International Journal of Clinical Medicine
Vol.4 No.4(2013), Article ID:30874,5 pages DOI:10.4236/ijcm.2013.44034
Primary Anaplastic Large Cell Lymphoma of the Nasal Cavity: A Case Report*
![]()
1Medical Oncology Department, National Oncology Institute Mohamed Ben Abdellah, Rabat, Morocco; 2Pathological Anatomy Department, Ibn Sina University Hospital, Hospital of Specialties, Rabat, Morocco.
Email: ouafkiimane@gmail.com
Copyright © 2013 Imane Ouafki et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 10th, 2013; revised February 25th, 2013; accepted April 12th, 2013
Keywords: Non-Hodgkin’s Lymphoma; Anaplastic Lymphoma Kinase; Chemotherapy
ABSTRACT
Introduction: Anaplastic large-cell lymphoma occurring in the nasal cavity is a rare disease. The latest World Health Organization (WHO) Classification recognizes three distinct subtypes: primary systemic anaplastic lymphoma kinase positive been our case, primary systemic anaplastic lymphoma kinase negative and primary cutaneous types. Through this case study, we focus on the clinical presentation, treatment and prognostic characteristics of this pathology. Case Presentation: We report the case of a patient aged 32 years, who presented for seven months a runny nose associated with swelling of the face on the left side, without peripheral lymphadenopathy or general signs. A Blondeau scanner objectified a total filling of the frontal and left maxillary sinus, and a filling of the left nasal cavity. Complete resection of the tumor was performed. Histological examination was in favor of anaplastic large T-cell lymphoma anaplastic lymphoma kinase positive. The patient was stage IE according to Ann Arbor classification, with an International Prognostic Index estimated at one. Thus, the patient received six cycles of CHOP chemotherapy. Currently, he is in good loco-regional control with a decline of three months. Conclusion: The rarity of this case lies partly in the lymphomatous localization in the nasal cavity, and secondly in the anaplastic histology. It poses a diagnostic problem. So, we conclude that in case of any symptom of nasal cavities, it is necessary to explore and possibly biopsy if tumor, before surgery, because lymphomas are chemosensitive disease.
1. Introduction
Anaplastic large-cell lymphoma (ALCL), is an uncommon disease, accounting for <5% of all cases of nonHodgkin’s lymphoma (NHL) [1]. Its location in the nasal cavity is extremely rare.To the best of our knowledge; no large series has documented the exact number of cases worldwide. Data are available in isolated case reports and small case series. ALCL was first described as a clinical entity in 1985 based upon its unique characteristic of cohesive proliferation of large pleomorphic cells expressing CD 30 (also known as Ki-1) [2]. It occurs as two distinct clinical entities, a localized cutaneous variant and widespread systemic disease been our case [3]. The latest WHO Classification recognizes three distinct subtypes of ALCL: primary systemic anaplastic lymphoma kinase positive (ALK+), primary systemic ALK negative (ALK−) and primary cutaneous types, which have differences in immunophenotype, genetics, and clinical behavior [4]. The majority of patients present with B symptoms (75%) including asthenia, anorexia, weight loss, night sweats and fever. However it may be asymptomatic in early stages. The diagnosis is made by a biopsy and histological examination. The most important prognostic indicator is ALK positivity, which confers a favorable prognosis with a 5-year disease free survival (DFS) and overall survival (OS) of 70.5% and 58% compared to ALK−, 49% and 36% respectively [5]. Through this case study, we focus on the clinical presentation, treatment and prognostic characteristics of this pathology.
2. Case Presentation
We report the case of a patient aged 32 years, who presented for seven months a runny nose associated with swelling of the face on the left side, without peripheral lymphadenopathy or general signs. A Blondeau scanner has objectified a total filling of the frontal and left maxillary sinus, a filling of the left nasal cavity with deviation of the nasal septum to the right and mucosal thickening of the right maxillary sinus (Figure 1). Excision of the tumor in the left nasal cavity, left maxillary sinus and the floor of the orbit was performed. The histological study with immunohistochemical complement was in favor of anaplastic large T-cell lymphoma ALK positive, CD30 positive, LCA positive, EMA positive and CD20 negative (Figures 2 and 3). Cervical thoracic abdominal and pelvic postoperatively computed tomography (CT) did not show lymph node enlargement. The bone marrow was not invaded. Sedimentation rate and lactate dehydrogenase were correct. Thus, the patient was classified as stage IE according to the Ann Arbor classification, with an International Prognostic Index (IPI) estimated at one (low risk). Echocardiography was normal, with an ejection fraction of the left ventricle estimated at 80%. The patient had adequate hepatic, hematological and renal function. CHOP chemotherapy was introduced, one cycle every three weeks for a total of six courses. Currently, he is in good loco-regional control with a decline of 3 months.
3. Discussion
Since ALCL was first recognized by Stein et al. in 1985 [2], significant achievements have been made in the study on the pathology of this disease over the years. However, the knowledge on the clinical characteristics and outcomes of ALCL was based on small case series [6]. Systemic ALCL is generally very aggressive. The majority of patients presents with Stage III or IV disease and have systemic symptoms [7]. Extra nodal disease occurs in 40% - 60% of patients, with skin, bone, soft tissue, and lung being common sites of involvement [8,9]. ALCL has a peak incidence in childhood and accounts for approximately 40% of NHL cases diagnosed in pediatric populations [10]. It shows a male predominance, particularly in ALK positive cases, in which the male/female ratio is approximately 3:1 [8]. Some reports have suggested that Epstein-Barr virus (EBV) is important in the pathogenesis of ALCL. However, a recent series of 64 ALCL cases revealed no EBV encoded RNA (EBER) or immunohistochemistry evidence for EBV-latent membrane protein type 1 [11]. The authors concluded that previous reports of EBV in Western patients with ALCL were probably a result of the inclusion of tumors no longer considered to be ALCL, such as CD30+ anaplastic tumors of B-cell origin. EBV, however, may be important in the pathogenesis of ALCL occurring in Asia and in immune-compromised hosts [12-13]. ALCL can occasionally mimic Hodgkin lymphoma (HL) because ALCL often presents in young patients and can have ReedSternberg-like cells, prominent sclerosis, and expression of CD30. However, HL is often CD15+ but negative for T-cell markers and ALK, whereas ALCL is generally CD15− and CD30+, and often ALK+ and/or expresses T-cell antigens. ALCL can also be confused with diffuse large B-cell lymphoma (DLBCL), particularly mediastinal DLBCL, which has a tendency to present in younger patients and which can be CD 30+. However, DLBCL expresses B-cell markers such as CD20 and/or CD79a and does not express T-cell markers and, with very rare exceptions, is not ALK+ [14,15]. The most important prognostic factor in systemic ALCL was the presence or absence of the ALK protein. It is known that approximately 60% of systemic ALCL are ALK+ and have a significantly superior survival to ALK− cases [16,17], justifying the separation of these two categories. In a series that included 57 patients with ALCL, the 5-year overall survival (OS) rate was 93% in patients who were ALK+ compared with 37% for those who were ALK− [17]. In that series, a normal lactate dehydrogenase (LDH) and an International Prognostic Index (IPI) score of ≤3 also were independent predictors of better outcome. The importance of the IPI was also demonstrated in an Italian series in which ALK+ patients with a low or low– intermediate IPI score had a 5-year OS rate of 94%, whereas ALK+ patients with a high-intermediate or high IPI score had a 5-year OS rate of only 41% [10]. However, in multivariate analysis, only high IPI and CD56 positivity were independent predictors of poor outcome. ALK+ patients in that series tended to be younger, with less advanced disease, a normal LDH, and a better performance status, suggesting that ALK status is a useful determinant of outcome but should be interpreted also in the context of the patient’s IPI. Some studies showed that ALK+ and ALK− patients had different gene expression profiles, clinical characteristics and prognosis [18]. ALK+ patients are characterized by younger age and better prognosis. Most ALK+ patients are below 30 years old, and their 5-year OS rates were 70% - 95%, while the 5-year OS rates of ALK− patients were 28% - 46% [19,20]. Current main therapeutic strategy for ALCL is combination chemotherapy. Most trials have been conducted in children. Some adult trials have been conducted, but most adult treatment series are retrospective, and patients were treated on a variety of protocols. In most studies, adult patients were treated with anthracycline-based regimens, such as CHOP and MACOP regimens [6]. An Italian multicentric trial randomized 40 ALCL-Hodgkin-like patients to frontline chemotherapy with MACOP-B (meth-

Figure 1. Blondeau scanner objectifying the tumor in cross sections.
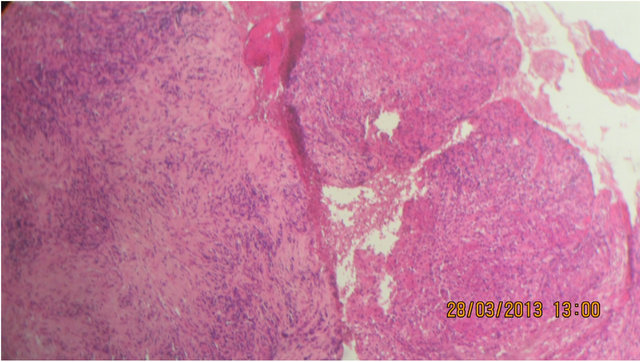
Figure 2. Hematoxylin eosin staining objectifying tumor proliferation made of layers of round cells of large size, with hyper chromatic nuclei, with a typical mitotic figures. Loose stroma with dense polymorphous inflammatory infiltrate with hemorrhagic suffusion.
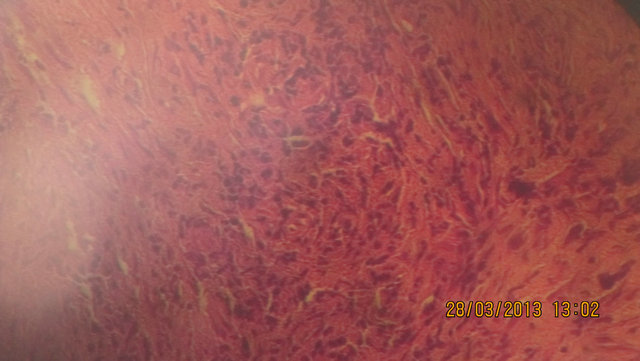
Figure 3. Microphotograph showing the immunohistochemical appearance.
otrexate with leucovorin, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin—a third-generation HL-NHL regimen) or ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine—a scheme specific for HL) [21]. Complete response (CR) rates in both arms were in excess of 90%, with an approximately 90% chance of being relapse free at 32 months. Unfortunately, we now know that most cases of ALCL-Hodgkin like are actually HL, making the results of that trial difficult to interpret. A French series contained a subset of patients with ALCL treated with three cycles of ACVBP (doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone) chemotherapy followed by a consolidation phase with high dose methotrexate, ifosfamide, etoposide, asparaginase, and cytosine-arabinoside or eight cycles of m-BACOD (methotrexate, bleomycin, cyclophosphamide, vincristine, dexamethasone) [22]. The CR rate was 69% for the T-cell subtype with an estimated 5-year OS rate of 63.2%. In an Italian series that stratified patients by ALK expression, the overall response rate (ORR) for ALK+ patients was 92.3% (77.3% achieved a CR), whereas the ORR in ALK− patients was 84%, with only 56% achieving a CR [10]. Because of the heterogeneity of reports in the literature and lack of systematic trials, the US standard has generally been to treat ALCL with CHOP chemotherapy. The choice of CHOP is largely an extrapolation from the DLBCL literature. Studies in DLBCL have failed to demonstrate a benefit to so-called thirdgeneration regimens (m-BACOD, ProMACECytaBOM [cyclophosphamide, doxorubicin, etoposide, prednisone, vincristine, bleomycin, cytosine arabinoside, methotrexate, and leucovorin], MACOP-B) over CHOP, and hence, most oncologists are hesitant to use these regimens in related diseases like ALCL [23]. Previous studies indicated that the overall response rates, CR rates and 5-year OS rates of primary systemic ALCL patients undergoing initial chemotherapy were 60% - 95%, 60% - 85% and 40% - 80%, respectively [8,24], and most recurrent patients were still sensitive to chemotherapy and achieved remission again [25,26]. Autologous stem cell transplantation (ASCT) has been studied prospectively and retrospectively in ALCL. In a retrospective series of patients transplanted for relapsed ALCL, the 3-year OS rate was 86%. ALK+ patients had an event-free survival (EFS) rate of 100% at 3 years, compared with 0% in ALK− cases [27].
4. Conclusion
The rarity of this case lies partly in the lymphomatous localization in the nasal cavity, and secondly in the anaplastic histology. This disease poses a diagnostic problem, insofar as the symptoms may be absent in the early stages. As is the case of our patient with nasal discharge considered initially as a simple rhinitis. ALCL may also be confused histologically with HL and DLBCL. This will lead us to conclude that in case of any symptom of nasal cavities, it is necessary to explore and possibly biopsy if tumor, before surgery, because lymphomas are chemosensitive disease.
REFERENCES
- M. E. Kadin and S. W. Morris, “The t(2;5) in Human Lymphomas,” Leukemia & Lymphoma, Vol. 29, No. 3-4, 1998, pp. 249-256. doi:10.3109/10428199809068562
- H. Stein, D. Y. Mason, J. Gerdes, et al., “The Expression of the Hodgkin’s Diseaseassociated Antigen Ki-1 in Reactive and Neoplastic Lymphoid Tissue: Evidence That Reed-Sternberg Cells and Histiocytic Malignancies Are Derived from Activated Lymphoid Cells,” Blood, Vol. 66, No. 4, 1985, pp. 848-858.
- N. L. Harris, E. S. Jaffe, H. Stein, et al., “A Revised European-American Classification of Lymphoid Neoplasms: A Proposal from the International Lymphoma Study Group,” Blood, Vol. 84, No. 5, 1994, pp. 1361-1392.
- S. H. Swerdlow, E. Campo, N. L. Harris, E. S. Jaffe, S. A. Pileri, H. Stein, J. Thiele and J. W. Vardiman, “WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues,” IARC, Lyon, 2008.
- K. J. Savage, N. L. Harris, J. M. Vose, et al., “ALK-Negative Anaplastic Large-Cell Lymphoma (ALCL) Is Clinically and Immunophenotypically Different from Both ALKPositive ALCL and Peripheral T-Cell Lymphoma, Not Otherwise Specified: Report from the International Peripheral T-Cell Lymphoma Project,” Blood, Vol. 111, No. 12, 2008, pp. 5496-5504. doi:10.1182/blood-2008-01-134270
- F.-H. Wang, Y.-H. Li, J. Zeng, H.-L. Rao, Z.-J. Xia, X.-F. Sun, H.-Q. Huang, T.-Y. Lin, W.-Q. Jiang and Z.-Z. Guan, “Clinical Analysis of Primary Systemic Anaplastic Large Cell Lymphoma: A Report of 57 Cases,” Chinese Journal of Cancer, Vol. 28, No. 1, 2009.
- D. A. Filippa, M. Ladanyi, N. Wollner, et al., “CD30 (Ki-1)-Positive Malignantlymphomas: Clinical, Immunophenotypic, Histologic, and Genetic Characteristics and Differences with Hodgkin’s Disease,” Blood, Vol. 87, No. 7, 1996, pp. 2905-2917.
- B. Falini, S. Pileri, P. L. Zinzani, et al., “ALK + Lymphoma: Clinico-Pathological Findings and Outcome,” Blood, Vol. 93, No. 8, 1999, pp. 2697-2706.
- M. E. Kadin and C. Carpenter, “Systemic and Primary Cutaneous Anaplasticlarge Cell Lymphomas,” Seminars in Hematology, Vol. 40, No. 3, 2003, pp. 244-256. doi:10.1016/S0037-1963(03)00138-0
- J. T. Sandlund, C. H. Pui, V. M. Santana, et al., “Clinical Features and Treatment Outcome for Children with CD30+ Large-Cell Non-Hodgkin’s Lymphoma,” Journal of Clinical Oncology, Vol. 12, No. 5, 1994, pp. 895-898.
- M. Herling, G. Z. Rassidakis, D. Jones, et al., “Absence of Epstein-Barr Virus Inanaplastic Large Cell Lymphoma: A Study of 64 Cases Classified According to World Health Organization Criteria,” Human Pathology, Vol. 35, No. 4, 2004, pp. 455-459. doi:10.1016/j.humpath.2003.10.013
- S. Noorali, S. Pervez, N. Yaqoob, et al., “Prevalence and Characterization of Anaplastic Large Cell Lymphoma and Its Association with Epstein-Barrvirus in Pakistani Patients,” Pathology—Research and Practice, Vol. 200, No. 10, 2004, pp. 669-679. doi:10.1016/j.prp.2004.08.004
- V. Costes-Martineau, C. Delfour, S. Obled, et al., “Anaplastic Lymphomakinase (ALK) Protein Expressing Lymphoma after Liver Transplantation: Case Report and Literature Review,” Journal of Clinical Pathology, Vol. 55, No. 11, 2002, pp. 868-871. doi:10.1136/jcp.55.11.868
- R. D. Gascoyne, L. Lamant, J. I. Martin-Subero, et al., “ALK-Positive Diffuse Large B-Cell Lymphoma Is Associated with Clathrin-ALK Rearrangements: Report of 6 Cases,” Blood, Vol. 102, No. 7, 2003, pp. 2568-2573. doi:10.1182/blood-2003-03-0786
- H. Ruchatz, A. M. Coluccia, P. Stano, et al., “Constitutive Activation of Jak2 Contributes to Proliferation and Resistance to Apoptosis in NPM/ALK Transformed Cells,” Experimental Hematology, Vol. 31, No. 4, 2003, pp. 309- 315. doi:10.1016/S0301-472X(03)00007-9
- R. L. Ten Berge, J. J. Oudejans, G. J. Ossenkoppele, et al., “ALK Expression in Extranodal Anaplastic Large Cell Lymphoma Favours Systemic Diseasewith (Primary) Nodal Involvement and a Good Prognosis and Occurs before Dissemination,” Journal of Clinical Pathology, Vol. 53, No. 6, 2000, pp. 445-450. doi:10.1136/jcp.53.6.445
- R. D. Gascoyne, P. Aoun, D. Wu, et al., “Prognostic Significance of Anaplastic Lymphoma Kinase (ALK) Protein Expression in Adults with Anaplasticlarge Cell Lymphoma,” Blood, Vol. 93, 1999, pp. 3913-3921.
- L. Lamant, A. de Reyniès, M. M. Duplantier, et al., “GeneExpression Profiling of Systemic Anaplastic Large-Cell Lymphoma Reveals Differences Based on ALK Status and Two Distinct Morphologic ALK+ Subtypes,” Blood, Vol. 109, No. 5, 2007, pp. 2156-2164. doi:10.1182/blood-2006-06-028969
- M. Shiota, S. Nakamura, R. Ichinohasama, et al., “Anaplastic Large Cell Lymphomas Expressing the Novel Chimeric Protein p80NPM/ALK: A Distinct Clinic Opathologic Entity,” Blood, Vol. 86, No. 5, 1995, pp. 1954-1960.
- K. J. Savage, N. L. Harris, J. M. Vose, et al., “ALK-Negative Anaplastic Large-Cell Lymphoma (ALCL) Is Clinically and Immunophenotypically Different from Both ALK-Positive ALCL and Peripheral T-Cell Lymphoma, Not Otherwise Specified: Report from the International Peripheral T-Cell Lymphoma Project,” Blood, Vol. 111, No. 12, 2008, pp. 5496-5504. doi:10.1182/blood-2008-01-134270
- P. L. Zinzani, M. Martelli, M. Magagnoli, et al., “Anaplastic Large Cell Lymphoma Hodgkin’s-Like: A Randomized Trial of ABVD versus Macop-B with and without Radiation Therapy,” Blood, Vol. 92, No. 3, 1998, pp. 790- 794.
- H. Tilly, P. Gaulard, E. Lepage, et al., “Primary Anaplastic Large-Cell Lymphomain Adults: Clinical Presentation, Immunophenotype, and Outcome,” Blood, Vol. 90, No. 9, 1997, pp. 3727-3734.
- R. I. Fisher, E. R. Gaynor, S. Dahlberg, et al., “Comparison of a Standard Regimen (CHOP) with Three Intensive Chemotherapy Regimens for Advanced Non-Hodgkin’s Lymphoma,” The New England Journal of Medicine, Vol. 328, No. 14, 1993, pp. 1002-1006. doi:10.1056/NEJM199304083281404
- K. Seidemann, M. Tiemann, M. Schrappe, et al., “ShortPulse B-Non-Hodgkin Lymphoma-Type Chemotherapy Is Efficacious Treatment for Pediatric Anaplastic Large Cell Lymphoma: A Report of the Berlin-Frankfurt-Münster Group Trial NHL-BFM 90,” Blood, Vol. 97, No. 12, 2001, pp. 3699-3706. doi:10.1182/blood.V97.12.3699
- S. R. Park, J. Y. Baek, D. W. Kim, et al., “Primary Systemic Anaplastic Large Cell Lymphoma in a Single Korean Institution: Clinical Characteristics and Treatment Outcome,” Journal of Korean Medical Science, Vol. 21, No. 4, 2006, pp. 633-638. doi:10.3346/jkms.2006.21.4.633
- T. Mori, T. Takimoto, N. Katano, et al., “Recurrent Childhood Anaplastic Large Cell Lymphoma: A Retrospective Analysis of Registered Cases in Japan,” British Journal of Haematology, Vol. 132, No. 5, 2006, pp. 594-597. doi:10.1111/j.1365-2141.2005.05910.x
- Jagasia, M., Morgan, D., Goodman, S., et al. (2004) Histology impacts the outcome of peripheral T-cell lymphomas after high dose chemotherapy and stem cell transplant. Leukemia & Lymphoma, 45, 2261-2267.
NOTES
*Competing Interests: The authors have declared that no potential conflicts of interest.
Authors’ Contributions: Imane Ouafki is the principal author of this work. She made the initial drafting of the article and process integration of all corrections. Tanae Sghiri contributed to the correction the form and substance of the paper. Saber Boutayeb enriched the substance of this work. Mohamed Mouanis has provided the microscopic image. All authors have read and approved the final manuscript.