cascade, including ERK, JNK, and p38, results in the phosphorylation and activation of transcriptional factors implicated in the induction of the expression of COX-2, while the activation of IKK converges on NF-κB pathway involved in the induction of iNOS gene [68] [69] [70] [71] . It is pertinent to note, however, that ERK signals also converge on IKK pathway activation through phosphorylation of its IKK-β catalytic subunit and hence are associated with the enhancement in NF-κB nuclear translocation and the induction in iNOS expression [37] [72] [73] .
In concordance with the current literature data, NF-κB is ubiquitously present in all types of mammalian cells, where it controls a variety of genes involved in immune, inflammatory, and proinflammatory responses, including those of TNF-α, IL-6, and NOS isozymes responsible for NO production [26] [56] [65] . Although NF-κB is known to consist of five homo- and heterodimers of Rel protein family, the most characterized complex is a p50/p65 heterodimer, which is sequestered in the cytoplasm of resting cells bound to a family of inhibitory IκB proteins [68] [69] . Activation of NF-κB via LPS elicited TLR4 ligation involves a rapid phosphorylation of the inhibitory IκB-α at two critical serine residues by IKK complex. This phosphorylation event signals IκB-α for degradation by ubiquitin-proteasomal pathway, and leads to nuclear translocation of the released NF-κB, its binding to promoter response elements, and activation of the transcription of genes implicated in immunoregulation and inflammation, including that of iNOS [30] [68] . Moreover, we have observed that ERK activation by P. gingivalis LPS, like the LPS of other bacteria (Figure 2), elicits an enhancement in IKK-β phosphorylation as well as the upsurge in NF-κB nuclear translocation that is accompanied by the induction in iNOS expression [42] [69] [73] . In addition, the LPS-induced upregulation in IKK-β activity displays susceptibility to suppression by the inhibitors of ERK phosphorylation, thus attesting further to the role of ERK in the processes of NF-κB-dependent induction of iNOS expression for the increase in NO generation [37] [42] . Interestingly, the effect of hormone ghrelin on the LPS-induced up-regulation in iNOS expression is manifested by the inhibition in IκB-α degradation and a decrease in NF-κB nuclear translocation [71] .
In contrast to the signaling events involved in the control of the mucosal expression of iNOS, the nature of factors implicated in transcriptional regulation and the induction of COX-2 expression by LPS is considerably more complex. Indeed, the current consensus is that the process may be maintained in the synchrony through several redundant systems involving at least four central response elements located on the COX-2 gene promoter. These include transcriptional factor, NF-κB, activator protein-1 (AP-1), cAMP response element-binding protein (CREB), and CCAAT/enhancer-binding protein (C/EBP) which plays an important role in COX-2 promoter induction mainly through interaction with its two-family members, C/EBPβ and C/EBPδ [37] [74] [75] [76] . Certainly, the available data clearly suggest that LPS-induced JNK, p38 and ERK activation through phosphorylation leads to the transcription factors, AP-1, CEB, and C/EBP activation, that results in the induction of COX-2 expression (Figure 2).
MAPK ERK, moreover, in addition to its well-defined involvement in the activation of IKK-β associated with the up-regulation in NF-kB nuclear translocation, also exerts its effect on the activation of transcription factor, cFos, involved in the assembly of AP-1complex associated with the induction in COX-2 gene expression [37] [42] [70] [75] [77] . There are furthermore, preponderant data demonstrating that LPS-induced ERK activation plays a principal role in the activation of cytosolic phospholipase A2 (cPLA2), an enzyme implicated in the AA release for PGE2 synthesis [58] [60] [73] [78] . Indeed, the activity pf cPLA2 is tightly regulated by posttranslational mechanism involving ERK-dependent enzyme phosphorylation that facilitates its translocation from cytosol to membrane to gain access to phospholipid substrates for the increase in AA generation [73] [78] . Interestingly, the literature data indicate that the gene locus of cPLA2 is located on chromosome I in the proximity of COX-2 gene, and the increased level of AA is known to affect the COX-2 expression [79] .
The systems implicated in transcription factors activation, furthermore, remain under additional regulatory control through S-nitrosylation (Figure 3). Indeed, S-nitrosylation has been implicated in the processes COX-2 and cPLA2 activation, as well as the regulation of IKK-β activity and the level of NF-kB nuclear translocation [29] [30] [67] [68] [69] . Moreover, we have demonstrated that P. gingivalis LPS-elicited induction in iNOS leads to up-regulation in COX-2

Figure 3. Schematic representation of the mechanism involved in the excessive oral mucosal NO and PGE2 generation in response to P. gingivalis LPS. Binding of the LPS to TLR4 triggers the activation of JNK, p38 and ERK1/2 MAPKs, and nuclear translocation of transcription factors involved in the induction of COX-2 (AP-1, CREB and C/EBP), and iNOS (NF-kB) genes transcription. Moreover, the activation of ERK by the LPS leads to phosphorylation and activation of IKK-β and cPLA2, which trigger up-regulation in arachidonic acid (AA) release and the induction in iNOS. The rise in iNOS-elicited NO production leads to COX-2 activation through S-nitrosylation that results in the excessive PGE2 generation. Engagement of the growth hormone secretagogue receptor (GHSR) by ghrelin leads to the inhibition of C/EBP and p38/JNK-mediated AP-1 activation, and hence results in the reduced COX-2 expression. The effect of ghrelin, moreover, is reflected in further enhancement in the LPS-induced ERK activation, and up-regulation in Src/Akt-dependent cNOS phosphorylation that leads to the inhibition of IKK-β and cPLA2 activation by cNOS-mediated S-nitrosylation. This results in the repression of iNOS gene induction and the inhibition of COX-2 activation through iNOS-dependent S-nitrosylation, as well as the suppression of AA release.
activation through S-nitrosylation that results in an excessive PGE2 generation [30] .
5. Factors Affecting Amplification in Proinflammatory Signal Propagation
5.1. P. gingivalis LPS Amplification in PLC Activation
One of the key elements of LPS-induced activation of TLR4 is the receptor-mediated recruitment of phosphoinositide-specific phospholipase C (PLC) [80] [81] . The mammalian phosphoinositide-specific PLC is a family of 13 isozymes divided into six subfamilies on the basis of their size, amino acid sequences, domain structure, and activation mechanisms [80] . The most ubiquitously expressed are the two isoforms of the PLCγ subfamily, PLCγ1 and PLCγ2, both activated following TLR4 ligation by LPS and resulting in generation of the second messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), from membrane phosphatidylinositol 4,5-bisphosphate (PIP2) [80] [81] [82] [83] . The generated second messengers have far-reaching regulatory and metabolic roles: DAG is known to stimulate the activity of a variety of enzymes, including PKC, while IP3 is recognized for its role in the regulation of the cytoplasmic calcium concentration [80] . Other data point to the existence of cross talk between PLC and PKC, and indicate that while DAG causes stimulation of PKC activation, the PKC in turn may be involved in the reciprocal modulation of PLC as well as PI3K activation [81] [83] .
Moreover, several PLC isozymes, including mucosal tissue PLCγ2, appear to be directly activated by Ras superfamily of small guanosine triphosphatases (GTPases) [84] [85] [86] . The GTPases specifically implicated in the regulation of PLCγ2 activation, Rac1 and Rac2, are members of Rho family and their activation status is controlled through the exchange of GDP for GTP, catalyzed by the guanine nucleotide exchange factors, referred to as Dock (dedicator of cytokinesis) 180-related family of GEFs [87] . Interestingly, the Dock180 facilitating GDP/GTP exchange in Rac1 responds to stimuli activating tyrosine kinase receptors and up-regulation in Rac1 activation has been observed in association with LPS-induced gastric mucosal and pulmonary inflammation [88] [89] .
Hence, considering the pivotal role of PLCγ2 in proinflammatory signal propagation in response to bacterial endotoxins, we explored the mechanism involved in P. gingivalis LPS-induced amplification in PLCγ2 activation [90] . Our findings revealed that stimulation of salivary gland acinar cells with P. gingivalis LPS leads to up-regulation in Dock180 and PLCγ2 activation and is associated with membrane translocation of Rac1 and PLCγ2. Apparently, in this process, the LPS-induced TLR4 activation triggers Src-dependent Dock180 phosphorylation on Tyr and the PLCγ2-mediated activation of PKCδ. This leads to the PKCδ-induced up-regulation in Dock180 and PLCγ2 activation through phosphorylation on Ser. The up-regulation in Dock180, in turn, stimulates the formation of Rac1-GTP and promotes its association with PLCγ2, thus resulting in the amplification in PLCγ2 activation. The modulatory effect of hormone, ghrelin, on the other hand, is manifested by the suppression in Rac1 membrane translocation as well as a distinct drop in Dock180 phosphorylation on Ser.
Taken together, the findings underscore the role of Src/PKCδ-mediated GEF Dock180 phosphorylation on Tyr/Ser in the amplification of salivary gland acinar cell PLCγ2 activation in response to P. gingivalis as well as in the modulation of PLCγ2 activity by ghrelin. The schematic representation of the signaling pathways involved in P. gingivalis LPS-elicited amplification in oral mucosal PLCγ2 activation and the modulatory influence of ghrelin is depicted in Figure 4.
5.2. P. gingivalis LPS and EGFR Transactivation
Investigations into the signaling cascades triggered byTLR4 activation indicate that proinflammatory signal propagation induced by LPS is also associated with the activation of EGFR [91] [92] [93] . Indeed, recent data show that LPS-induced EGFR transactivation is an essential part of the inflammatory and repair process, and involves TACE-dependent release of inflammatory regulators, including EGFR ligand, TGF-α [93] [94] .
The EGFR is a transmembrane glycosylated protein consisting of an extracellular ligand binding domain, a single membrane-spanning region, and a cytoplasmic tyrosine kinase domain, that has been implicated in growth and repair processes of a variety of epithelial cells, including those of oral mucosa and salivary glands [46] [95] [96] . Upon ligand binding to the extracellular domain, EGFR undergoes dimerization, and the intrinsic tyrosine kinase activation through phosphorylation of key tyrosine residues within the cytoplasmic region of the receptor [95] [97] . Subsequently, these phosphorylated tyrosine residues serve as docking sites for a variety of the recruited signal transducer molecules

Figure 4. Diagram of the regulatory role of GEF Dock180 phosphorylation on Tyr/Ser in modulation of Rac-1 activation in response to P. gingivalis LPS and ghrelin. Ligation by ghrelin of salivary gland acinar cell GHS-R1a activates several G protein-dependent signal transduction pathways, including that of Src kinase-dependent Dock180 phosphorylation on Tyr that maintains the regulatory level of Rac1-GTP formation. Binding of the LPS to TLR4 triggers Src kinase-dependent Dock180 phosphorylation on Tyr and the PLCγ2-mediated PKCδ activation that leads to the PKCδ-induced up-regulation in Dock180 and PLCγ2 activation through phosphorylation on Ser. The up-regulation in Dock180, in turn, stimulates the formation of Rac1-GTP and promotes its association with PLCg2, thus resulting in the amplification in PLCγ2 activation. G, heterotrimeric G-protein; pS, phosphoserine; pY, phosphotyrosine.
[95] . Furthermore, in addition to its cognate epidermal growth factor (EGF) ligand, the EGFR activation occurs in response to transforming growth factor-α (TGF-α), heparin binding EGF-like growth factor (HB-EGF), epiregulin, amphiregulin, and epigen [95] [97] [98] . All these ligands are expressed as inactive membrane-anchored proteins that in response to a specific cellular stimulus undergo proteolytic cleavage, termed ectodomain shedding, to release the particular mature growth factor [95] [99] .
The cleavage of EGFR ligands occurs with the involvement of membrane-anchored family of disintegrin-metalloproteases (ADAMs), which show preference with respect to stimulus response and exhibit various degree of substrate specificity [95] . Among the metalloproteinases that respond to LPS stimulation, the primary role is assigned to TNF-α converting enzyme (TACE), also known as ADAM17 [93] [94] [98] [99] . Indeed, studies show that LPS-induced TACE activation leads to the enhancement in TGF-α shedding and EGFR transactivation [93] [94] . Furthermore, EGFR transactivation by TACE-dependent TGF-α shedding has been linked to LPS-induced TLR4 signaling through p38 MAPK [94] [98] , and there are strong indications for the involvement of Rac1 GTPase in the activation process [89] [98] [100] .
As oral mucosal inflammatory responses to P. gingivalis LPS are characterized by up-regulation in MAPK and Rac1 activation, and the role of Rac1 has been suggested in p38 and ADAM17 activation in association with pulmonary, oral, and gastric mucosal inflammatory responses to LPS [70] [88] [89] [90] , we explored the nature of factors associated with the induction in salivary gland acinar cell EGFR transactivation by P. gingivalis LPS. Our findings revealed that stimulation of the acinar cells with the LPS leads to p38 MAPK-dependent increase in TGF-α ligand shedding and EGFR transactivation [31] . We further demonstrated that the LPS-induced TGF-α ectodomain shedding and EGFR transactivation involves the activation of membrane-anchored TACE through phosphorylation by p38 that requires Dock 180 mediated up-regulation in Rac1-GTP formation (Figure 5). Moreover, we established that the LPS-elicited Rac1 membrane translocation serves as an essential platform for the localization of p38 with the TACE, its activation through phosphorylation on Thr735, and subsequent increase in TGF-α ectodomain shedding for EGFR transactivation [31] .
Although the detailed functional paradigm of the role of Rac1 in p38 membrane recruitment and TACE activation requires further defining, it is becoming increasingly apparent that Rac GTPases operate as sophisticated modulators of a remarkably complex and diverse range of cellular processes, including regulation of MAPK expression and PLCγ2 membrane localization, and metalloproteinase activation [22] [70] [80] [85] . Hence, therapeutic targeting Rac GTPases may prove to be useful approach for developing more effective treatments of periodontal disease. A noteworthy step in this direction is the recent use of Rac inhibitor, NSC23766, to alleviate LPS-induced acute pulmonary inflammation [89] .
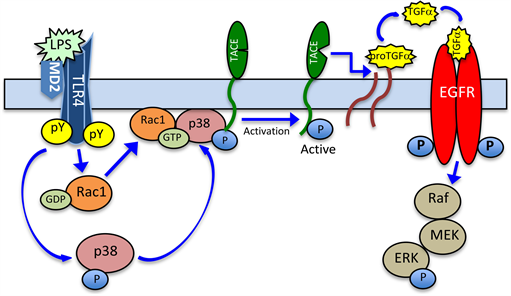
Figure 5. Schematic representation of the signaling mechanism leading to P. gingivalis LPS-induced EGFR transactivation by TGF-α. Ligation by the LPS of salivary gland acinar cell TLR4 triggers up-regulation in p38 MAPK phosphorylation, as well as prompts the activation and translocation of Rac1-GTP to the vicinity of membrane-anchored latent TACE. This, in turn, leads to the recruitment and localization of p38 to the complex, and TACE activation through phosphorylation. The subsequent cleavage by the TACE of pro-TGF-α results in a soluble TGF-α ligand that binds to and activates EGFR, thus causing the increased signaling to ERK/MAPK.
5.3. Amplification of P. gingivalis LPS Inflammatory Signals by Syk
Careful dissection of the events following TLR4 ligation by LPS indicates that TLR4 activation and the ensuing phosphorylation of its intracellular tyrosine domain by Src-family kinases not only leads to recruitment to the cytoplasmic domain of the receptor of several different adaptor molecules involved in the propagation of signaling cascades converging on MAPK and IKK [32] [36] [43] , but also provides a convenient docking site for spleen tyrosine kinase (Syk), a non-receptor tyrosine kinase the activation of which is known to increase the expression of proinflammatory genes [101] [102] [103] .
Indeed, Syk initially found in hematopoietic cells and recognized for its role in adaptive immune responses, has emerged recently as a major effector in TLR4-mediated inflammatory reaction to LPS [102] . This 72 kDa non-receptor tyrosine kinase comprises of two tandem N-terminal Src homology 2 (SH2) domains, a linker region, and a C-terminal kinase domain [101] [102] . The first step in Syk activation is its binding through SH2 domains to the intracellular Toll-IL-1 receptor (TIR) domain of TLR4 or signaling proteins containing phosphorylated immunoreceptor tyrosine-based activation motifs (ITAMs) in their cytoplasmic regions [103] . This results in conformational changes in Syk and its activation through phosphorylation on several tyrosine residues, which leads to the activation of the PLC, PI3K, MAPK and ERK signaling cascades, and amplification in the induction of inflammatory response [103] [104] .
Therefore, to gain further leads into the pathways utilized by P. gingivalis endotoxin, LPS, in triggering up-regulation in oral mucosal inflammatory responses, we investigated the nature of factors involved in the recruitment and interaction of Syk with TLR4 in salivary gland acinar cells in response to stimulation by P. gingivalis LPS [42] . By following the acinar cell TLR4 activation and its interaction with Syk, we found that stimulation with the LPS led to a rapid, time-dependent induction in the phosphorylation of TLR4 and Syk on Tyr, and that the association between Syk and TLR4 induced by the LPS required phosphorylation of both proteins on Tyr. These findings thus support the results obtained with neutrophils and macrophages demonstrating the increase in phosphotryrosine-dependent binding of Syk to the cytoplasmic region of TLR4 in response to stimulation by LPS [102] [105] . Furthermore, we revealed that stimulation of the acinar cells with the LPS leads to PKCδ-mediated Syk phosphorylation on Ser which is essential for its localization with the membrane associated TLR4 complex and the activation through phosphorylation on Tyr. Thus, PKCδ appears to be a primary linchpin affecting Syk recruitment to TLR4, as well as influencing the efficiency of its activation and the magnitude of inflammatory responses. This assertion is certainly in line with the literature reports as to the involvement of PKCδ in Syk activation associated with Detectin-1 signaling and thrombin-induced NF-κB activity involved in ICAM-1 expression [106] [107] [108] .
In further efforts to reveal the role of Syk in the amplification of inflammatory responses of the acinar cells to P. gingivalis LPS, we have turned our attention to the involvement of Syk in regulation of the expression of transcription factors affected byTLR4-mediated signaling cascades converging on IKK and MAPK kinase systems. Our results revealed that the LPS-induced ERK activation, in addition to its well-defined involvement in the activation of IKK associated with up-regulation in the expression of iNOS gene [37] [69] [73] , also exerts its effect on the activation through phosphorylation on transcription factor, c-Fos, involved in the assembly of AP1 transcription complex associated with the induction in COX-2 gene expression. Furthermore, in agreement with the reported data [71] , we have found that MAPK/p38 is involved in phosphorylation of ATF2, while Syk-mediated activation of JNK along with p38 are involved in the phosphorylation of transcriptional factor, c-Jun.
As phosphorylation of transcription factors alters their stability and the extent of dimerization with other members of AP1 complex, giving rise to complexes with different transcriptional potential [77] , it is apparent that the Syk-induced changes in transcription factors activation plays a major role in the transcriptional outcome of proinflammatory genes expression (Figure 6). Therefore, pharmacological agents targeting Syk activation offer tempting alternative for the therapeutic intervention in the treatment of chronic periodontitis. It should be noted, however, that most of the current Syk inhibitors suffer from low therapeutic efficacies, cytotoxicity, and multiple kinase targeting [41] [103] .
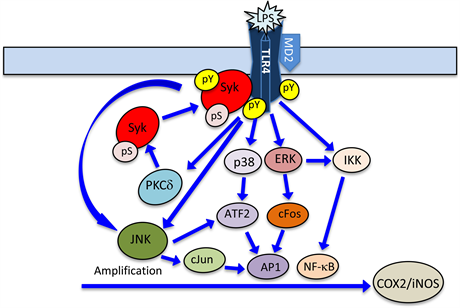
Figure 6. Illustration of the signaling mechanism involved Syk-induced amplification of salivary gland acinar cell inflammatory responses to P. gingivalis LPS. Engagement of TLR4 by the LPS initiates the signal transduction cascades converging on PLC-dependent pathway of PKCδ activation, MAPKs, and IKK. The induction in PKCδ activation leads to Syk phosphorylation on Ser that results in up-regulation of its membrane colocalization with TLR4 and the activation through phosphorylation on Tyr. The activated Syk, in turn, amplifies the JNK, p38 and ERK cascades that lead to up-regulation in cJun, cFos and ATF2 transcription factors involved in the regulation of AP-1 complex assembly and the induction of COX-2 gene, while the IKK signals converge on NFκB pathway involved in the induction of iNOS gene. pS, phosphoserine; pY, phosphotyrosine.
6. Conclusions
P. gingivalis is the most prominent member of the bacterial flora associated with pathogenesis of periodontitis and its role as primary etiological agent of periodontal diseases has been firmly entrenched for over 50 years. However, the role of the bacterium LPS as the principal virulence factor implicated in triggering oral mucosal inflammatory responses leading to the development of periodontitis has gained the acceptance more recently. Indeed, the engagement by the LPS of oral mucosal TLR4 has been convincingly linked to signal transduction events characterized by the activation of MAPK and IKK cascades, induction of PLC/PKC/PI3K pathway, up-regulation in TGF-α ectodomain shedding and EGFR transactivation, and the amplification in proinflammatory signal propagation by Syk. Moreover, considerable progress has been made in the identification and elucidation of the function role of endogenous peptide hormones, leptin and ghrelin, in the modulation of oral mucosal inflammatory responses to P. gingivalis.
The rise and the excessive oral mucosal generation of NO and PGE2 in response to P. gingivalis challenge has been demonstrated to relay on the LPS-induced MAPK activation and the nuclear translocation of transcription factors involved in the induction of COX-2 gene, while the LPS-elicited activation of IKK converges on NF-κB pathway implicated in the induction of iNOS gene. The ERK activation signals, moreover, induce further activation of IKK-β and cPLA2, and hence are associated with up-regulation in AA release for PGE2 synthesis and the upsurge in NF-kB nuclear translocation and further induction in iNOS expression. The systems implicated in transcription factors activation, furthermore, remain under additional regulatory control through S-nitrosylation. Indeed, S-nitrosylation has been implicated in P. gingivalis LPS-induced processes of COX-2 and cPLA2 activation, and the regulation of IKK-β activity. Moreover, the induction in iNOS leads to up-regulation in COX-2 activation through S-nitrosylation that results in an excessive PGE2 generation.
Another aspect of P. gingivalis elicited proinflammatory signal propagation is the role of TLR4 activation in the recruitment of phosphoinositide-specific PLCγ2 which catalyzes formation of the second messengers, IP3 and DAG. These messengers in association with two members of Rho family of small GTPases, Rac1 and Rac2, facilitate the cross-talk between PLC/PKC and PI3K and, hence, affect the level of the proinflammatory signal induction by the LPS. Indeed, P. gingivalis LPS-elicited TLR4 activation triggers the PLCγ2-mediated activation of PKCδ that leads to the PKCδ-induced up-regulation in Rac1 membrane translocation and the amplification in PLCγ2 activation. Interestingly, the modulatory influence of peptide hormone, ghrelin on the excessive LPS-induced PLCγ2 activation is manifested in the suppression in Rac1 membrane translocation.
Remarkably, up-regulation in oral mucosal Rac1 membrane translocation, induced by P. gingivalis LPS, appears to play a major role in recruitment of the activated p38 MAPK to the cytosolic aspect of the membrane for TACE activation and the subsequent increase in TGF-α ectodomain shedding and the EGFR transactivation. Thus, LPS-induced proinflammatory signal propagation through MAPK cascade clearly affects the inflammatory and repair processes associated with EGFR activation. Hence, Rac GTPases, although referred to as “small GTPases”, appear to function as “full-size” and dominant modulators of a complex and diverse range of cellular events, including regulation of MAPK expression, TACE activation and TGF-α shedding, and the amplification in PLCγ2 activation.
Equally interesting are the recent developments as to the role of non-receptor tyrosine kinase, Syk in the amplification of oral mucosal inflammatory responses to P. gingivalis LPS. The findings indicate that the LPS-elicited phosphorylation of the intracellular tyrosine domain of TLR4 not only leads directly to propagation of signals converging on MAPK and IKK, but also provides a convenient docking site for Syk. The ensuing Syk activation through phosphorylation on tyrosine results in the kinase binding to a number of downstream signaling effectors, and amplifies the inflammatory signal propagation by affecting transcription factors activation and their assembly to transcriptional complexes involved in proinflammatory genes expression. Hence, the development of new pharmacological agents selectively targeting Syk activation could be of therapeutic value in the treatment of chronic periodontitis.
Cite this paper
Slomiany, B.L. and Slomiany, A. (2018) Proinflammatory Signaling Cascades of Periodontopathic Oral Pathogen Porphyromonas gingivalis. Journal of Biosciences and Medicines, 6, 63-88. https://doi.org/10.4236/jbm.2018.65009
References
- 1. Colombo, A.P., Boches, S.K. and Cotton, S.L. (2009) Comparisons of Subgingival Microbial Profiles of Refractory Periodontitis, Severe Periodontitis, and Periodontal Health Using the Human Oral Microbe Identification Microarray. Journal of Periodontology, 80, 1421-1432. https://doi.org/10.1902/jop.2009.090185
- 2. How, K.Y., Song, K.P. and Chan, K.G. (2016) Porphyromonas gingivalis: An Overview of Periodontopathic Pathogen below the Gum Line. Frontiers of Microbiology, 7, 53. https://doi.org/10.3389/fmicb.2016.00053
- 3. Eloe-Fadrosh, E.A. and Rosko, D.A. (2013) The Human Microbiome: From Symbiosis to Pathogenesis. Annual Review of Medicine, 64, 145-163. https://doi.org/10.1146/annurev-med-010312-133513
- 4. Slomiany, B.L., Murty, V.L.N. and Slomiany, A. (1993) Structural Features of Carbohydrate Chains in Human Salivary Mucins. International Journal of Biochemistry, 25, 259-265. https://doi.org/10.1016/0020-711X(93)90015-7
- 5. Slomiany, B.L., Murty, V.L.N., Piotrowski, J. and Slomiany, A. (1996) Salivary Mucins in Oral Mucosal Defense. General Pharmacology: The Vascular System, 27, 761-771. https://doi.org/10.1016/0306-3623(95)02050-0
- 6. Groschl, M., Rauch, M., Wagner, R., et al. (2001) Identification of Leptin in Human Saliva. Journal of Clinical Endocrinology and Metabolism, 86, 5234-5239.https://doi.org/10.1210/jcem.86.11.7998
- 7. Groschl, M., Topf, H.G., Bohlender, J., et al. (2005) Identification of Ghrelin in Human Saliva: Production by the Salivary Glands and Potential Role in Proliferation of Oral Keratinocytes. Clinical Chemistry, 51, 997-1006.https://doi.org/10.1373/clinchem.2004.040667
- 8. Fabian, T.K., Hermann, P., Beck, A., Fejerdy, P. and Fabian, G. (2012) Salivary Defense Proteins: Their Network and Role in Innate and Acquired Oral Immunity. International Journal of Molecular Sciences, 13, 4295-4320.https://doi.org/10.3390/ijms13044295
- 9. Dorn, B.R., Burks, J.N., Seifert, K.N. and Progulske-Fox, A. (2000) Invasion of Endothelial and Epithelial Cells by Strains of Porphyromonas gingivalis. FEMS Microbiology Letters, 187, 139-144. https://doi.org/10.1111/j.1574-6968.2000.tb09150.x
- 10. Wiebe, C.B. and Putnins, E.E. (2000) The Periodontal Disease Classification System of the American Academy of Periodontology—An Update. Journal of Canadian Dental Association, 66, 594-597.
- 11. Bodet, C., Chandad, E. and Grenier, D. (2007) Pathogenic Potential of Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia, the Red Bacterial Complex Associated with Periodontitis. Pathologie Biologie, 55, 154-162.https://doi.org/10.1016/j.patbio.2006.07.045
- 12. Mysak, J., Pdzimek, S., Sommerova, P., Lyuya-Mi, Y., Bartova, J., Janatova, T., Prochazkova, J. and Duskova, J. (2014) Porphyromonas gingivalis: Major Periodontopathic Pathogen Overview. Journal of Immunology Research, 2014, Article ID: 476068. https://doi.org/10.1155/2014/476068
- 13. Slot, J. (1976) The Predominant Cultivable Organism in Juvenile Periodontitis. Scandinavian Journal of Dental Research, 84, 1-10.https://doi.org/10.1111/j.1600-0722.1976.tb00454.x
- 14. Saxen, L. (1980) Juvenile Periodontitis. Journal of Periodontology, 7, 1-19.https://doi.org/10.1111/j.1600-051X.1980.tb01944.x
- 15. Datta, H.K., Ng, W.F., Walker, J.A., Tuck, S.P. and Varanasi, S.S. (2008) The Cell Biology of Bone Metabolism. Journal of Clinical Pathology, 61, 577-587.https://doi.org/10.1136/jcp.2007.048868
- 16. Bostanci, N. and Belibasakis, G.N. (2012) Porphyromonas gingivalis: An Invasive and Evasive Opportunistic Oral Pathogen. FEMS Microbiology Letters, 333, 1-9. https://doi.org/10.1111/j.1574-6968.2012.02579.x
- 17. Amano, A. (2007) Disruption of Epithelial Barrier and Impairment of Cellular Function by Porphyromonas gingivalis. Frontiers in Bioscience, 12, 3965-3974. https://doi.org/10.2741/2363
- 18. Nagano, K., Hasegawa, Y., Yoshida, T. and Yoshimura, F. (2015) A Major Fimbrilin Variant of Mfa1 Fimbriae in Porphyromonas gingivalis. Journal of Dental Research, 94, 1143-1148. https://doi.org/10.1177/0022034515588275
- 19. Kristoffersen, A.K., Solli, S.J., Nguyen, T.D. and Ensersen, M. (2015) Association of the rgpB Gigipain Genotype to the Major Fimbriae (fimA) Genetype in Clinical Isolates of the Periodontal Pathogen Porphyromonas gingivalis. Journal of Oral Microbiology, 7, Article 29124. https://doi.org/10.3402/jom.v7.29124
- 20. Salminen, A., Gursoy, U.K., Paju, S., Hyvarinen, K., Mantyla, P., Buhlin, K., Kononen, E., Nieminen, M.S., Sorsa, T., Sinisalo, J. and Pussinen, P.J. (2014) Salivary Biomarkers of Bacterial Burden, Inflammatory Response, and Tissue Destruction in Periodontitis. Journal of Clinical Periodontology, 41, 442-450.https://doi.org/10.1111/jcpe.12234
- 21. DeLeon-Pannell, K.Y., de Castro Bras, L.E. and Lindsey, M.L. (2013) Circulating Porphyromonas gingivalis Lipopolysaccharide Resets Cardiac Homeostasis in Mice through Matrix Metalloproteinase-9-Dependent Mechanism. Physiological Reports, 1, e00079. https://doi.org/10.1002/phy2.79
- 22. Slomiany, B.L. and Slomiany, A. (2016) Role of Rac1/p38 and ERK-Dependent Cytosolic Phospholipase A2 Activation in Porphyromonas gingivalis-Evoked Induction in Matrix Metalloproteinase-9 (MMP-9) Release by Salivary Gland Cells. Journal of Biosciences and Medicines, 4, 68-79. https://doi.org/10.4236/jbm.2016.44010
- 23. Sochalska, M. and Potempa, J (2017) Manipulation of Neutrophils by Porphyromonas gingivalis in the Development of Periodontitis. Frontiers in Cellular and Infection microbiology, 7, 197. https://doi.org/10.3389/fcimb.2017.00197
- 24. Slomiany, B.L., Murty, V.L.N., Piotrowski, J., Liau, Y.H. and Slomiany, A. (1993) Glycosulfatase Activity of Porphyromonas gingivalis. A Bacterium Associated with Periodontal Disease. Biochemistry and Molecular Biology International, 29, 973-980.
- 25. Slomiany, B.L. and Slomiany, A. (2002) Porphyromonas gingivalis Lipopolysaccharide Interferes with Salivary Mucin Synthesis through Inducible Nitric Oxide Synthase Activation by ERK and p38 Kinase. Biochemical and Biophysical Research Communication, 297, 1149-1153. https://doi.org/10.1016/S0006-291X(02)02354-9
- 26. Wang, P.L. and Ohura, K. (2002) Porphyromonas gingivalis Lipopolysaccharide Signaling in Gingival Fibroblasts-CD14 and Toll-Like Receptors. Critical Reviews in Oral Biology and Medicine, 13, 132-142. https://doi.org/10.1177/154411130201300204
- 27. Slomiany, B.L. and Slomiany, A. (2003) Activation of Peroxisome Proliferator-Activated Receptor γ Impedes Porphyromonas gingivalis Lipopolysaccharide Interference with Salivary Mucin Synthesis through Phosphatidylinositol 3-Kinase/ERK Pathway. Journal of Physiology and Pharmacology, 54, 3-15.
- 28. Slomiany, B.L. and Slomiany, A. (2005) Role of Modulation of Porphyromonas gingivalis Lipopolysaccharide-Induced Up-Regulation of Endothelin-1 in Salivary Gland Acinar Cells. IUBMB Life, 57, 591-595. https://doi.org/10.1080/15216540500215598
- 29. Slomiany, B.L. and Slomiany, A. (2010) Suppression by Ghrelin of Porphyromonas gingivalis-Induced Constitutive Nitric Oxide Synthase S-Nitrosylation and Apoptosis in Salivary Gland Acinar Cells. Journal of Signal Transduction, 2010, Article ID: 643642. https://doi.org/10.1155/2010/643642
- 30. Slomiany, B.L. and Slomiany, A. (2011) Cyclooxygenase-2 S-Nitrosylation in Salivary Gland Acinar Cell Inflammatory Responses to Porphyromonas gingivalis: Modulatory Effect of Ghrelin. Advances in Bioscience and Biotechnology, 2, 434-442. https://doi.org/10.4236/abb.2011.26064
- 31. Slomiany, B.L. and Slomiany, A. (2015) Porphyromonas gingivalis-Stimulated TACE Activation for TGF-α Ectodomain Shedding and EGFR Transactivation in Salivary Gland Cells Requires Rac1-Dependent p38 MAPK Membrane Localization. Journal of Biosciences and Medicines, 3, 42-53. https://doi.org/10.4236/jbm.2015.311005
- 32. Carpenter, S. and O’Neill, L.A.J. (2009) Recent Insights into the Structure of Toll-Like Receptors and Posttranslational Modifications of their Associated Signaling Proteins. Biochemical Journal, 422, 1-10. https://doi.org/10.1042/BJ20090616
- 33. Kawai, T. and Akira, S. (2010) The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nature Immunology, 11, 373-384. https://doi.org/10.1038/ni.1863
- 34. Farrugia, M. and Baron, B. (2017) The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. International Journal of Inflammation, 2017, Article ID: 8391230. https://doi.org/10.1155/2017/8391230
- 35. Wang, J.E., Dahle, M.K., McDonald, M., Foster, S.J., Aasen, A.O. and Thiemermann, C. (2003) Peptidoglycan and Lipoteichoic Acid in Gram-Positive Bacterial Sepsis: Receptors, Signal Transduction, Biological Effects, and Synergism. Shock, 20, 402-414. https://doi.org/10.1097/01.shk.0000092268.01859.0d
- 36. Smith, S.M. (2014) Role of Toll-Like Receptors in Helicobacter pylori Infection and Immunity. World Journal of Gastrointestinal Physiology, 5, 133-146.https://doi.org/10.4291/wjgp.v5.i3.133
- 37. Slomiany, B.L. and Slomiany, A. (2017) Role of LPS-Elicited Signaling in Triggering Gastric Mucosal Inflammatory Responses to H. pylori: Modulatory Effect of Ghrelin. Inflammopharmacology, 25, 415-429. https://doi.org/10.1007/s10787-017-0360-1
- 38. Amith, S.R., Abdulkhalek, S. and Szewczuk, M.R. (2016) Role of Glycosylation in Toll-Like Receptor Activation and Pro-Inflammatory Responses. In: Weiderschain, G., Ed., Glycobiology and Human Diseases, CRC, Boca Raton, FL, 165-184.
- 39. Park, B.S. and Lee, J.O. (2013) Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Experimental & Molecular Medicine, 45, e66.https://doi.org/10.1038/emm.2013.97
- 40. Chen, Z.J. (2012) Ubiquitination in Signaling to and Activation of IKK. Immunological Reviews, 246, 95-106. https://doi.org/10.1111/j.1600-065X.2012.01108.x
- 41. Miller, Y.I., Choi, S.H., Wiesner, P. and Bae, Y.S. (2012) The SYK Side of TLR4: Signaling Mechanism in Response to LPS and Minimally Oxidized LDL. British Journal of Pharmacology, 167, 990-999. https://doi.org/10.1111/j.1476-5381.2012.02097.x
- 42. Slomiany, B.L. and Slomiany, A. (2018) Role of Protein Kinase Cδ-Mediated Spleen Tyrosine Kinase (SYK) Phosphorylation on Ser in the Amplification of Oral Mucosal Inflammatory Responses to Porphyromonas gingivalis. Journal of Biosciences and Medicines, 6, 70-85. https://doi.org/10.4236/jbm.2018.63005
- 43. Coats, S.R., Jones, J.W., Do, C.T., Braham, P.M., Bainbridge, B.W., To, T.T., Goodlett, D.R., Ernst, R.K. and Darveau, R.P. (2009) Human Tool-Like Receptor 4 Responses to P. gingivalis Are Regulated by Lipid A 1- and 4’-Phosphatase Activities. Cellular Microbiology, 11, 1587-1599. https://doi.org/10.1111/j.1462-5822.2009.01349.x
- 44. Trent, M.S., Stead, C.M., Tran, A.X. and Hankins, J.V. (2006) Diversity of Endotoxin and Its Impact on Pathogenesis. Journal of Endotoxin Research, 12, 205-223.
- 45. Cohen, S. (1962) Isolation of a Mouse Submaxillary Gland Protein Accelerating Incisors Eruption and Eyelid Opening in the Newborn Animal. Journal of Biological Chemistry, 237, 1555-1562.
- 46. Carpenter, G. and Cohen, S. (1979) Epidermal Growth Factor. Annual Review of Biochemistry, 48, 193-216. https://doi.org/10.1146/annurev.bi.48.070179.001205
- 47. Matarese, G., Moschos, S. and Mantzoros, S. (2005). Leptin in Immunology. Journal of Immunology, 174, 3137-3142. https://doi.org/10.4049/jimmunol.174.6.3137
- 48. Hallas, J.L. and Friedman, J.M. (1997) Leptin and Its Receptor. Endocrinology, 155, 215-216.
- 49. Breidert, M., Miehlke, S., Glasgow, A., et al. (1999) Leptin and Its Receptors in Normal Human Gastric Mucosa and in Helicobacter pylori-Associated Gastritis. Scandinavian Journal of Gastroenterology, 34, 954-961.https://doi.org/10.1080/003655299750025039
- 50. Slomiany, B.L. and Slomiany, A. (2003) Leptin Suppresses Porphyromonas gingivalis Lipopolysaccharide Interference with Salivary Mucin Synthesis. Biochemical and Biophysical Research Communications, 312, 1099-1103. https://doi.org/10.1016/j.bbrc.2003.11.035
- 51. Ogunwobi, O., Mutungi, G. and Beales, I.L.P. (2006) Leptin Stimulates Proliferation and Inhibits Apoptosis in Barrett’s Esophageal Adenocarcinoma Cells by Cyclooxygenase-2-Dependent, Prostaglandin-E2-Medited Transactivation of the Epidermal Growth Factor Receptor and c-Jun NH2-Terminal Kinase Activation. Endocrinology, 147, 4505-4516. https://doi.org/10.1210/en.2006-0224
- 52. Slomiany, B.L. and Slomiany, A. (2008) Leptin Protection of Salivary Gland Acinar Cells against Ethanol Cytotoxicity Involves Src Kinase-Mediated Parallel Activation of Prostaglandin and Constitutive Nitric Oxide Synthase Pathways. Inflammopharmacology, 16, 76-82. https://doi.org/10.1007/s10787-007-1650-9
- 53. Kojima, M. and Kangawa, K. (2005) Ghrelin: Structure and Function. Physiological Reviews, 85, 495-555. https://doi.org/10.1152/physrev.00012.2004
- 54. Xu, X., Jhun, B.S., Ha, C.H. and Jin, Z.G. (2008) Molecular Mechanism of Ghrelin-Mediated Endothelial Nitric-Oxide Synthase Activation. Endocrinology, 149, 4183-4192. https://doi.org/10.1210/en.2008-0255
- 55. Lodeiro, P., Theodoropoulous, M., Pardo, M., Casanueva, F.F. and Camina, J.P. (2009) c-Src Regulates Akt Signaling in Response to Ghrelin via β-Arrestin Signaling-Independent and -Dependent Mechanism. PLoS ONE, 4, e4686. https://doi.org/10.1371/journal.pone.0004686
- 56. Slomiany, B.L. and Slomiany, A. (2011) Ghrelin Protects against the Detrimental Consequences of Porphyromonas gingivalis-Induced Akt Inactivation through S-nitrosylation on Salivary Mucin Synthesis. International Journal of Inflammation, 2011, Article ID: 807279. https://doi.org/10.4061/2011/807279
- 57. Slomiany, B.L. and Slomiany, A. (2011) Ghrelin-Induced cSrc Activation through Constitutive Nitric Oxide Synthase-Dependent S-nitrosylation in Modulation of Salivary Gland Acinar Cell Inflammatory Responses to Porphyromonas gingivalis. American Journal of Molecular Biology, 2, 43-51. https://doi.org/10.4236/ajmb.2011.12006
- 58. Slomiany, B.L. and Slomiany, A. (2006) Leptin Modulates the Detrimental Effect of Porphyromonas gingivalis Lipopolysaccharide-Induced Cytosolic Phospholipase A2 Activation on Salivary Mucin Synthesis via ERK-Signal Transduction. InflammoPharmacology, 14, 250-255. https://doi.org/10.1007/s10787-006-1525-5
- 59. Cuzzocrea, S. and Salvemini, D. (2007) Molecular Mechanisms Involved in Reciprocal Regulation of Cyclooxygenase and Nitric Oxide Synthase Enzymes. Kidney International, 71, 290-297.
- 60. Slomiany, B.L. and Slomiany, A. (2010) Ghrelin Protection against Cytotoxic Effect of Ethanol on Rat Salivary Mucin Synthesis Involves Cytosolic Phospholipase A2 Activation through S-Nitrosylation. International Journal of Biomedical Sciences, 6, 37-44.
- 61. Korhonen, R., Lahti, A., Kankaanranta, H. and Moilanen, E. (2005) Nitric Oxide Production and Signaling in Inflammation. Current Drug Targets: Inflammation & Allergy, 4, 471-479. https://doi.org/10.2174/1568010054526359
- 62. Sibilia, V., Pagani, F., Rindi, G., et al. (2008) Central Ghrelin Gastroprotection Involves Nitric Oxide/Prostaglandin Cross-Talk. British Journal of Pharmacology, 154, 688-697. https://doi.org/10.1038/bjp.2008.120
- 63. Ricciotti, E. and FitzGerald, G.A. (2011) Prostaglandins and Inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology, 31, 986-1000. https://doi.org/10.1161/ATVBAHA.110.207449
- 64. Agard, M., Asakrah, S. and Morici, L.A. (2013) PGE2 Suppression of Innate Immunity during Mucosal Bacterial Infection. Frontiers in Cellular and Infection Microbiology, 3, Article 45. https://doi.org/10.3389/fcimb.2013.00045
- 65. Slomiany, B.L. and Slomiany, A. (2010) Role of Ghrelin in Modulation of S-Nitrosylation-Dependent Akt Inactivation Induced in Salivary Gland Acinar Cells by Porphyromonas gingivalis. Health, 12, 1448-1455.https://doi.org/10.4236/health.2010.212215
- 66. Slomiany, B.L. and Slomiany, A. (2010) Constitutive Nitric Oxide Synthase-Mediated Caspase-3 S-Nitrosylation in Ghrelin Protection against Porphyromonas gingivalis-Induced Salivary Gland Apoptosis. Inflammopharmacology, 18, 119-125. https://doi.org/10.1007/s10787-010-0035-7
- 67. Xu, L., Han, C. and Wu, T. (2008) Activation of Cytosolic Phospholipase A2α through Nitric Oxide-Induced S-Nitrosylation. Involvement of Inducible Nitric-Oxide Synthase and Cyclooxygenase-2. Journal of Biological Chemistry, 283, 3077-3087. https://doi.org/10.1074/jbc.M705709200
- 68. Slomiany, B.L. and Slomiany, A. (2011) Ghrelin Suppression of Helicobacter pylori-Induced Gastric Mucosal iNOS Is Mediated through the Inhibition of IKK-β Activation by c-NOS-Dependent S-Nitrosylation. Open Journal of Cell Biology, 1, 1-10. https://doi.org/10.4236/ojcb.2011.11001
- 69. Ye, Y., Martinez, J.D., Perez-Polo, R.J., Lin, Y., Uretsky, B.F. and Birnbaum, Y. (2008) The Role of eNOS, iNOS, and NF-κB in Upregulation and Activation of Cyclooxygenase-2 and Infarct Size Reduction by Atorvastin. American Journal of Physiology-Heart and Circulatory Physiology, 295, H343-H351.https://doi.org/10.1152/ajpheart.01350.2007
- 70. Cuadrado, A. and Nebreda, A.R. (2010) Mechanism and Function of p38 MAPK Signaling. Biochemistry Journal, 429, 403-417. https://doi.org/10.1042/BJ20100323
- 71. Slomiany, B.L. and Slomiany, A. (2013) Involvement of p38 MAPK-Dependent Activation Protein (AP-1) Activation in Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori by Ghrelin. Inflammopharmacology, 21, 67-78.https://doi.org/10.1007/s10787-012-0141-9
- 72. Rieke, C., Papendieck, A., Sokolova, O. and Neumann, M. (2011) Helicobacter pylori-Induced Tyrosine Phosphorylation of IKKβ Contributes to NF-κB Activation. Journal of Biological Chemistry, 392, 387-393.
- 73. Slomiany, B.L. and Slomiany, A. (2013) Induction in Gastric Mucosal Prostaglandin and Nitric Oxide by Helicobacter pylori Is Dependent on MAPK/ERK-Mediated Activation of IKK-β and cPLA2: Modulatory Effect of Ghrelin. Inflammopharmacology, 21, 241-251. https://doi.org/10.1007/s10787-013-0169-5
- 74. Caivano, M., Gorgoni, B., Cohen, P. and Poli, V. (2001) The Induction of Cyclooxygenase-2 mRNA in Macrophages Is Biphasic and Requires Both CCAAT Enhancer-Binding Protein β (C/EBPβ) and C/EBPδ Transcription Factors. Journal of Biological Chemistry, 276, 48693-48701. https://doi.org/10.1074/jbc.M108282200
- 75. Grishin, A.V., Wang, J., Potoka, D.A., et al. (2006) Lipopolysaccharide Induces Cyclooxygenase-2 in Intestinal Epithelium via a Non-Canonical p38 MAPK Pathway. Journal of Immunology, 176, 580-588. https://doi.org/10.4049/jimmunol.176.1.580
- 76. Kang, Y.J., Wingerd, B.A., Arakawa, T. and Smith, W.L. (2006) Cyclooxygenase-2 Gene Transcription in a Macrophage Model of Inflammation. Journal of Immunology, 177, 8111-8122. https://doi.org/10.4049/jimmunol.177.11.8111
- 77. Lopez-Bergami, P., Lau, E. and Ronai, Z. (2010) Emerging Roles of ATF2 and the Dynamic of AP1 Network in Cancer. Nature Reviews Cancer, 10, 65-76. https://doi.org/10.1038/nrc2681
- 78. Slomiany, B.L. and Slomiany, A. (2007) Alteration by Indomethacin in Proinflammatory Consequences of Salivary Gland Cytosolic Phospholipase A2 Activation by Porphyromonas gingivalis: Role of Leptin. Journal of Applied Research, 7, 127-136.
- 79. Lin, C.C., Lin, W.N., Wang, W.J., et al. (2009) Functional Coupling of COX-2 and cPLA2 Induced by ATP in Rat Vascular Smooth Muscle Cells: Role of ERK1/2, p38 MAPK, and NF-κB. Cardiovascular Research, 82, 522-531.
- 80. Kadamur, G. and Ross, E.M. (2013) Mammalian Phospholipase C. Annual Review of Physiology, 75, 127-154. https://doi.org/10.1146/annurev-physiol-030212-183750
- 81. Slomiany, B.L. and Slomiany, A. (2014) Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori via Ghrelin-Induced Protein Kinase Cδ Tyrosine Phosphorylation. Inflammopharmacology, 22, 251-262.https://doi.org/10.1007/s10787-014-0206-z
- 82. Gong, P., Angelini, D.J., Yang, S., et al. (2008) TLR4 Signaling Is Coupled to SRC Family Kinase Activation, Tyrosine Phosphorylation of Zonula Adherens Proteins, and Opening of the Paracellular Pathway in Hlung Microvascular Endothelia. Journal of Biological Chemistry, 283, 13437-13449. https://doi.org/10.1074/jbc.M707986200
- 83. Deason-Towne, F., Perraud, A.L. and Schmitz, C. (2012) Identification of Ser/Thr Phosphorylation Sites in the C2-Domain of Phospholipase Cγ2 (PLCγ2) Using TRPM7-Kinase. Cell Signaling, 24, 2070-2075. https://doi.org/10.1016/j.cellsig.2012.06.015
- 84. Harden, T.K., Hicks, S.N. and Sondek, J. (2009) Phospholipase C Isozymes as Effectors of Ras Superfamily GTPases. Journal of Lipid Research, 50, S243-S248.https://doi.org/10.1194/jlr.R800045-JLR200
- 85. Parri, M. and Chiarugi, P. (2010) Rac and Rho GTPases in Cancer Cell Motility Control. Cell Communication and Signaling, 8, 23-37. https://doi.org/10.1186/1478-811X-8-23
- 86. Boulter, E., Estrach, S., Gracia-Mata, R. and Feral, C.C. (2012) Off the Beaten Paths: Alternative and Crosstalk Regulation of Rho GTPases. FASEB Journal, 26, 469-479.
- 87. Jun, J.E., Rubio, I. and Roose, J.P. (2013) Regulation of Ras Exchange Factors and Cellular Localization of Ras Activation by Lipid Messengers in T Cells. Frontiers in Immunology, 4, 239. https://doi.org/10.3389/fimmu.2013.00239
- 88. Slomiany, B.L. and Slomiany, A. (2015) Mechanism of Rac1-Induced Amplification in Gastric Mucosal Phospholipase Cγ2 Activation in Response to Helicobacter pylori: Modulatory Effect of Ghrelin. Inflammopharmacology, 23, 101-109.https://doi.org/10.1007/s10787-015-0231-6
- 89. Yao, H.Y., Chen, L., Wang, J., et al. (2011) Inhibition of Rac Activity Alleviates Lipopolysaccharide-Induced Acute Pulmonary Injury in Mice. Biochimica et Biophysica Acta (BBA)-General Subjects, 1810, 666-674. https://doi.org/10.1016/j.bbagen.2011.03.020
- 90. Slomiany, B.L. and Slomiany, A. (2015) Porphyromonas gingivalis-Induced GEF Dock180 Activation by Src/PKCδ-Dependent Phosphorylation Mediates PLCγ2 Amplification in Salivary Gland Acinar Cells: Effect of Ghrelin. Journal of Biosciences and Medicines, 3, 66-77. https://doi.org/10.4236/jbm.2015.37008
- 91. McElroy, S.J., Hobbs, S., Kallen, M., et al. (2012) Transactivation of EGFR by LPS Induces COX-2 Expression in Enterocytes. PLoS ONE, 7, e38373.https://doi.org/10.1371/journal.pone.0038373
- 92. Slomiany, B.L. and Slomiany, A. (2013) Role of EGFR Transactivation in the Amplification of Helicobacter pylori-Elicited Induction in Gastric Mucosal Expression of COX-2 and iNOS. OA Inflammation, 1, 1. https://doi.org/10.13172/2052-787X-1-1-412
- 93. Trussoni, C.E., Tabibian, J.H., Splinter, P.L. and O’Hara, S.P. (2015) Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR). PLoS ONE, 10, e0125793.https://doi.org/10.1371/journal.pone.0125793
- 94. Finzi, L., Shao, M.X.G., Paye, F., Housset, C. and Nadel, J.A. (2009) Lipopolysaccharide Initiates a Positive Feedback of Epidermal Growth Factor Receptor Signaling by Prostaglandin E2 in Human Biliary Carcinoma Cells. The Journal of Immunology, 182, 2269-2276. https://doi.org/10.4049/jimmunol.0801768
- 95. Ohsu, H., Dempsey, P. and Eguchi, S. (2006) ADAMs as Mediators of EGF. Receptor Transactivation by G Protein-Coupled Receptors. American Journal of Cell Physiology, 291, C1-C10.
- 96. Slomiany, B.L and Slomiany, A. (2004) Porphyromonas gingivalis Lipopolysaccharide-Induced Up-Regulation in Endothelin-1 Interferes with Salivary Mucin Synthesis via Epidermal Growth Factor Receptor Transactivation. IUBMB Life, 56, 601-607. https://doi.org/10.1080/15216540400020361
- 97. Bergin, D.A., Greene, C.M., Sterchi, E.E., et al. (2008) Activation of the Epidermal Growth Factor Receptor (EGFR) by a Novel Metalloprotease Pathway. Journal of Biological Chemistry, 283, 31736-31744. https://doi.org/10.1074/jbc.M803732200
- 98. Xu, P. and Derynck, R. (2010) Direct Activation of TACE-Mediated Ectodomain Shedding by p38 MAP Kinase Regulates EGF Receptor-Dependent Cell Proliferation. Molecular Cell, 37, 551-566.
- 99. Slomiany, B.L. and Slomiany, A. (2000) Aspirin Ingestion Impairs Oral Mucosal Ulcer Healing by Inducing Membrane-Bound Tumor Necrosis Factor-α Release. IUBMB Life, 50, 391-395.
- 100. Kong, L. and Ge, B.X. (2008) MyD88-Independent Activation of a Novel Actin-Cdc42/Rac Pathway Is Required for Toll-like Receptor-Stimulated Phagocytosis. Cell Research, 18, 745-755. https://doi.org/10.1038/cr.2008.65
- 101. Lin, Y.C., Huang, D.Y., Chu, C.L. and Lin, W.W. (2010) Anti-Inflammatory Actions of Syk Inhibitors in Macrophages Involve Non-Specific Inhibition of Toll-Like Receptors-Mediated JNK Signaling Pathway. Molecular Immunology, 47, 1569-1578. https://doi.org/10.1016/j.molimm.2010.01.008
- 102. Choi, S.H., Wiesner, P., Almazan, F., Kim, J. and Miller, Y.I. (2012) Spleen Tyrosine Kinase Regulates AP-1 Dependent Transcriptional Response to Minimally Oxidized LDL. PLoS ONE, 7, e32378. https://doi.org/10.1371/journal.pone.0032378
- 103. Yi, Y.S., Son, Y.J., Ryou, C., Sung, G.G., Kim, J.H. and Cho, J.Y. (2014) Functional Roles of Syk in Macrophage-Mediated Inflammatory Responses. Mediators of Inflammation, 2014, Article ID: 270302. https://doi.org/10.1155/2014/270302
- 104. Wang, J.G. and Aikawa, M. (2015) Toll-Like Receptors and Src-Family Kinases in Atherosclerosis—Focus on Macrophages. Circulation Journal, 79, 2332-2334.
- 105. Bohnenberger, H., Oellerich, T., Engelke, M., Hsiao, H.H., Urlaub, H. and Wienands, J. (2011) Complex Phosphorylation Dynamics Control the Composition of the Syk Interactome in B Cells. European Journal of Immunology, 41, 1550-1562.https://doi.org/10.1002/eji.201041326
- 106. Elsori, D.H., Yakubenko, V.P., Roome, T., Thiagarajan, P.S., Bhattacharjee, A., Yadav, S.P. and Cathcart, M.K. (2011) Protein Kinase Cδ Is a Critical Component of Detectin-1 Signaling in Primary Human Monocytes. Journal of Leukocyte Biology, 90, 599-611.
- 107. Bijli, K.M., Fazal, F., Minhajuddin, M. and Rahman, A. (2008) Activation of Syk by Protein Kinase C-δ Regulates Thrombin-Induced Intracellular Adhesion Molecule-1 Expression in Endothelial Cells via Tyrosine Phosphorylation of RelA/p65. Journal of Biological Chemistry, 283, 14674-14684. https://doi.org/10.1074/jbc.M802094200
- 108. Kazi, J.U. (2011) The Mechanism of Protein Kinase C Regulation. Frontiers in Biology, 6, 328-336.