Open Journal of Pathology
Vol.3 No.2(2013), Article ID:29947,5 pages DOI:10.4236/ojpathology.2013.32011
Solitary Retinal Astrocytoma: A Case Series*
![]()
1Department of Pathology, Nagasaki University Hospital, Nagasaki, Japan; 2Department of Ophthalmology and Visual Sciences, Graduate School of Biomedical Sciences, Nagasaki University, Nagasaki, Japan.
Email: #toma@nagasaki-u.ac.jp
Copyright © 2013 Takafumi Fuchino et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received February 23rd, 2013; revised March 19th, 2013; accepted April 15th, 2013
Keywords: Retina; Astrocytoma; Pathology
ABSTRACT
Background: Astrocytic tumors of the retina are rare. We report and discuss the clinical features of two cases of retinal astrocytoma along with presenting a current literature review. Case Presentation: Case 1 was a 46-year-old Japanese female who became aware of her decreased visual acuity. A fundus photograph indicated the presence of a 5 mm hemispherical yellow-white tumor, retinal edema and hard exudate around the tumor. In case 2, a 36-year-old Japanese female became aware of her myodesopsia, and presented with a retinal tumor and vitreous hemorrhage in her right eye. Since the tumors occurred in the peripheral retina in both cases, endoresections were performed. Histological examination showed that the tumors were composed of spindle-shaped cells with small nuclei, which was consistent with astrocytes. Conclusion: Pathologically, it can be difficult to differentiate astrocytic tumors. Therefore, when making a diagnosis, it is important that comprehensive examinations be done in conjunction with the clinical findings. Since retinal astrocytoma has a favorable prognosis, provided proper treatment is administered, utilization of endoresection for peripheral astrocytoma may be advantageous in this patient group.
1. Introduction
A variety of benign and malignant tumors are known to involve the retina. The more common tumors include retinoblastoma, primary intraocular lymphoma and angiomatosis retinae [1]. However, astrocytic tumors of the retina are quite rare [2], with these tumors sometimes found to be solitary in patients without stigmata of phakomatosis. Conversely, multiple tumors are sometimes associated with tuberous sclerosis or neurofibromatosis patients [3,4].
When present, glial tumors primarily tend to be hamartomas in these phakomatoses [3]. In one review, 71% of the astrocytic tumors occurred in patients with tuberous sclerosis (57%) or neurofibromatosis (14%), while the remaining 29% of the tumors occurred in patients without either tuberous sclerosis or neurofibromatosis [4]. Here, we report and discuss the clinical features of two cases of retinal astrocytoma along with presenting a current literature review.
2. Case Report
2.1. Case 1
A 46-year-old Japanese female with no systemic disease or any significant medical or family history became aware of decreased visual acuity in her eyes. The patient stated that while she had no visual distortion, she felt the presence of a foreign body in addition to experiencing eye fatigue. Epiretinal membrane (ERM) and cataract were found in her right eye. At the time of admission, visual acuity was 20/200 and 20/30 in her right and left eyes, respectively. A fundus photograph indicated there was a 5 mm hemispherical yellow-white tumor arising from the inferior peripheral fundus that was accompanied by retinal edema and hard exudate around the tumor (Figure 1). The tumor appeared to be a hemangioma of the retina rather than a metastatic tumor.
Fluorescein angiography found hyperfluorescence during the arterial phase and leakage in the late-phase, suggesting there was a diffuse increased permeability of the blood vessels of the retina. The patient underwent vitreous surgery for ERM, and after performing endo-
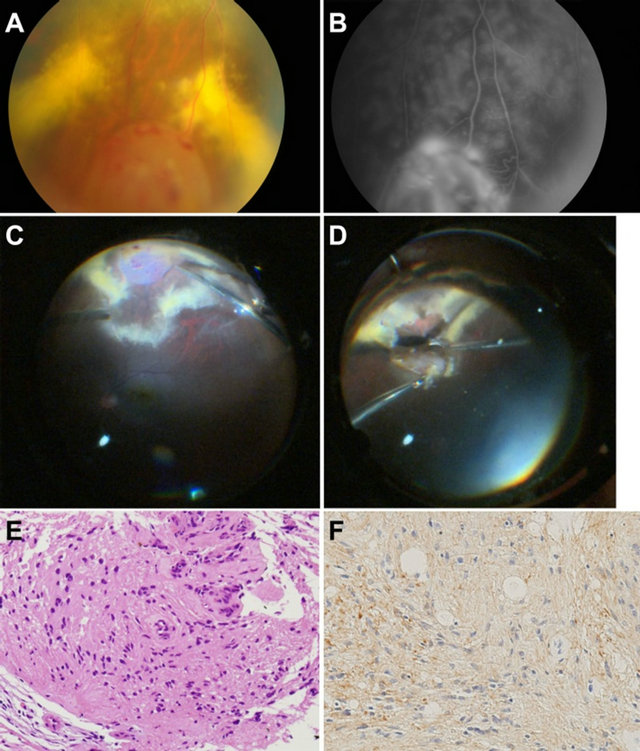
Figure 1. (A) Fundus photograph. A yellow-white mass is arising from the inferior peripheral fundus, and there is a hard exudate surrounding the tumor; (B) Fluorescein angiogram. The hyperfluorescence supports the diagnosis of a tumor; (C) Operative findings. Photo taken after the vitrectomy and ERM removal; (D) Operative findings. Resection of the tumor using a bimanual technique; (E) Hematoxylin-eosin, ×200. Histopathologic analysis shows spindle-shaped cells that have fibrillar cytoplasm with small nuclei; (F) GFAP × 200. Positive staining of the cells for GFAP is seen.
photocoagulation around the retinal tumor, the tumor was then completely resected.
Histological examination showed that the tumor was composed of spindle-shaped cells that had short, spindleshaped, densely-stained nuclei and fibrillary eosinophilic cytoplasm. Cell borders were indistinct, with the cells forming vague fascicular patterns. No calcification, mitoses or necrosis were noted. While hyalinization in the vascular wall was seen, the cells were negative for amyloid. Since immunohistochemistry staining of the tumor cells was positive for glial fibrillary acidic protein (GFAP), we diagnosed astrocytoma of the retina. At 3 months after the surgery, no improvement of the visual acuity was noted. However, there was no recurrence of the tumor in the right retina.
2.2. Case 2
A 36-year-old Japanese female without systemic disease or any significant medical or family history became aware of her myodesopsia. At the initial examination, the patient was found to have a retinal tumor and vitreous hemorrhage in her right eye. Upon admission, visual acuity was 20/800 and 20/80 in her right and left eyes, respectively.
During follow-up for her vitreous hemorrhage, fundus examination showed membrane tissue proliferation and retinal traction around the tumor, and there was growth of the tumor and vitreous opacity (Figures 2(A) and (B)). To treat the symptoms, the patient underwent surgery.
A 5 mm tumor was located inferior to the ora serrata and the tumor bleeding. After achieving coagulation around the tumor by using diathermy, the tumor was completely resected, followed by photocoagulation and cryocoagulation. Although severe intraocular inflammation and deposition of fibrin initially occurred after the surgery, these gradually disappeared and the patient’s visual acuity recovered to 20/160 in her right eye at the 6-year follow-up. Subsequently, the patient has had a favorable outcome and there has been no recurrence of the tumor.
Histological examination showed that the tumor was composed of spindle-shaped cells, which had short nuclei that were either round or spindle-shaped. These cells also had fibrillary, eosinophilic cytoplasm that formed small cystic cavities. While some vessels were dilated due to hyalinization (Figure 2(C)), there was no calcification, mitoses or necrosis noted. Immunohistochemistry staining showed the tumor cells were positive for GFAP and S-100, and negative for AE1/AE3 (Figure 2(D)). Based on these findings, the patient was diagnosed with astrocytoma of the retina.
3. Discussion
Retinal astrocytic tumors are considered to be rare [2], and when found, they are classified as massive retinal gliosis (MRG), astrocytic hamartoma, or acquired
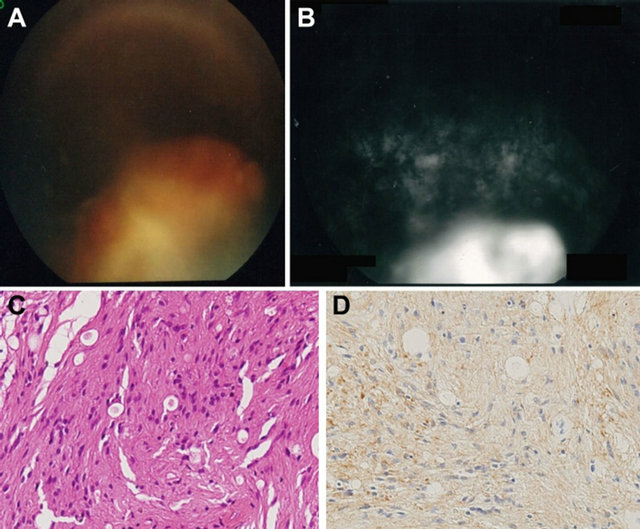
Figure 2. (A) Fundus photograph. A yellow-white tumor is seen arising from the inferior peripheral fundus; (B) Fluorescein angiogram. The hyperfluorescence supports the diagnosis for a tumor; (C) Hematoxylin-eosin × 200. Histopathologic analysis shows spindle-shaped cells with fibrillar cytoplasm with small nuclei. These cells form small cystic cavities; (D) GFAP × 200. Positive staining of the cells for GFAP is seen.
astrocytoma [2]. Since it is impossible to distinguish the tumor pathologically, the clinical features play an important role in the diagnosis. MRG is a non-neoplastic tissue that occurs in response to eye disorders such as inflammation, trauma or infection [5]. The presence of MRG pathology indicates previous retinal inflammation, retinal pigment epithelial proliferation and calcification [2,5].
Astrocytic hamartoma is a benign, stable tumor. While the tumor can be either solitary or multiple, it is most often associated with tuberous sclerosis or neurofibromatosis [2]. The hamartoma contains giant astrocytes that are often nonreactive or weakly positive for GFAP, and there is a high frequency of calcification [6]. Acquired retinal astrocytoma is a benign tumor that occurs sporadically and which is unassociated with tuberous sclerosis or neurofibromatosis [2].
In the current cases, there was no association found with systemic tuberous sclerosis or neurofibromatosis, and neither of the patients had any previous medical histories of eye disorders. Since the histopathology for both patients indicated there was neither calcification nor giant cells present, we diagnosed the tumors as acquired retinal astrocytoma.
While MRG and astrocytic hamartoma tend to occur in the peripheral retina, acquired retinal astrocytoma generally arises from the optic disc or retinal juxtapapillary area [5,7,8]. In the current patients, the tumor in case 1 arose from the periphery of the retina, while in case 2, it arose from the ora serrata. Since onlyone of the patients had solitary acquired retinal astrocytoma located at the periphery of the retina [8] (Table 1), the present cases
Table 1. Review of published cases of retinal astrocytoma not associated with tuberous sclerosis or neurofibromatosis.
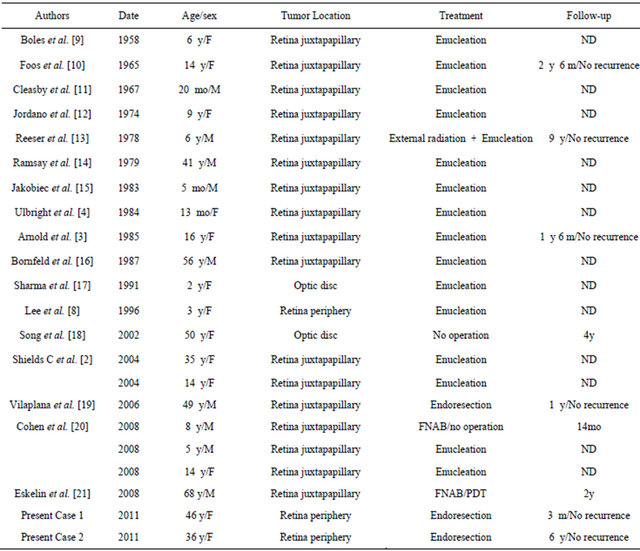
F = female, M = male, ND = no data, as the follow-up was not mentioned in the report. The dates in this table were collected from the papers published in English in Pubmed.
were viewed as being extraordinarily rare.
Differentiating acquired retinal astrocytoma from retinoblastoma, choroidal melanoma or other malignant tumors can be clinically difficult to perform. Previous studies have reported performing enucleation of the eye after a presumptive diagnosis for retinoblastoma [3,4,11, 15] or choroidal melanoma [2,16]. Retinal traction, retinal pigment epithelial alterations, and extensive yellow retinal exudation are typically found with astrocytic tumor but not with retinoblastoma [2]. Additionally, astrocytic tumor is totally amelanotic, produces yellow exudation, and is located in the sensory retina rather than the choroid, which is in direct contrast to that seen for choroidal melanoma [6]. Overall, these findings suggest that acquired retinal astrocytoma should be able to be differentiated from intraocular malignancy, and especially from retinoblastoma and choroidal melanoma.
There are two different types of acquired retinal astrocytoma, with one being progressive while the other is stationary [3]. The former causes intraocular damage that includes exudative retinal detachment, neovascular glaucoma, central retinal vein occlusion and tumor necrosis with secondary intraocular inflammation, which ultimately results in a blind and painful eye [3,20].
Until the 21st century, enucleation was normally performed in patients with retinal astrocytoma [3,13]. However, eyeball preserving therapies such as brachytherapy [22], endoresection [19], and photodynamic therapy (PDT) [21], have been recently introduced. Unfortunately, all of these therapies are associated with some risk, with potential visual acuity damage occurring due to the radiation used during brachytherapy [21]. When PDT is utilized, the tumor may not be completely removed [21], while with endoresection, there is a risk of a defect occurring in the visual area [21]. In the present study, however, the procedure was successfully used to treat the peripheral tumor in both of our cases.
4. Conclusion
We reported two cases of peripherally located solitary acquired retinal astrocytoma that did not involve the optic disc. While it may normally be difficult to differentiate astrocytic tumors pathologically, comprehensive examinations that take into account the clinical findings, along with the medical histories and the type of complications, can assist in making the correct diagnosis. Since the administration of proper treatment leads to a favorable prognosis for retinal astrocytoma, this suggests that the utilization of endoresection for peripheral astrocytoma may provide distinct advantages in these types of patients.
5. Ethics and Conflict of Interest
We prepared the manuscript not to be identified the patient’s personal information. This case report was approved by the Institutional Review Board of Nagasaki University Hospital.
REFERENCES
- R. J. Font, J. O. Croxatto and N. A. Rao, “AFIP Atlas of Tumor Pathology Series 4, Tumors of the Eye and Ocular Adnexa,” American Registry of Pathology, Washington DC, 2006.
- C. L. Shields, J. A. Shields, R. C. Eagle Jr. and F. Cangemi, “Progressive Enlargement of Acquired Retinal Astrocytoma in 2 Cases,” Ophthalmology, Vol. 111, No. 2, 2004, pp. 363-368. doi:10.1016/j.ophtha.2003.05.009
- A. C. Arnold, R. S. Hepler, R. W. Yee, J. Maggiano, L. F. Eng and R. Y. Foos, “Solitary Retinal Astrocytoma,” Survey of Ophthalmology, Vol. 30, No. 3, 1985, pp. 173-181. doi:10.1016/0039-6257(85)90061-X
- T. M. Ulbright, K. H. Fulling and E. M. Helveston, “Astrocytic Tumors of the Retina. Differentiation of Sporadic Tumors from Phakomatosis-Associated Tumors,” Archives of Pathology & Laboratory Medicine, Vol. 108, No. 2, 1984, pp. 160-163.
- S. D. Deshmukh, A. V. Ashturkar, S. V. Babanagare, S. K. Gokhale and A. A. Deshpande, “Massive Retinal Gliosis: An Unusual Case with Immunohistochemical Study,” Indian Journal of Ophthalmology, Vol. 59, No. 3, 2011, pp. 246-248. doi:10.4103/0301-4738.81050
- J. A. Shields, R. C. Eagle Jr., C. L. Shields and B. P. Marr, “Aggressive Retinal Astrocytomas in Four Patients with Tuberous Sclerosis Complex,” Transactions of the American Ophthalmological Society, Vol. 102, 2004, pp. 139- 147, Discussion 147-148.
- E. de Juan Jr., W. R. Green, P. K. Gupta and E. C. Barañano, “Vitreous Seeding by Retinal Astrocytic Hamartoma in a Patient with Tuberous Sclerosis,” Retina, Vol. 4, No. 2, 1984, pp. 100-102. doi:10.1097/00006982-198400420-00005
- J. A. Lee, P. Harvey, R. D. Finlay and P. J. Berry, “Solitary Astrocytoma of the Retina in a Child,” British Journal of Ophthalmology, Vol. 80, 1996, pp. 674-675. doi:10.1136/bjo.80.7.674-a
- W. M. Boles, T. C. Naugle and C. L. M. Samson, “Glioma of the Optic Nerve,” Archives of Ophthalmology, Vol. 59, No. 2, 1958, pp. 229-231. doi:10.1001/archopht.1958.00940030097008
- R. Y. Foos, B. R. Straatsma and R. A. Allen, “Astrocytoma of the Optic Nerve Head,” Archives of Ophthalmology, Vol. 74, No. 3, 1965, pp. 319-326. doi:10.1001/archopht.1965.00970040321006
- G. W. Cleasby, W. E. Fung and W. B. Shekter, “Astrocytoma of the Retina. Report of Two Cases,” American Journal of Ophthalmology, Vol. 64, No. 3, 1967, pp. 633-637.
- J. Jordano, H. Galera, M. Toro and B. Carreras, “Astrocytoma of the Retina. Report of a Case,” British Journal of Ophthalmology, Vol. 58, No. 5, 1974, pp. 555-559. doi:10.1136/bjo.58.5.555
- F. H. Reeser, T. M. Aaberg and D. L. Van Horn, “Astrocytichamartoma of the Retina Not Associated with Tuberous Sclerosis,” American Journal of Ophthalmology, Vol. 86, No. 5, 1978, pp. 688-698.
- R. C. Ramsay, J. L. Kinyoun, C. W. Hill, U. P. Aturaliya and W. H. Knobloch, “Retinal Astrocytoma,” American Journal of Ophthalmology, Vol. 88, No. 1, 1979, pp. 32- 36.
- F. A. Jakobiec, S. E. Brodie, B. Haik and T. Iwamoto, “Giant Cell Astrocytoma of the Retina. A Tumor of Possible Mueller Cell Origin,” Ophthalmology, Vol. 90, No. 12, 1983, pp. 1565-1576.
- N. Bornfeld, E. P. Messmer, G. Theodossiadis, G. MeyerSchwickerath and A. Wessing, “Giant Cell Astrocytoma of the Retina. Clinicopathologic Report of a Case Not Associated with Bourneville’s Disease”, Retina, Vol. 7, No. 3, 1987, pp. 183-189. doi:10.1097/00006982-198700730-00009
- A. Sharma, J. Ram and A. Gupta, “Solitary Retinal Astrocytoma,” Acta Ophthalmologica (Copenhagen), Vol. 69, No. 1, 1991, pp. 113-116. doi:10.1111/j.1755-3768.1991.tb02006.x
- S. Song and M. S. Seo, “Spontaneous Regression of a Solitary Astrocytoma of the Optic Disk,” Retina, Vol. 2, No. 4, 2002, pp. 502-503. doi:10.1097/00006982-200208000-00020
- D. Vilaplana, M. Castilla, V. Poposki, F. Alameda and C. L. Shields, “Acquired Retinal Astrocytoma Managed with Endoresection,” Retina, Vol. 26, No. 9, 2006, pp. 1081- 1082. doi:10.1097/01.iae.0000254888.63519.3c
- V. M. Cohen, C. L. Shields, M. Furuta and J. A. Shields, “Vitreous Seeding from Retinal Astrocytoma in Three Cases,” Retina, Vol. 28, No. 6, 2008, pp. 884-888. doi:10.1097/IAE.0b013e3181669781
- S. Eskelin, P. Tommila, T. Palosaari and T. Kivelä, “Photodynamic Therapy with Verteporfin to Induce Regression of Aggressive Retinal Astrocytomas,” Acta Ophthalmologica, Vol. 86, No. 7, 2008, pp. 794-799. http://onlinelibrary.wiley.com/doi/10.1111/j.1755-3768.2007.01151.x/pdf doi:10.1111/j.1755-3768.2007.01151.x
- S. R. Drummond and E. G. Kemp, “Retinal Astrocytoma Managed by Brachytherapy,” Ophthalmology, Vol. 116, No. 3, 2009, pp. 597-597. doi:10.1016/j.ophtha.2008.12.008
NOTES
*The authors declare that they have no competing interests.
#Corresponding author.