Open Journal of Genetics
Vol.07 No.01(2017), Article ID:74603,11 pages
10.4236/ojgen.2017.71002
Ultra-Fast Next Generation Human Genome Sequencing Data Processing Using DRAGENTM Bio-IT Processor for Precision Medicine
Amit Goyal, Hyuk Jung Kwon, Kichan Lee, Reena Garg, Seon Young Yun, Yoon Hee Kim, Sunghoon Lee, Min Seob Lee*
EONE-DIAGNOMICS Genome Center Co. Ltd., Inchon, Korea

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: February 14, 2017; Accepted: March 6, 2017; Published: March 9, 2017
ABSTRACT
Slow speed of the Next-Generation sequencing data analysis, compared to the latest high throughput sequencers such as HiSeq X system, using the current industry standard genome analysis pipeline, has been the major factor of data backlog which limits the real-time use of genomic data for precision medicine. This study demonstrates the DRAGEN Bio-IT Processor as a potential candidate to remove the “Big Data Bottleneck”. DRAGENTM accomplished the variant calling, for ~40× coverage WGS data in as low as ~30 minutes using a single command, achieving the over 50-fold data analysis speed while maintaining the similar or better variant calling accuracy than the standard GATK Best Practices workflow. This systematic comparison provides the faster and efficient NGS data analysis alternative to NGS-based healthcare industries and research institutes to meet the requirement for precision medicine based healthcare.
Keywords:
NGS Data Analysis, BWA-GATK, DRAGEN Bio-IT Processor, Genomics, INDEL, Mapping

1. Introduction
With the emergence of the 2nd generation high throughput Next Generation Sequencing (NGS) platforms as well as accurate and consistent identification of the genomic variants, the use of the personal genome sequencing information for the diagnostic and prognostic purpose has become the reality [1] [2] . Furthermore, fast sequencing turnaround time and roughly $1000 NGS whole genome cost is encouraging more institutes and individuals to opt for NGS based personalized medicine [3] - [8] . However, the “Big Data Bottleneck” is still the largest obstacle to use the NGS-based precision medicine in the real time disease and healthcare management. For instance, high throughput NGS HiSeq X Ten System has around 18,000 humans’ whole genome sequencing capacity at 30× genome coverage annually which translates into just ~30 - 40-minute turnaround time for each genome sequencing. The most commonly used Genome Analysis Toolkit (GATK) best practice pipelines requires several hours to several days to analyze one human whole genome sequencing data, depending on the available processors. At commercial level, NGS-based data analysis time can be reduced significantly using the clusters of hundreds or thousands of CPUs. Also, several cloud-based solutions, such as GenomePilot by Appistry [9] , etc., to accelerate NGS-data analysis platform to speed-up the analysis has been introduced. However, this conventional cluster approach requires expensive computer system, maintenance and monitoring. Similarly, cloud-based platforms require massive data upload and download which is a limitation/burden for many research institutes and small to medium scale companies, especially in low bandwidth supported countries. Overall, the data processing strategy is not suitable for real time guidance/management of many medical diseases/condi- tions such as tolerance and rejection monitoring in organ transplant recipients, etc. Considering the routine use of the NGS-based diagnostic and prognostic in clinical setting, the need for the fast turnaround time, easy operation and accurate NGS-based data analysis platform has become prominent.
In this study, we assessed the performance of the world’s first bioinformatics processor DRAGEN Bio-IT Processor [10] [11] which is designed to analyze the NGS data. The DRAGEN (Dynamic Read Analysis for Genomics) Processor uses a field-programmable gate array (FPGA), implemented on a PCIe card embedded in a pre-configured server, to provide hardware-accelerated implementations of genome pipeline algorithms, such as BCL conversion, compression, mapping, alignment, sorting, duplicate marking and haplotype variant calling.
This study was carried out in two steps. First, run time performance of the DRAGEN Bio-IT Genome pipelines with the most commonly used GATK’s best-practice guidelines were analyzed for the 2 replicates of NA12878 Whole Genome Sequencing (WGS) dataset. Second, the variant calling efficiencies of the two pipelines were evaluated by comparing variants with the GIABv2.19 high confidence (truth) call-set [12] [13] . These studies demonstrate that the employment of the DRAGEN Bio-IT processor decreased the WGS NGS-data analysis time to just ~40 minute while achieving the equivalent or better genotype variant calling accuracy than the standard GATK Best Practices workflow.
2. Methods
2.1. Sequence Data-Set and GIAB Validation Call-Set
Two WGS replicates of the Coriell Cell Repository NA12878 reference sample NA12878 were downloaded from the Garvan NA12878 HiSeqX datasets [18] . These datasets have been sequenced on the Illumina HiSeq X platform using the Illumina’s TruSeq Nano kit using 350 bp inserts. Each dataset contains over 120 GB of fastq data yield, with > 87% bases with quality > Q30. These replicates are sequenced to assess the reproducibility and has been provided freely for research purpose by the The Garvan Institute of Medical Research, DNA nexus and AllSeq.
As a gold standard practice to validate the variant calling platform’s performance, the high confidence reference variant calls for the 1000 Genome project individual (sample NA12878), published by the Genome in a Bottle (GIAB) consortium [12] using hg19 coordinates, were utilized. The highly confident variant call-set in the Variant Call Format (NISTIntegratedCalls_14datasets_ 131103_allcall_UGHapMerge_HetHomVarPASS_VQSRv2.19_2mindatasets_5minYesNoRatio_all_nouncert_excludesimplerep_excludesegdups_excludedecoy_ excludeRepSeqSTRs_noCNVs.vcf.gz, GIAB v2.19) as well as the high confidence genomic region file (union13callableMQonlymerged_addcert_nouncert_exclu- desimplerep_excludesegdups_excludedecoy_excludeRepSeqSTRs_noCNVs_ v2.19_2mindatasets_5minYesNoRatio.bed.gz) were downloaded for the validation purpose.
2.2. GATK Best Practices Workflow
GATK Best Practices workflow is used most commonly to analyze the genomic data. The complete best practice pipeline [19] can be basically divided into two phase. First, preprocessing the raw data which includes, alignment the raw fastq data to the hg19 reference genome using mapping by BWA (version 0.7.12- r1039) [20] , sorting by samtools (version 1.2 using htslib 1.2.1) [21] , MarkDuplicate and addRG by steps using picard-tools (version 1.119), and Base Recalibration using GATK (version 3.6-0-g89b7209) [19] . Second, Variants calling using GATK HaplotypeCaller. This study followed the GATK best practice work- flow recommended commands and arguments at each step which were executed on 48 core (using-nt and -nct arguments) the Intel Xeon E5-2697v2 12C server with 2.7 GHz processors,128 GB RAM and 3.2 TB capacity SSD running on CentOS 6.6.
2.3. DRAGEN Bio-IT Processor and DRAGEN Genome Pipelines
Unlike the traditional Genome analysis workflows, DRAGEN Bio-IT processor is the hardware accelerated platform which comes equipped with a custom Peripheral Component Interconnect Express (PCIe) board with a field-program- mable gate array (FPGA) which has been bundled with two 24 core Intel Xeon E5-2697v2 12C, 2.7 GHz processors with 128 GB RAM and 3.2 TB capacity SSD running on CentOS 6.6. DRAGEN system is supplied with DRAGEN Genome Pipeline which utilizes the DRAGEN Bio-It Platform with the improved and highly optimized mapping, aligning, sorting, duplicate marking and haplotype variant calling algorithms 11. A single DRAGEN run for WGS data, from fastq files to vcf files, can be completed in just one simple command. Also, DRAGEN can be run to output the intermediate alignment BAM file to be used with other variant caller (alignment mode) and vice versa. More details about the DRAGEN Bio-IT Platform and DRAGEN Genome Pipeline has been recently published recently by Miller NA et al. [10] [11] .
DRAGEN Command
“dragen--num-threads 48-r/path/to/reference_Dir/--output-directory/path/to/ Output_Dir/--output-file-prefix PREFIX-1 Sequence_R1.fastq-2 Sequence_ R2.fastq--enable-variant-caller true--vc-reference/path/to/hg19.fa--vc-sample-name SampleID--enable-duplicate-marking true--remove-duplicates true―enable-bam- indexing true--enable-map-align-output true--intermediate-results-dir/staging/ tmp”.
2.4. Performance Assessment of the Two Variant Calling Pipelines
This study utilized below mentioned two WGS data analysis pipelines to process the dataset consist of two replicate of the NA12878.
Pipeline 1. DRAGEN Alignment and DRAGEN Variant Caller (DRAGEN Genome Pipeline)
Pipeline 2 GATK Best Practices workflow (BWA alignment, BAM file preprocessing and HaplotypeCaller).
All the analyses were performed on the server equipped with two 48 core Intel Xeon E5-2697v2 12C, 2.7 GHz processors with 128 GB RAM and 3.2 TB capacity SSD running on CentOS 6.6. Variant called using the pipelines were compared with the GIAB variants truth-set. For WGS dataset, a subset of variants in the GIAB high confidence genomic region bed file was extracted for each pipeline and compared with the GIAB’s high confident variant call-set to assess the performance.
To draw receiver operating characteristic (ROC) curve and calculate the sensitivity and specificity of SNPs and INDELs, we defined the true positive (TP), true negative (TN), false positive (FP), and false negative (FN) variants as follows:
TP: Correctly called ALT genotype which is also listed in GIAB truth-set.
TN: Correctly called REF genotype which is also not listed in GIAB-truth-set
FP: Incorrectly called ALT genotype which is not listed in GIAB truth-set.
FN: Incorrectly missed ALT genotype which is listed in GIAB truth-set.
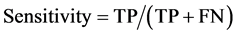 (1)
(1)
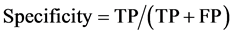 (2)
(2)
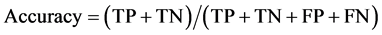 (3)
(3)
3. Results
3.1. Research Scheme
Figure 1 shows the research scheme to assess the variant calling pipelines performance for the whole genome sequencing data. This research employed two genome analysis pipelines, i.e. the DRAGEN genome pipelines and GATK Best Practice Pipelines, to assess the read-alignment and variant calling accuracy (as

Figure 1. Scheme of assessment of NGS data analysis pipelines. Flow chart shows the steps to assess the variant calling performance of the various pipelines using DRAGEN and GATK-best practices guidelines.
described in Method section). Two replicates of the NA12878 WGS sample, labelled as NA12878D and NA12878J18 with the coverage of 39.00× and 38.65× respectively, were used to assess the consistency and reproducibility of the variant calling workflows. GIAB high confidence truth-set, along with the high confidence genomic region bed file, was used to assessed the performance of the both variant calling pipelines and suggest the best pipeline to the NGS-based researcher. More details about the sequence dataset and validation call-set can be found in Method section.
3.2. Runtime Performance of the Genome Analysis Pipelines
One of the main object of this study is the Run-time assessment of the DRAGEN against the GATK Best Practice pipelines. Run time matrices were divided into, mapping time (i.e. fastq to Bam file generation time) and variant calling time (Bam to VCF generation). Table 1 lists the run time matrices for both the pipelines. DRAGEN alignment includes the sorting, duplicate marking, ReadGroup information adding, etc. GATK best practice pipeline includes the mapping by BWA and preprocessing by samtools, Picard-tools, GATK, etc. All the command utilized the multithreaded option with maximum of 48 core, except the Picard-tools which utilized single core.

Table 1. Performance comparison: run time assessment of variant calling pipelines.
Here, table lists the run time profile of the two pipelines measured on the 2 replicates of NA12878 WGS dataset. For each pipeline, run-time for individual step, i.e. Mapping/Alignment (FastQ2BAM), Variant Calling (Bam2VCF) and complete pipeline run (FastQ2VCF), is listed.
As listed in Table 1, DRAGEN competed alignment and BAM preprocessing for the NA12878D dataset in ~18 minute while GATK best practice pipeline took over 23 hours for the same (Figure 2). Likewise, variant calling using the GATK HaplotypeCaller completed in over 9 hours while DRAGEN Haplotype aware variant caller took just 23 minutes. All over, DRAGEN NGS data run was completed in ~37 minutes while the GATK tools over 32 hours. A similar run-time was obtained while analyzing the another WGS dataset (NA12878J). In a nutshell, around 50-fold NGS data processing speed can be obtained by utilized the DRAGEN Genome Pipeline as compared to the GATK best practice recommendations.
3.3. Variant Calling Accuracy of the WGS Variant Calling Pipelines
Variant calling accuracy of the two pipelines were assessed against the standard GIAB high confidence region truth-set (v2.19). For this, both the WGS data analysis pipelines were executed for the two replicates of NA12878 WGS data. For each pipeline, a high-confident subset of variants, in the GIAB high confidence genome region (bed file), were selected and compared with the GIAB truth-set to calculate the sensitivity, specificity and accuracy of each pipelines.
Both the pipelines showed ~99% and ~90% variant calling sensitivities for SNPs and INDELs, respectively while maintaining over 98% detection specificity in both the cases. As listed in Table 2 and shown in Figure 3, DRAGEN Genome pipelines showed slightly higher SNP detection accuracy than GATK best practice workflow. On the other hand, GATK best practice pipelines showed high INDEL detection accuracy for NA12878D dataset while DRAGEN pipeline for other (NA12878J) dataset. Further, as shown in Figure 4, ROC curve of SNPs and INDELs for DRAGEN genome pipeline showed high variant detection sensitivity at low False Positive Rate, but gradually with the in-crease of false positive hits the curve become similar (or lag behind) to that of the GATK HaplotypeCaller. Overall, DRAGEN Genome Pipelines showed the comparable or better variant calling accuracy than the GATK best practice workflow.

Figure 2. Genome analysis pipelines run-profile statistics. The figure shows the run-profile statistics for each steps of the NGS-data analysis, i.e. the Alignment step (FastQ2BAM), Variant Calling step (BAM2VCF) and total run-time (FastQ2VCF) for each dataset, for the two NGS data analysis pipelines in this study.

Figure 3. Variant calling performance assessment for WGS dataset. The figure shows the performance assessment of the genome analysis pipelines against the GIAB truth-set for the NA12878 sample. The Venn diagram shows the concordant SNPs (a) and (c) and the INDELs call (b) and (d) obtained by two pipelines against the GIAB truth-set.
4. Discussion
The major focus on the NGS-data analysis workflow is how to speed-up the analysis time without sacrificing the variant calling accuracy to utilize the NGS- based diagnosis more effectively, especially in a real-time disease management,
 (a) (b)
(a) (b) (c) (d)
(c) (d)
Figure 4. ROC curves of SNPs and INDELs for WGS dataset. ROC curves showing sensitivity vs. false positive rate for two replicates of the whole genome, (a) and (c) SNPs and (b) and (d) INDELs, for the NA12878 data set. Variant quality and true positive/false positive variants were identified as described in the Online Methods section.
Table 2. Performance comparison: accuracy of the variants calling pipelines.
Here, table lists the variant calling sensitivity and specificity profile of the 2 pipelines measured on the 2 replicated of NA12878 WGS dataset. For each pipeline, total number of SNP/INDEL, false positives, Sensitivity, Specificity and Accuracy is listed. Definition and formula of FP, sensitivity, specificity and accuracy is described in Method section. For SNP calling, DRAGEN Pipeline is shown to be more efficient than the BWA + HC pipelines. Similarly, DRAGEN pipeline shows comparable or better INDEL calling accuracy than GATK best practice workflow.
outbreaks of infectious disease and disaster situations, etc. This study assessed the analysis speed of sequencing data and variant calling accuracy of two genome analysis pipeline. The results showed that the ~40× coverage human WGS data processing using the DRAGEN Bio-IT Genome Pipelines can be completed in less than 40 minutes while obtaining the comparable accuracy with the standard GATK best practice workflow.
One of the main objects of this study is to identify the fast, accurate and efficient genomic analysis solution which can deal with the high computing demand in the era of massive NGS data analysis. Utilization of the DRAGEN Bio-IT processor with DRAGEN Genome Pipeline can provide an efficient solution to the “Big-data bottleneck” since it can complete the standard human whole genome sequencing data analysis (fastq to vcf) in less than 40 minutes. The DRAGEN system processing time is sufficient to support ~30 - 40 minutes sequencing time for a single WGS sample in currently available high throughput sequencer. Therefore, one DRAGEN-system is enough to analyze the raw data generated from the high throughput sequencing system such as Illumina HiSeq X 10 sequencing center.
In the recent time, several modifications of the GATK best practice pipelines have been published, e.g. Churchill [14] , SpeedSeq [15] , etc. Churchill pipeline claims to accomplish the 30× WGS sample in ~11 hours on a 48-core single CPU or ~1 hour 50 minutes on Ohio Supercomputer Center’s Glenn Cluster (768 cores over 96 nodes). Similarly, SpeedSeq claims 13-hour run-time for 50× NA12878 WGS using default software parameters and a single 16-core server (allowing 32 threads) with 128 GB of RAM. Even though, this study doesn’t compare the DRAGEN Genome pipeline’s speed and accuracy with such pipelines, but ~40 minutes WGS data analysis time is much less than above mentioned pipelines which makes the DRAGEN system highly promising at industrial scale.
One important observation in our study is that DRAGEN Genome Pipelines is highly sensitive at low False Positive Rate. As shown in ROC curve of SNPs and INDELs for WGS dataset in the Figure 4, with the increase in the variant calling sensitivity (over 92% for SNPs and over 80% for INDEL case) the false positive hits increased significantly which reduces the overall DRAGEN variant callers’ accuracy. For example, as shown in Table 2, NA12878D and NA12878J samples have 1% and 0.5% lower INDEL calling specificity than the GATK HaplotypeCaller, respectively. Accurate detection of INDEL from the NGS-data has been challenging due to the varying size and difficulty to map to the correct position in the genome (especially in the case of longer INDEL), etc. [16] [17] . These are the well-known issues which are caused by the technical limitation of NGS- platforms and analysis workflows. In the current study, we only compared the result of INDEL calling from the available resource/software without additional INDEL detection accuracy improvement.
Altogether, this study focused on demonstrating the proficiency and comparison of DRAGEN Bio-IT software and DRAGEN Genome Pipelines with traditional approaches. These results implicate that the DRAGEN system can be used as a single platform to analyze the genomic data accurately in quicker time at industrial scale. We expect, this research will help the scientist to make an informed choice to set-up a new (or modify the existing) genome analysis platform in their laboratory and/or institute.
Cite this paper
Goyal, A., Kwon, H.J., Lee, K., Garg, R., Yun, S.Y., Kim, Y.H., Lee, S. and Lee, M.S. (2017) Ultra-Fast Next Generation Human Genome Sequencing Data Processing Using DRAGENTM Bio-IT Processor for Precision Medicine. Open Jour- nal of Genetics, 7, 9-19. https://doi.org/10.4236/ojgen.2017.71002
References
- 1. Hayden, E.C. (2014) Technology: The $1,000 Genome. Nature, 507, 295-295.
- 2. Watson, M. (2014) Illuminating the Future of DNA Sequencing. Genome Biology, 15, 108-108.
https://doi.org/10.1186/gb4165 - 3. Petric, R.C., Pop, L.-A., Jurj, A., Raduly, L., Dumitrascu, D., Dragos, N. and Neagoe, I.B. (2015) Next Generation Sequencing Applications for Breast Cancer Research. Clujul Medical, 88, 278-287.
https://doi.org/10.15386/cjmed-486 - 4. George, A. (2015) UK BRCA Mutation Testing in Patients with Ovarian Cancer. British Journal of Cancer, 113, S17-S21.
https://doi.org/10.1038/bjc.2015.396 - 5. Vivante, A. and Hildebrandt, F. (2016) Exploring the Genetic Basis of Early-Onset Chronic Kidney Disease. Nature Reviews Nephrology, 12, 133-146.
https://doi.org/10.1038/nrneph.2015.205 - 6. Zutt, R., van Egmond, M.E., Elting, J.W., van Laar, P.J., Brouwer, O.F., Sival, D.A., Kremer, H.P., de Koning, T.J. and Tijssen, M.A. (2015) A Novel Diagnostic Approach to Patients with Myoclonus. Nature Reviews Neurology, 11, 687-697.
https://doi.org/10.1038/nrneurol.2015.198 - 7. Hoyle, J.C., Isfort, M.C., Roggenbuck, J., Arnold, D. and Hoyle, C. (2015) The Genetics of Charcot-Marie-Tooth Disease: Current Trends and Future Implications for Diagnosis and Management. Application of Clinical Genetics, 8, 235-243.
- 8. Ono, S., Lam, S., Nagahara, M. and Hoon, D. (2015) Circulating microRNA Biomarkers as Liquid Biopsy for Cancer Patients: Pros and Cons of Current Assays. Journal of Clinical Medicine, 4, 1890-1907.
https://doi.org/10.3390/jcm4101890 - 9. Appistry. AppistryGenomePilot&trade.
http://www.appistry.com/genomepilot/ - 10. DRAGEN. Edicogenome.
http://www.edicogenome.com/ - 11. Miller, N.A., Farrow, E.G., Gibson, M., Willig, L.K., Twist, G., Yoo, B., Marrs, T., Corder, S., Krivohlavek, L., Walter, A., Petrikin, J.E., Saunders, C.J., Thiffault, I., Soden, S.E., Smith, L.D., Dinwiddie, D.L., Herd, S., Cakici, J.A., Catreux, S., Ruehle, M. and Kingsmore, S.F. (2015) A 26-Hour System of Highly Sensitive Whole Genome Sequencing for Emergency Management of Genetic Diseases. Genome Medicine, 7, 100.
https://doi.org/10.1186/s13073-015-0221-8 - 12. Zook, J.M., Chapman, B., Wang, J., Mittelman, D., Hofmann, O., Hide, W. and Salit, M. (2014) Integrating Human Sequence Data Sets Provides a Resource of Benchmark SNP and Indel Genotype Calls. Nature Biotechnology, 32, 246-251.
https://doi.org/10.1038/nbt.2835 - 13. Hwang, S., Kim, E., Lee, I. and Marcotte, E.M. (2015) Systematic Comparison of Variant Calling Pipelines Using Gold Standard Personal Exome Variants. Scientific Reports, 5, 17875-17875.
https://doi.org/10.1038/srep17875 - 14. Kelly, B.J., Fitch, J.R., Hu, Y., Corsmeier, D.J., Zhong, H., Wetzel, A.N., Nordquist, R.D., Newsom, D.L. and White, P. (2015) Churchill: An Ultra-Fast, Deterministic, Highly Scalable and Balanced Parallelization Strategy for the Discovery of Human Genetic Variation in Clinical and Population-Scale Genomics. Genome Biology, 16, 6.
https://doi.org/10.1186/s13059-014-0577-x - 15. Chiang, C., Layer, R.M., Faust, G.G., Lindberg, M.R., Rose, D.B., Garrison, E.P., Marth, G.T., Quinlan, A.R. and Hall, I.M. (2015) SpeedSeq: Ultra-Fast Personal Genome Analysis and Interpretation. Nature Methods, 12, 966-968.
https://doi.org/10.1038/nmeth.3505 - 16. Jiang, Y., Turinsky, A.L. and Brudno, M. (2015) The Missing Indels: An Estimate of Indel Variation in a Human Genome and Analysis of Factors That Impede Detection. Nucleic Acids Research, 43, 7217-7228.
https://doi.org/10.1093/nar/gkv677 - 17. Fang, H., Wu, Y., Narzisi, G., O’Rawe, J.A., Barrón, L.T.J., Rosenbaum, J., Ronemus, M., Iossifov, I., Schatz, M.C. and Lyon, G.J. (2014) Reducing INDEL Calling Errors in Whole Genome and Exome Sequencing Data. Genome Medicine, 6, 89-89.
https://doi.org/10.1186/s13073-014-0089-z - 18. HiSeq X-Ten Test Data.
https://dnanexus-rnd.s3.amazonaws.com/NA12878-xten.html - 19. Van der Auwera, G.A., Carneiro, M.O., Hartl, C., Poplin, R., del Angel, G., Levy-Moonshine, A., Jordan, T., Shakir, K., Roazen, D., Thibault, J., Banks, E., Garimella, K.V., Altshuler, D., Gabriel, S. and DePristo, M.A. (2013) From fastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics, 43, 1-33.
https://doi.org/10.1002/0471250953.bi1110s43 - 20. Li, H. and Durbin, R. (2010) Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics, 26, 589-595.
https://doi.org/10.1093/bioinformatics/btp698 - 21. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078-2079.
https://doi.org/10.1093/bioinformatics/btp352