Open Journal of Medicinal Chemistry
Vol.2 No.4(2012), Article ID:25626,8 pages DOI:10.4236/ojmc.2012.24014
Recent Development in Thrombin Receptor Antagonist as Novel Antithrombotic Agent
School of Environmental and Municipal Engineering, Qingdao Technological University, Qingdao, China. Email: *yanxia08820@yahoo.com
Received October 31, 2012; revised November 14, 2012; accepted November 16, 2012
Keywords: Thrombin Receptor Antagonist; PAR-1 Antagonist; Vorapaxar; Antithrombotic; Review
ABSTRACT
Significant progress was achieved in the search of a thrombin receptor antagonist as a novel antithrombotic treatment since the thrombin receptor (protease-activated receptor-1, PAR-1) was cloned 20 years ago. Previous works have shown that it is possible to develop potent thrombin receptor antagonists to compete effectively with the receptor’s internal “tethered” ligand to block platelet activation. Vorapaxar (SCH 530348) from Schering-Plough (now Merck) and atopaxar (E5555) from Eisai have been advanced to human clinical trials. Recently, the pivotal phase III clinical trial results for vorapaxar were published. In this article we review these results plus the phase II results from atopaxar. Several newly described thrombin receptor antagonists from the literature will also be discussed. The phase III results from vorapaxar demonstrated that a thrombin receptor antagonist can achieve efficacy in addition to current standardof-care in treating atherothrombotic patients, especially those with previous myocardial infarction (MI). However, the increased moderate and severe bleeding, especially intracranial bleeding, point to the limitations of current thrombin receptor antagonists. Future thrombin receptor antagonists that can improve on the efficacy and bleeding profile of current ones should have a promising place in meeting the unmet medical need in treating atherothrombotic patients using current standard therapy.
1. Introduction
Thrombin is well known as the enzyme for blood coagulation and plays a key role for the formation of thrombus (clot) at the site of injury to prevent blood loss. Its normal function is essential for hemostasis and human health. However, under pathological conditions such as atherosclerosis, thrombin’s over-activity triggered by plaque rupture often results in arterial or venous thrombosis which can lead to serious diseases such as myocardial infarction (MI), unstable angina, or stroke. Thrombin’s importance for thrombosis consists of two aspects (Figure 1) [1,2]. First, it plays a central role in the coagulation cascade in both the intrinsic and extrinsic pathways to catalyze the cleavage of fibrinogen to form fibrin. These fibrin fibers then cross link and trap platelet aggregates and other particles to form the blood clot (thrombus). Secondly, thrombin activates the quiescent or resting platelets through receptors on the platelet surface. These activated platelets change shape, aggregate, and combine with fibrin fibers to strengthen and stabilize the blood clot1. Drugs blocking thrombin’s enzymatic action on fibrin formation either directly or indirectly have been used for decades as antithrombotic agents (e.g. heparins and hirudins). For these drugs, however, their therapeutic use was often hindered by the need to properly balance their antithrombotic activity and their bleeding side effects [3]. Thrombin’s second role on platelets was not explored in great detail until the early 1990s when the platelet thrombin receptor was cloned by Prof. Shaun Coughlin’s group at University of California at San Francisco [4]. Thrombin receptor belongs to the G-protein coupled receptors (GPCRs) super family and was discovered to have a novel mechanism of receptor activation. Thrombin activates the platelet receptor by enzymatically cleaving the extracellular N-terminal at the arginine 41 and serine 42 bond. The newly formed N-terminal binds intramolecularly to the receptor body initiating intracellular signal transduction which leads to platelet activation.
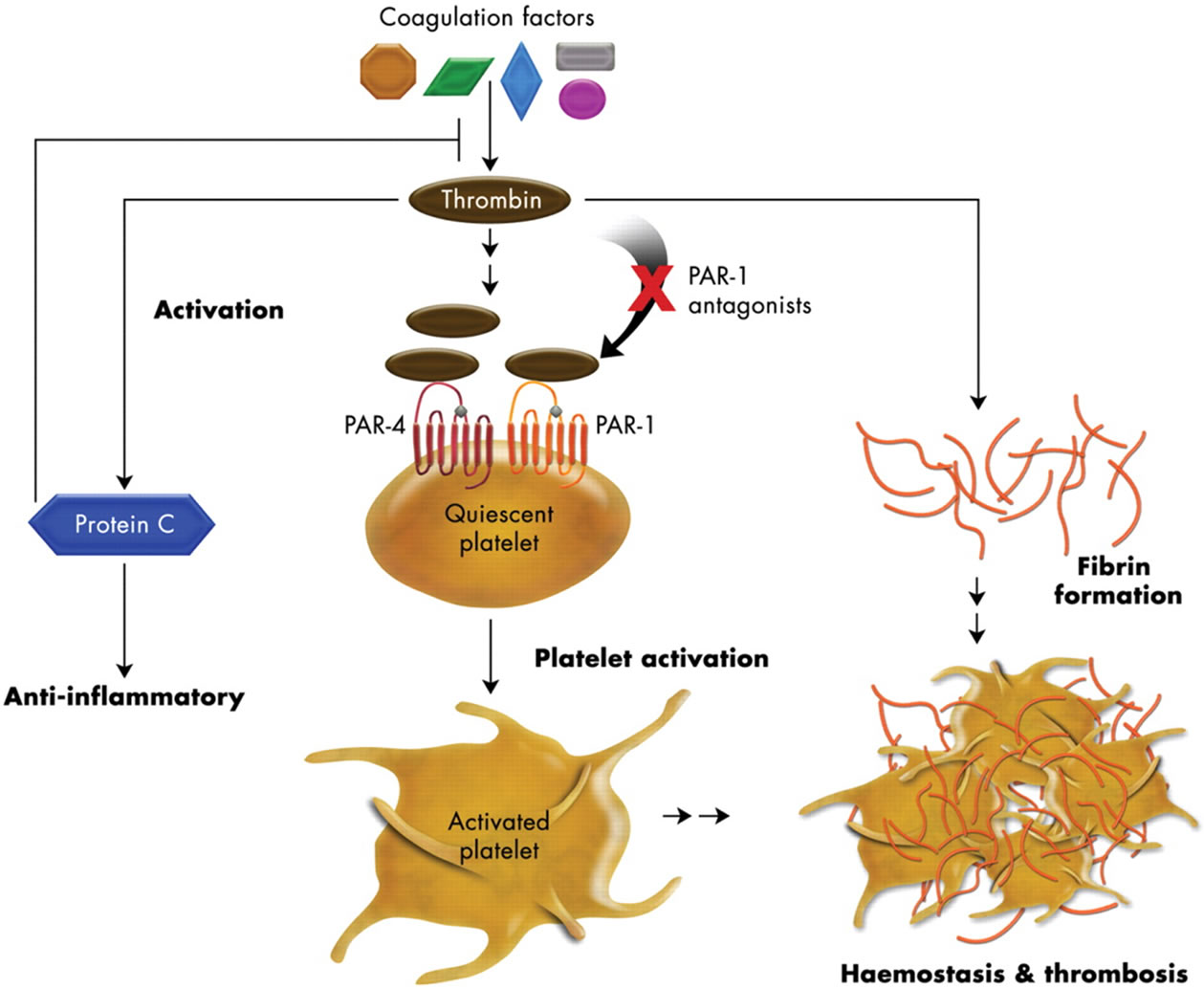
Figure 1. Role of thrombin in haemostasis and thrombosis. Thrombin catalyzes fibrin formation, activates PARs on platelets, and facilitates protein C activation. PAR antagonists block thrombin’s activation of platelets without affecting its activity on coagulation which is essential for haemostasis [1] (Copyright from the Oxford University Press, used with permission from the publisher).
The first six amino acid peptide (serine-phenylalanineleucine-leucine-arginine-asparagine or SFLLRN) itself is capable of activating the thrombin receptor although at a higher concentration than thrombin. It is named thrombin receptor activating peptide (TRAP) and used as a tool in in vitro and in vivo thrombin receptor activation studies. Thrombin receptor was the first example of such a protease activated receptor and was named protease-activated receptor-1 (PAR-1). Since the discovery of PAR-1, three more protease activated receptors, PAR-2, PAR-3, and PAR-4, were discovered. PAR-1 and PAR-4 are involved in human platelet aggregation whereas PAR-3 and PAR-4 are involved in rodent platelet aggregation. PAR-2 is activated by trypsin. It was reported that PAR-1 and PAR-3 are more important at low thrombin concentrations whereas the PAR-4 receptor plays a role at high thrombin concentrations [2].
PAR-1 generated great interests as a new target for antiplatelet drug discovery, especially in light of the success and wide application of other antiplatelet drugs such as aspirin and clopidogrel (Plavix). Since thrombin is the most potent activator of platelets, it was hoped that PAR-1 antagonists would achieve robust antiplatelet efficacy. Moreover, since PAR-1 antagonists do not affect thrombin’s enzymatic activity on fibrin formation and other mechanisms of platelet activation (such as through the ADP or thromboxane A2 receptors), it was hoped PAR-1 antagonists would have less bleeding side effects [5,6]. After overcoming early challenges in finding a good lead that can compete effectively against PAR-1’s internal ligand, two compounds have been advanced to human clinical trials. The thrombin receptor antagonist vorapaxar (SCH 530348) from ScheringPlough (now Merck) recently finished two large phase III trials. Another thrombin receptor antagonist atopaxar (E5555) from Eisai finished phase II trials. In this review, we examine these recent developments in the thrombin receptor antagonist drug discovery and development field in detail. We also discuss several new thrombin receptor antagonists that have appeared recently from the literature. These developments laid a good foundation in finding an efficacious and safe thrombin receptor antagonist to treat atherothrombotic patients.
2. Vorapaxar (SCH 530348)
Vorapaxar was discovered and developed from a natural product (himbacine) derived lead (Figure 2) [7]. It is a potent PAR-1 antagonist with a Ki of 8 nM. Besides, it has a slow dissociation from the thrombin receptor with a t1/2 of ca 20 h. This slow dissociation from the receptor may explain why it is an effective blocker of the receptor’s internal tethered ligand in a number of in vitro and in vivo assays. In human platelet-rich plasma, it inhibited thrombin (10 nM) induced platelet aggregation with an IC50 of 47 nM and high-affinity TRAP (15 μM) induced platelet aggregation with an IC50 of 25 nM. Vorapaxar
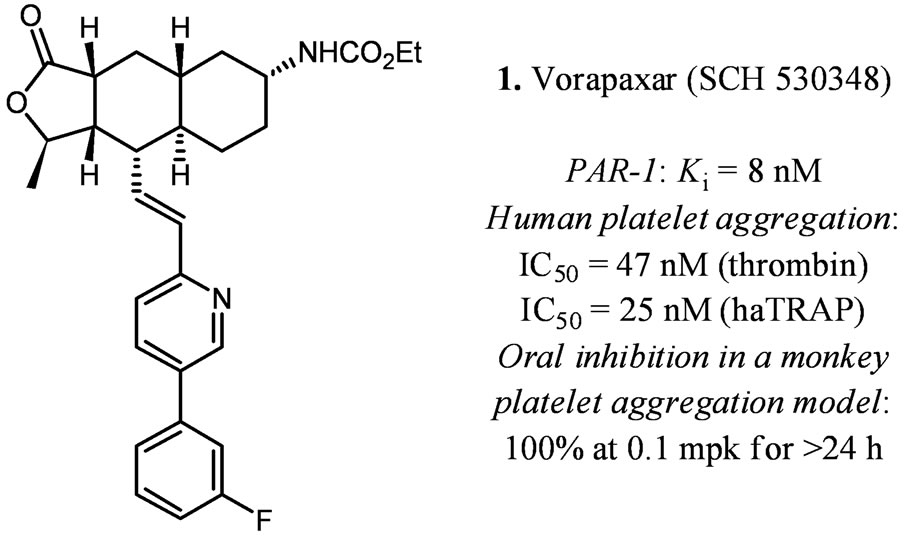
Figure 2. In vitro and in vivo data for vorapaxar [7].
showed no inhibition of platelet aggregation induced by ADP (20 μM), collagen (5 μM), U46619 (a thromboxane mimetic), or a PAR-4 agonist peptide. It also did not affect clotting parameters suggesting its activity is not from active site inhibition of thrombin.
In a dose-finding clinical trial, it was determined that a loading dose of 20 or 40 mg and a daily maintenance dose of ≥1 mg of vorapaxar were needed to achieve rapid (1 h) and long lasting inhibition (>80%) of platelet aggregation [8,9]. Vorapaxar underwent two phase II clinical trials to determine safety and tolerability [10,11]. The results were encouraging: at loading doses of 20 or 40 mg and daily maintenance doses of 0.5, 1.0, and 2.5 mg, vorapaxar did not show significant increase in major or minor bleeding by the thrombolysis in MI (TIMI) scale. In one phase II trial with Japanese patients [11], vorapaxar plus standard-of-care (aspirin, ticlopidine, and heparin) gave significant reduction of periprocedural MI compared to standard-of-care (16.9% vs. 42.9%, P = 0.013). Therefore, vorapaxar was progressed to two large phase III trials to evaluate efficacy: The Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRACER) trial [12] and the Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2˚P)-Thrombolysis in Myocardial Infarction (TIMI) 50 trial [13- 15].
The TRACER trial involved 12,944 patients with acute coronary syndromes without ST-segment elevation [12]. It was to compare vorapaxar in addition to standard-of-care (aspirin and a P2Y12 inhibitor) versus placebo which only received standard-of-care. The dosage for vorapaxar was a 40 mg loading dose and a daily maintenance dose of 2.5 mg thereafter. The primary efficacy end points are cardiovascular events such as death from cardiovascular causes, MI, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization. A planned interim analysis found no reason to discontinue the trial. However, an unplanned safety review ca six month thereafter resulted in the termination of the trial ca five months before the planned date. Even though the protocol-defined number of primary end points had been reached, vorapaxar did not show statistically significant reduction in cardiovascular events versus the placebo group. Moreover, the vorapaxar group had more moderate and severe bleeding than the placebo group (7.2% vs. 5.2%) and the result is statistically significant (P < 0.001). Intracranial bleeding was also significantly higher for the vorapaxar group vs. placebo (1.1% vs. 0.2%; P < 0.001).
The TRA 2˚P-TIMI 50 trial involved 26,449 patients with a history of MI, ischemic stroke, or peripheral arterial disease and compares vorapaxar (2.5 mg/day) with placebo in reducing cardiovascular death or ischemic events (Table 1) [13]. Both the vorapaxar group and the placebo group also received standard therapy (aspirin and thienopyridine). At 3 years, the vorapaxar group showed statistically significant decrease in cardiovascular death, MI, or stroke vs. the placebo group (1028 or 9.3% vs. 1176 or 10.5%; hazard ratio [HR] 0.87, 95% confidence interval [CI], 0.80 - 0.94; P < 0.001). The vorapaxar group also showed statistically significant decrease in cardiovascular death, MI, stroke, or recurrent ischemia leading to revascularization vs. the placebo group (1259 or 11.2% vs. 1417 or 12.4%; HR 0.88, 95% CI, 0.82 - 0.95; P = 0.001). Similar to the TRACER trial, vorapaxar produced more moderate or severe bleeding (4.2% vs. 2.5%; P < 0.001) and more intracranial hemorrhage (1.0% versus 0.5%; P < 0.001) than the placebo group. The TRA 2˚P trial result is encouraging in that it is the first time a thrombin receptor antagonist demonstrated statistically significant efficacy in a large phase III trial. However, the moderate or severe bleeding, especially intracranial bleeding, will likely limit vorapaxar’s potential therapeutic application. A subgroup analysis of the TRA 2˚P-TIMI 50 trial showed that for the subgroup of patients (17,779) with a history of previous MI (within 2 weeks to 12 months) vorapaxar (2.5 mg/day) significantly decreased cardiovascular death, MI, or stroke vs. the placebo group (610 or 8.1% vs. 750 or 9.7%; HR 0.80; 95% CI 0.72 - 0.89; P < 0.0001) after an average treatment of 2.5 years (Table 2) [14]. Vorapaxar produced more moderate or severe bleeding than the placebo group (241 or 3.4% vs. 151 or 2.1%; HR 1.61; 95% CI 1.31 - 1.97; P < 0.0001). However, the intracranial hemorrhage was not significantly different (43 or 0.6% versus 28 or 0.4%; P = 0.076). Therefore, vorapaxar appears to give better results for this subgroup of patients in that it demonstrated significant reduction (20%) on top of standard-of-care in cardiovascular death or ischaemic events without producing significantly more intracranial bleeding. It is interesting to note that vorapaxar compared to placebo seemed to reduce acute limb ischemia (2.3% vs. 3.9%, HR 0.58, 95% CI 0.39 - 0.86, P = 0.006) and peripheral arterial revascularizations (18.4% vs. 22.2%, HR
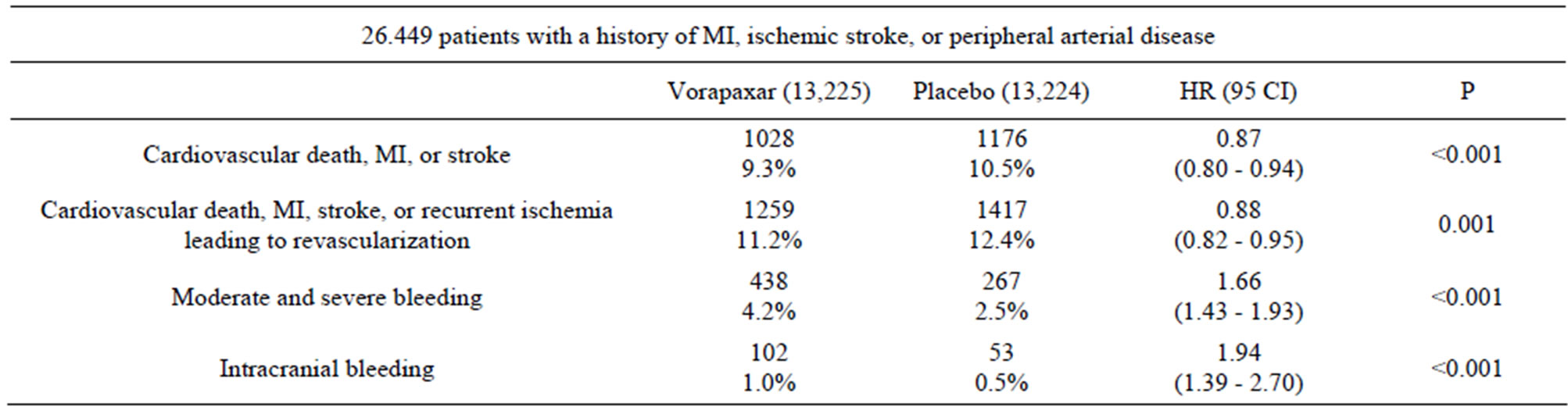
Table 1. Vorapaxar phase-III trial (TRA 2˚P-TIMI 50) [13].
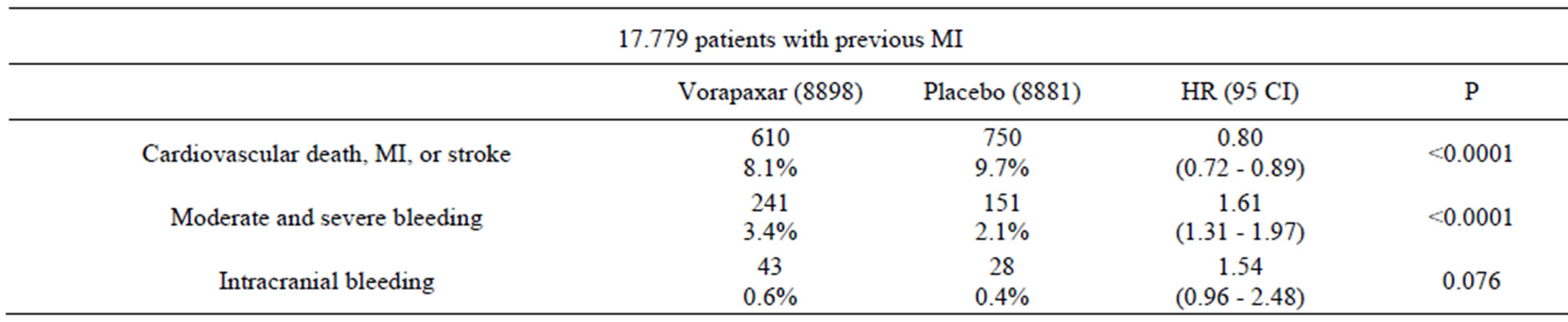
Table 2. Vorapaxar phase-III trial (TRA 2˚P-TIMI 50) subgroup analysis [14].
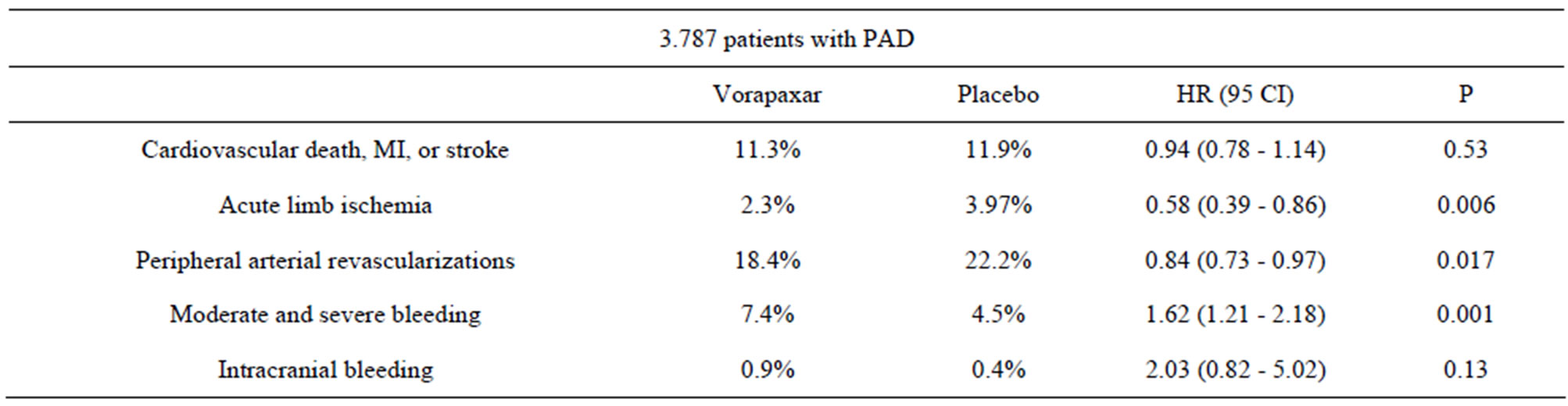
Table 3. Vorapaxar phase-III trial (TRA 2˚P-TIMI 50) PAD cohort [15].
0.84, 95% CI 0.73 - 0.97, P = 0.017) for the patient cohort (N = 3787) with peripheral arterial disease (PAD) (Table 3) [15]. Although moderate or severe bleeding was also higher (7.4% vs. 4.5%, P = 0.001), the results are encouraging since currently there seemed to be no other therapy that has such efficacy and acute limb ischemia is a serious disease that often leads to limb amputations.
Based on the TRA 2˚P results, it is clear that a thrombin receptor antagonist can be beneficial to patients with prior MI, ischemic stroke, or peripheral arterial disease, especially with prior MI. However, care must be taken to monitor the moderate and severe bleedings, especially intracranial bleeding. In its update made on August 26, 2012, Merck stated that it is planning to file for FDA and European approvals for vorapaxar in 2013 as an indication for the prevention of cardiovascular events in patients with a history of heart attack (MI) and no history of TIA (transient ischemic attack) or stroke. The outcome of Merck’s NDA application will have important implications to the future of thrombin receptor antagonist drug discovery and development research.
Atopaxar (E5555)
Atopaxar (E5555) was developed at Eisai from a benzimidazoledione lead [16]. It has good in vitro and in vivo activites as shown in Figure 3. Atopaxar underwent two phase II trials to determine safety and effects on major adverse cardiac events (MACE, such as cardiovascular death, MI, stroke, or recurrent ischemia) [17,18]. Similar to vorapaxar’s phase II results, atopaxar did not produce statistically significant bleeding in phase II trials. There was a trend toward reducing MACE but the trial was not designed to demonstrate efficacy [17]. Atopaxar generated good inhibition of human platelet aggregation (90% at 100 mg and 200 mg, and 20% - 60% at 50 mg). In vitro studies of atopaxar in healthy volunteers and coronary artery disease (CAD) patients treated with aspirin with or without clopidogrel showed that atopaxar inhibited moderately but significantly platelet activity beyond PAR-1 blockade. The antiplatelet activity of aspirin, aspirin combination with clopidogrel seemed to be enhanced by atopaxar providing rationale for their synergistic use [19]. However, it was reported that hepatic abnormalities were noticed for atopaxar in a phase II trial [18]. Currently, no further development was known for atopaxar.
3. Other Thrombin Receptor Antagonists
The early thrombin receptor antagonists have been thoroughly reviewed previously [5,6]. Since then, several new and interesting thrombin receptor antagonists have emerged in the literature or patents (Figure 4). The Chinese pharmaceutical company Hengrui published a
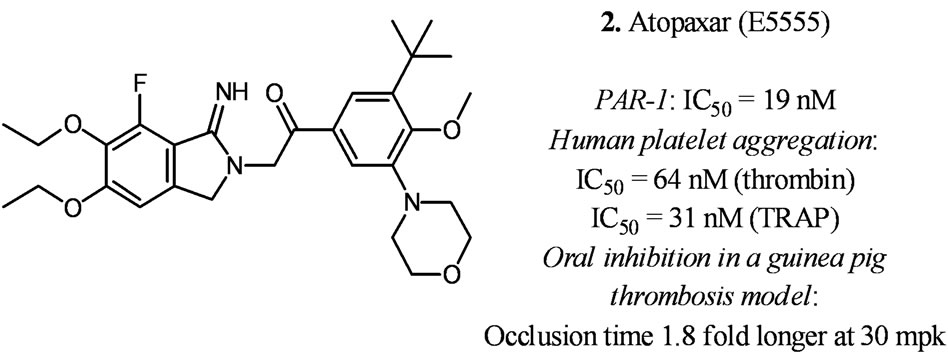
Figure 3. In vitro and in vivo data for atopaxar [16].
recent patent describing thrombin receptor antagonists similar to atopaxar as exemplified by structure 3 in Figure 4 [20]. Besides good in vitro receptor binding and platelet aggregation inhibition activities, other claimed compounds were shown to have oral bioavailability in the rat and oral platelet inhibition activity in a guinea pig model. Bayer published several patents recently describing thrombin receptor antagonists with a piperidine scaffold as exemplified by 4 in Figure 4 [21]. Some of the claimed compounds were listed as having nanomolar IC50s at the thrombin receptor. The French research institute Pierre-Fabre described a series of thrombin receptor antagonists as exampled by F16618 (5, Figure 4) [22]. F16618 was described as having antiplatelet and antithrombotic activities in vivo [23,24] and active in reduceing arterial restenosis [25]. Researchers at the Broad institute of Harvard-MIT recently described a new series of thrombin receptor antagonists with a 1,3-diaminobenzene scaffold as exemplified by ML161 (6, Figure 4) [26]. It is interesting that these compounds appear to be allosteric modulators of the thrombin receptor which may have implications for different efficacy and bleeding profiles.
4. Conclusion
Twenty years after the cloning of the thrombin receptor,
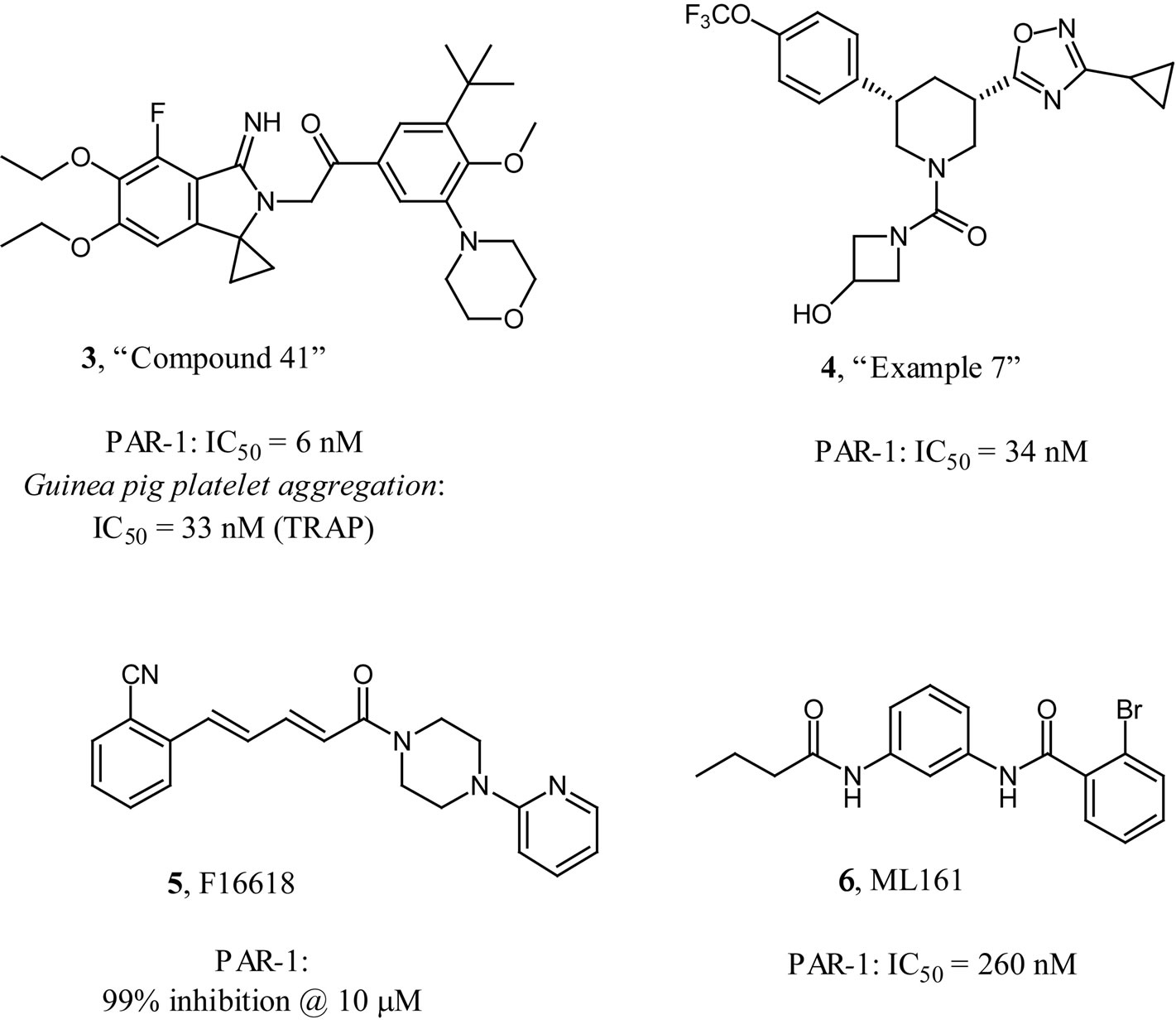
Figure 4. Other thrombin receptor antagonists.
tremendous amount of progress and knowledge were accumulated in using thrombin receptor antagonist as a novel antithrombotic agent to treat patients with cardiovascular disease such as MI, atherothrombosis, or peripheral vascular disease. One begins to have a clearer idea about the benefits and risks involved in using thrombin receptor antagonists. As with any type of medicines, future success is built on the improvements of efficacy and side effects of previous generation of drugs. The road to a successful thrombin receptor antagonist will undoubtedly remain a challenging one, as demonstrated by the clinical outcomes of vorapaxar and atopaxar. One needs to properly balance the efficacy and bleeding profile of the thrombin receptor antagonist besides the usual considerations in developing a new drug. However, the unmet medical need and the commercial market is huge. As researchers gather more experimental and clinical data around thrombin receptor antagonists, hopefully newer generations of thrombin receptor antagonists that are efficacious and less prone to cause bleeding, especially intracranial bleeding, will successfully make it to the medicine cabinet of cardiovascular patients worldwide.
5. Acknowledgements
We thank the Qingdao Technological University and the Shandong Taishan Scholar Administration for encouragement and financial supports.
REFERENCES
- L. D. J. Angiolillo, D. Capodanno and S. Goto, “Platelet Thrombin Receptor Antagonism and Atherothrombosis,” European Heart Journal, Vol. 31, No. 1, 2010, pp. 17-28. Hdoi:10.1093/eurheartj/ehp504
- S. R. Coughlin, “Protease-Activated Receptors in Hemostasis, Thrombosis and Vascular Biology,” Journal of Thrombosis and Haemostasis, Vol. 3, No. 8, 2005, pp. 1800- 1814. Hdoi:10.1111/j.1538-7836.2005.01377.x
- G. Bisacchi, “Anticoagulants, Antithrombotics, and Hemostatics,” In: D. J. Abraham, Ed., Burger’s Medicinal Chemistry and Drug Discovery, 6th Edition, John Wiley and Sons, Inc., Hoboken, 2003, pp. 283-338. Hdoi:10.1002/0471266949
- T. K. Vu, D. T. Hung, V. I. Wheaton and S. R. Coughlin, “Molecular Cloning of a Functional Thrombin Receptor Reveals a Novel Proteolytic Mechanism of Receptor Activation,” Cell, Vol. 64, No. 6, 1991, pp. 1057-1068. Hdoi:10.1016/0092-8674(91)90261-V
- S. Chackalamannil, “Thrombin Receptor (Protease Activated Receptor-1) Antagonists as Potent Antithrombotic Agents with Strong Antiplatelet Effects,” Journal of Medicinal Chemistry, Vol. 49, No. 18, 2006, pp. 5389-5403. Hdoi:10.1021/jm0603670
- S. Chackalamannil and Y. Xia, “Thrombin Receptor (PAR- 1) Antagonists as Novel Antithrombotic Agents,” Expert Opinion on Therapeutic Patents, Vol. 16, No. 4, 2006, pp. 493-505.
- S. Chackalamannil, Y. Wang, W. J. Greenlee, Z. Hu, Y. Xia, H. S. Ahn, et al. “Discovery of a Novel, Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) with Potent Antiplatelet Activity,” Journal of Medicinal Chemistry, Vol. 51, No. 11, 2008, pp. 3061- 3064. Hdoi:10.1021/jm800180e
- T. Kosoglou, L. Reyderman, R. Tiessen, R. R. Fales, D. L. Cutler, R. Keller, et al. “TRAP-Induced Platelet Aggregation Following Single and Multiple Rising Doses of SCH 530348, a Novel Thrombin Receptor Antagonist, in Healthy Volunteers (Abstract),” Clinical Pharmacology & Therapeutics, Vol. 85 Supplement 1, 2009, p. S21.
- Y. Abdulsattar, T. Ternas and D. Garcia, “Vorapaxar: Targeting a Novel Antiplatelet Pathway,” Pharmacy and Therapeutics, Vol. 36, No. 9, 2011, pp. 564-568.
- R. C. Becker, D. J. Moliterno, L. K. Jennings, et al., “Safety and Tolerability of SCH530348 in Patients Undergoing Non-Urgent Percutaneous Coronary Intervention: A Randomised, Double-Blind, Placebo Controlled Phase II study,” Lancet, Vol. 373, No. 9667, 2009, pp. 919-928. Hdoi:10.1016/S0140-6736(09)60230-0
- S. Goto, T. Yamaguchi, Y. Ikeda, et al., “Safety and Exploratory Efficacy of the Novel Thrombin Receptor (PAR- 1) Antagonist SCH530348 for Non-ST-Segment Elevation Acute Coronary Syndrome,” Journal of Atherosclerosis & Thrombosis, Vol. 17, No. 2, 2010, pp. 156-164. Hdoi:10.5551/jat.3038
- P. Tricoci, Z. Huang, C. Held, D. J. Moliterno, P. W. Armstrong, F. V. de Werf, et al., for the TRACER Investigators, “Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes,” The New England Journal of Medicine, Vol. 366, No. 1, 2012, pp. 20-33. Hdoi:10.1056/NEJMoa1109719
- D. A. Morrow, E. Braunwald, M. P. Bonaca, S. F. Ameriso, A. J. Dalby, M. P. Fish, et al., for the TRA 2P-TIMI 50 Steering Committee and Investigators, “Vorapaxar in the Secondary Prevention of Atherothrombotic Events,” The New England Journal of Medicine, Vol. 366, No. 15, 2012, pp. 1404-1413. Hdoi:10.1056/NEJMoa1200933
- B. M. Scirica, M. P. Bonaca, E. Braunwald, G. M. De Ferrari, D. Isaza, B. S. Lewis, et al., for the TRA 2°PTIMI 50 Steering Committee Investigators, “Vorapaxar for Secondary Prevention of Thrombotic Events for Patients with Previous Myocardial Infarction: A Prespecified Subgroup Analysis of the TRA 2°P-TIMI 50 trial,” Lancet, Vol. 380, No. 9850, 2012, pp. 1317-1324. Hdoi:10.1016/S0140-6736(12)61269-0
- M. P. Bonaca, D. A. Morrow and E. Braunwald, “Vorapaxar for Secondary Prevention in Patients with Peripheral Artery Disease: Results from the Peripheral Artery Disease Cohort of the TRA 2°P-TIMI 50 Trial,” Circulation, Vol. 126, No. 4, 2012, pp. 520-521. Hdoi:10.1161/CIR.0b013e3182611cc2
- M. Kogushi, T. Matsuoka, T. Kawata, H. Kuramochi, S. Kawaguchi, K. Murakami, et al., “The Novel and Orally Active Thrombin Receptor Antagonist E5555 (Atopaxar) Inhibits Arterial Thrombosis without Affecting Bleeding Time in Guinea Pigs,” European Journal of Pharmacology, Vol. 657, No. 1, 2011, pp. 131-137. Hdoi:10.1016/j.ejphar.2011.01.058
- M. L. O’Donoghue, D. L. Bhatt, S. D. Wiviott, S. G. Goodman, D. J. Fitzgerald, D. J. Angiolillo, S. Goto, et al., LANCELOT-ACS Investigators, “Safety and Tolerability of Atopaxar in the Treatment of Patients with Acute Coronary Syndromes: The Lessons from Antagonizing the Cellular Effects of Thrombin-Acute Coronary Syndromes Trial,” Circulation, Vol. 123, No. 17, 2011, pp. 1843- 1853. Hdoi:10.1161/CIRCULATIONAHA.110.000786
- S. Goto, H. Ogawa, M. Takeuchi, M. D. Flather and D. L. Bhatt, on Behalf of the J-LANCELOT (Japanese-Lesson from Antagonizing the Cellular Effect of Thrombin) Investigators, “Double-Blind, Placebo-Controlled Phase II Studies of the Protease-Activated 1 Antagonist E5555 (Atopaxar) in Japanese Patients with Acute Coronary Syndrome or High-Risk Coronary Disease,” European Heart Journal, Vol. 31, No. 21, 2010, pp. 2601-2613. Hdoi:10.1093/eurheartj/ehq320
- V. L. Serebruany, M. Kogushi, D. Dastros-Pitei, M. Flather and D. L. Bhatt, “The In-Vitro Effects of E5555, a Protease-Activated Receptor (PAR)-1 Antagonist, on Platelet Biomarkers in Healthy Volunteers and Patients with Coronary Artery Disease,” Journal of Thrombosis and Haemostasis, Vol. 102, No. 1, 2009, pp. 111-119.
- H. Lu, P. C. Tang, Y. Chen, S. Wang, H. Wang, L. Zhang and J. Li, “5,5-Disubstituted-2-imino-pyrrolidine Derivatives, Preparation Methods and Pharmaceutical Uses Thereof,” Patent Cooperation Treaty International Application, 2011, Publication No. WO/2011/140936.
- D. Heimbach, S. Roehrig, Y. Cancho Grande, D. Schneider, U. Rester, E. Bender, et al., “Substituted 3-(1,2,4-Oxadiazol-3-yl)-5-phenylpiperidines as PAR-1 Antagonists and Their Preparation and Use in the Treatment of Cardiovascular and Tumor Diseases,” Patent Cooperation Treaty International Application, 2010, Publication No. WO 2010/136127.
- M. Perez, M. Lamothe, C. Maraval, E. Mirabel, C. Loubat, B. Planty, et al., “Discovery of Novel Protease Activated Receptors 1 Antagonists with Potent Antithrombotic Activity in Vivo,” Journal of Medicinal Chemistry, Vol. 52, No. 19, 2009, pp. 5826-5836. Hdoi:10.1021/jm900553j
- R. Létienne, A. Leparq-Panissié, Y. Calmettes, F. NadalWollbold, M. Perez and B. Le Grand, “Antithrombotic Activity of F 16618, a New PAR1 Antagonist Evaluated in Extracorporeal Arterio-Venous Shunt in the Rat,” Biochemical Pharmacology, Vol. 79, No. 11, 2010, pp. 1616- 1621. Hdoi:10.1016/j.bcp.2010.02.006
- M. Dumas, F. Nadal-Wollbold, P. Gaussem, M. Perez, T. Mirault, R. Letienne, et al., “Antiplatelet and Antithrombotic Effect of F 16618, a New Thrombin Proteinase-Activated Receptor-1 (PAR1) Antagonist,” British Journal of Pharmacology, Vol. 165, No. 6, 2012, pp. 1827-1835. Hdoi:10.1111/j.1476-5381.2011.01668.x
- P. Chieng-Yane, A. Bocquet, R. Létienne, T. Bourbon, S. Sablayrolles, M. Perez, et al., “Protease-Activated Receptor-1 Antagonist F 16618 Reduces Arterial Restenosis by Down-Regulation of Tumor Necrosis Factor a and Matrix Metalloproteinase 7 Expression, Migration, and Proliferation of Vascular Smooth Muscle Cells,” Journal of Pharmacology and Experimental Therapeutics, Vol. 336, No. 3, 2011, pp. 643-651. Hdoi:10.1124/jpet.110.175182
- C. Dockendorff, O. Aisiku, L. VerPlank, J. R. Dilks, D. A. Smith, S. F. Gunnink, et al., “Discovery of 1,3-Diaminobenzenes as Selective Inhibitors of Platelet Activation at the PAR1 Receptor,” ACS Medicinal Chemistry Letters, Vol. 3, No. 3, 2012, pp. 232-237. Hdoi:10.1021/ml2002696
NOTES
*Corresponding author.
1Thrombin’s other roles such as activation of protein C are beyond the scope of this article.