Open Journal of Genetics
Vol.05 No.04(2015), Article ID:61839,14 pages
10.4236/ojgen.2015.54011
Assessment of Genetic Relationship and Application of Computational Algorithm to Assess Functionality of Non-Synonymous Substitutions in DQA2 Gene of Cattle, Sheep and Goats
Steven B. Ugbo1*, Abdulmojeed Yakubu2*, Jude N. Omeje3, Bwaseh S. Bibinu1, Ibrahim S. Musa2, Joseph O. Egahi1, N. I. Dim1
1Department of Animal Breeding and Physiology, University of Agriculture, Makurdi, Nigeria
2Department of Animal Science, Faculty of Agriculture, Nasarawa State University, Keffi, Shabu-Lafia Campus, Lafia, Nigeria
3Department of Veterinary Medicine and Surgery, University of Abuja, Abuja, Nigeria

Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 March 2015; accepted 8 December 2015; published 11 December 2015
ABSTRACT
The major histocompatibility complex (MHC) is a fundamental part of the immune system in nearly all vertebrates. DQA2 is a member of the MHC complex and an important candidate gene involved in susceptibility/resistance to various diseases. Therefore, the present study aimed at investigating computationally molecular genetic variation of DQA2 gene of cattle, sheep and goats especially on its evolution and differentiation within and among species as well as the attendant effects of the polymorphism on the function of DQA2 gene. A total of thirty three DQA2 nucleotide sequences comprising cattle (10), sheep (12) and goats (11) were retrieved from the GenBank. Forty seven amino acid substitutions of the wild type alleles located in the putative peptide coding region of caprine DQA2 alleles were obtained from the alignment of deduced amino acid sequences of goats. Out of these, eleven amino acid substitutions (H14L, H14R, L34M, E35L, G56S, G56R, 161V, A62E, D69Q, T72N and T72G) were returned neutral; an indication that they did not impair protein function. The Expected Accuracy (EA) ranged from 53% - 87%. For sheep, sixteen amino acid substitutions (A11P, A11T, A11G, A11M, L14S, L14T, V27L, V27S, G35S, S46T, D55E, L57T, L57A, L57G, K65Q and V68I) appeared beneficial while the rest forty seven appeared harmful (EA ranged from 53% - 93%). Twenty four amino acid substitutions did not impair the function of protein while seventy seven substitutions appeared to have a negative effect on the function of protein of cattle (EA ranged from 53% - 94%). The phylogeny based on nucleotide and amino acid sequences of DQA2 gene revealed the close relatedness of the caprine, ovine and bovine species. The present knowledge would be relevant for performing further genotype-phenotype research as well as pharmacogenetics studies in order to show association between caprine, ovine and bovine DQA2 allelic variation and the clinical progression of infectious diseases especially in a developing country such as Nigeria.
Keywords:
DQA2, Genetic Relationship, Amino Acid Substitution, Phylogeny, Ruminants

1. Introduction
Genetic variation in parasite and host and relative distribution across space and time is of great interest and serves as a basis for adaptive change. Spatial population structure can strongly influence the process of co- adaptation between parasite and host and the evolution of virulence [1] . The major histocompatibility complex (MHC) is a large genomic region or gene family found in most vertebrates that encodes MHC molecules. MHC molecules play an important role in the immune system and autoimmunity. They are cellular glycoproteins involved in antigen presentation to CD4+ T cells. The genes encoding these molecules are polymorphic [2] [3] . DQ genes of MHC class II region encode for α (DQA) chain of the molecule [4] . The second exon has been shown to be highly polymorphic and under positive selection, and the class II DQA gene has recently attracted more attention [5] . In cattle, there are two or possibly three DQA genes [6] . The ovine DQ region encompasses 130 kb, with the DQ1 and DQ2 subregions located 22 kb apart. According to McKenzie et al. [7] , DQA2 of sheep is found on chromosome 20.
Because MHC genes must defend against a great diversity of microbes in the environment, the MHC molecules (coded for by the MHC genes) must be able to present a wide range of peptides. MHC genes achieve this through several mechanisms: 1) the MHC locus is polygenic, 2) MHC genes are highly polymorphic and numerous alleles have been described, and 3) several MHC genes arecodominantly expressed [8] [9] . DQ genes of MHC class II region encode for (DQA) and (DQB) chains of the molecule and are highly polymorphic [4] .
Recent advances in high-throughput technologies have generated massive amounts of genome sequence and genotype data for a number of species. The method to identify functional SNPs from a pool, containing both functional and neutral SNPs is challenging by experimental protocols [10] . Therefore, computational predictions have become indispensable for evaluating the disease-related impact of nonsynonymous single-nucleotide variants discovered in exome sequencing [11] . A number of computational methods have been developed to predict the functional effect of a non-synonymous single-nucleotide polymorphism (nsSNP), a single-nucleotide change in a protein-coding region of a gene that causes an amino acid substitution (AAS) in the resulting protein [12] . Many such methods have their roots in molecular evolution, as they use information derived from multiple sequence alignments. Most computational prediction tools for amino acid variants rely on the assumption that protein sequences observed among living organisms have survived natural selection. Therefore, evolutionarily conserved amino acid positions across multiple species are likely to be functionally important, and amino acid substitutions observed at conserved positions will potentially lead to deleterious effects on gene functions [13] .
The increasing information on genetics of host and parasites and their interaction at molecular level can lead to insights into disease emergence and control. Plasticity in the host genome especially for the genes responsible for disease resistance gives an advantage to host against pathogens with respect to protection. Therefore, studying the variability in the host population for disease resistance genes such as DQA2 is of utmost importance in practicing genetics of disease resistance.
The general objective of the study was to investigate computationally molecular genetic variation of DQA2 gene of some selected mammalian species (cattle, sheep and goats) especially on its evolution and differentiation within and among species as well as the attendant effects of the polymorphism on the function of DQA2 gene.
2. Materials and Methods
2.1. Sequences of Species
A total of thirty three (33) DQA2 nucleotide sequences comprising cattle (10), sheep (12) and goats (11) were retrieved from the GenBank (www.ncbi.nlm.nih.gov). The GenBank accession numbers of the sequences were AY829359, AY829358.1, AY829357, AY829356.1, AY829355.1, AY829354.1, AY829353.1, AY829352, AY829351, AY829350 and AY829349.1 (caprine); HG798789.1, HG798790.1, HG798791.1, HG798794.1, JX484834.1, FJ179558.1, FJ179557.1, FJ179551.1, HG798796.1, HG798795.1, HG798793.1, U65906.1 (ovine); D50045.1, D50046.1, D50049.1, D50048.1, D50047.1, AB098906.1, NM_001012681.1, JN225517.1, AY442305.1 and AY442304.1 (bovine).
2.2. Sequence Alignment and Translation
Sequences alignment, translation and comparison were done with ClustalW as described by Larkin et al. [14] using IUB substitution matrix, gap open penalty of 15 and gap extension penalty of 6.66.
2.3. Functional Analysis
In silico functional analysis of missense mutations was obtained using SNAP, which is a neural network based method for identifying from sequence functionally disruptive single amino acid substitutions [15] . The inputs to SNAP include secondary structure and solvent accessibility predictions, evolutionary and family information, biophysical differences between the wild type and mutant amino acids, statistical likelihoods of observing residue triplets around the mutation site, SIFT [16] and SwissProt [17] annotation if available. For each mutant, SNAP returns three values: the binary prediction (neutral/non-neutral), the RI (range 0 - 9) and the expected accuracy that estimates accuracy [Equation (1)] on a large dataset at the given RI (i.e. accuracy of test set predictions calculated for each neutral and non-neutral RI [18] .
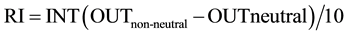 (1)
(1)
2.4. Phylogenetic Trees Analysis
Neighbor-Joining NJ trees were constructed each using P-distance model and pairwise deletion gap/missing data treatment. The construction was done on the basis of genetic distances, depicting phylogenetic relationships among the DQA2 nucleotide sequences of the investigated species. For the nucleotide sequences, the evolutionary distances were computed using the Maximum Composite Likelihood method. In the case of the amino acid however, the evolutionary distances were computed using the Poisson correction method. The reliability of the trees was calculated by bootstrap confidence values [19] , with 1000 bootstrap iterations using MEGA 5.1 software [20] . Similarly, UPGMA trees for the DQA2 gene were constructed with consensus nucleotide and amino acid sequences. All the nucleotide sequences were trimmed to equal length of 236 bp corresponding to same region before generating the trees.
3. Results
The predicted amino acid sequences of caprine, ovine and bovine DQA2 orthologous alleles are shown in Figure 1.
Forty seven amino acid substitutions of the wild type alleles located in the putative peptide coding region of caprine DQA2 alleles were obtained from the alignment of deduced amino acid sequences of goats. Out of these, eleven amino acid substitutions (H14L, H14R, L34M, E35L, G56S, G56R, 161V, A62E, D69Q, T72N and T72G) were returned neutral (Table 1); an indication that they do not impair protein function. The Expected Accuracy (EA) ranged from 53% - 87%.
For sheep, sixteen amino acid substitutions (A11P, A11T, A11G, A11M, L14S, L14T, V27L, V27S, G35S, S46T, D55E, L57T, L57A, L57G, K65Q and V68I) appeared beneficial while the rest forty seven appeared harmful (Table 2). In this case, the EA ranged from 53% - 93%.
Twenty four amino acid substitutions did not impair the function of protein while seventy seven substitutions

Table 1. Functional analysis of coding nsSNPs of the DQA2 gene of goat using SNAP.
I = isoleucine, L = Leucine, V = valine, C = cysteine, A = alanine, G = glycine, P = proline, T = threonine, S = serine, Q = glutamine, N = asparagine, H = histidine, E = glutamic acid, D = aspartic acid, K = lysine, R = arginine, Y = tyrosine, M = methionine, F = phenylalanine. Including only predictions with: RI ≥ 0; Expected Accuracy (EA) ≥ 50%.
Table 2. Functional analysis of coding nsSNPs of the DQA2 gene of sheep using SNAP.
I = isoleucine, L = Leucine, V = valine, C = cysteine, A = alanine, G = glycine, P = proline, T = threonine, S = serine, Q = glutamine, N = asparagine, H = histidine, E = glutamic acid, D = aspartic acid, K = lysine, R = arginine, Y = tyrosine, M = methionine, W = tryptophan. Including only predictions with: RI ≥ 0; Expected Accuracy (EA) ≥ 50%.

Figure 1. Comparison of the predicted amino acid sequences DQA2 alleles of goat, sheep and cattle. Dot indicates amino acid identity; Missing = ? Amino acid positions included in the peptide binding region according to Reche and Reinherz [31] .
appeared to have a negative effect on the function of protein of cattle (Table 3). The EA ranged from 53% - 94%, respectively.
The phylogeny based on nucleotide and amino acid sequences of DQA2 gene revealed the close relatedness of the caprine, ovine and bovine species, although there were some intermingling among the sequences of the three species investigated (Figure 2 and Figure 3).
The genetic relationships of DQA2 Bovidae subfamily members of goats, sheep, and cattle shown in the UPGMA phylogenetic trees (Figure 4 and Figure 5) revealed that goats and sheep were closer at this locus compared to cattle.
4. Discussion
MHC genes are the most polymorphic genes described in vertebrates, with polymorphisms occurring predominantly at residues involved in peptide binding (antigen binding sites) [21] . Variation at these sites may affect the antigen binding groove and antigenic-peptide binding ability, and hence peptide specificity [2] . The present findings indicate that the caprine DQA2 gene is highly polymorphic. Similar patterns were observed in the case of ovine and bovine species. Current concerns about food security highlight the importance of maintaining productive and disease-resistant livestock populations. Susceptibility to monogenic and complex diseases has a strong impact on the economic output of livestock farms. Disentangling the genetic factors that modulate disease progression might be useful to implement selection schemes aimed at eradicating or decreasing the incidence of pathological conditions [22] [23] . The genetic analysis of production and disease-related traits in goats has been rarely done at a genome-wide scale. In this regard, the lack of well-established microsatellite panels covering the whole genome hindered, to a significant extent, the implementation of genome scans aimed at detecting QTL
Table 3. Functional analysis of coding nsSNPs of the DQA2 gene of cattle using SNAP.
I = isoleucine, L = Leucine, V = valine, C = cysteine, A = alanine, G = glycine, P = proline, T = threonine, S = serine, Q = glutamine, N = asparagine, H = histidine, E = glutamic acid, D = aspartic acid, K = lysine, R = arginine, Y = tyrosine, M = methionine, F = phenylalanine, W = tryptophan. Including only predictions with: RI ≥ 0; Expected Accuracy (EA) ≥ 50%.

Figure 2. Phylogenetic relationships of caprine, ovine, and bovine DQA2 nucleotide sequences.

Figure 3. Phylogenetic relationships of caprine, ovine, and bovine DQA2 amino acid sequences.

Figure 4. Phylogenetic relationships of caprine, ovine, and bovine DQA2 consensus nucleotide sequences using UPGMA.

Figure 5. Phylogenetic relationships of caprine, ovine, and bovine DQA2 consensus amino acid sequences using UPGMA.
[22] . Although, identifying single gene markers associated with resistance to gastro-intestinal parasites is difficult as resistance to parasites is considered to be polygenic with hundreds to thousands mutations responsible for the resistant phenotype [24] [25] , research continues in the area of genetic markers as they have the advantage over phenotypic markers of measurement prior to birth, meaning that producers can make productivity decisions early [26] . A high level of diversity in MHC genes allows populations to survive despite exposure to rapidly evolving pathogens [27] . It plays major role in determining whether transplanted tissue will be accepted as self or rejected as foreign. Also the study of the MHC can aid in the development and the design of vaccines based on synthetic peptides comprising one or more T-cell epitopes of the pathogen. Using footrot marker screening of DQA2 gene of sheep, the potential to select a high resistant flock is possible within three to five breeding seasons [28] .
The presence of numerous alleles at a particular MHC locus is evidence of the long-term evolutionary persistence of the locus. This is suggested by the frequency with which alleles in one species are more closely related to the alleles in a closely related species than to the other alleles in the same species [29] . This could be exploited in the development and the design of vaccines as well as drug production. The close similarity of a gene among ruminants may be ascribed to recent separation in evolutionary process and/or similar selection pressure which the ruminants have suffered during evolution [30] . The genetic relationships of DQA2 Bovidae subfamily members of goat, sheep, and cattle shown in the UPGMA phylogenetic trees were in accordance with the well- known evolutionary history of Bovidae subfamily speciation, although more expressed using the amino acid sequences than the nucleotide sequences. Here, goats are more closely related to sheep than cattle; which is congruous to the submission of Zhou et al. [2] .
In developing countries, such as Nigeria, some quantitative and qualitative measurements have been used for selection and breeding purposes against disease infestation with little or no meaningful improvement in the stocks. This has necessitated the paradigm shift to computational genomics to facilitate the analysis and interpretation of the vast array of molecular data. Therefore, the present beneficial SNPs could be exploited in the genetic improvement of Nigerian native livestock for increased disease resistance using information emanating from both wet and dry laboratories in future studies.
5. Conclusion
The study revealed high genetic diversity at the DQA2 locus of goats, sheep and cattle. Some beneficial non- synonymous amino acid substitutions at putative peptide binding sites were also found at this locus. This knowledge would be relevant for performing further genotype-phenotype research as well as pharmacogenetics studies in order to show association between caprine, ovine and bovine DQA2 allelic variation and the clinical progression of infectious diseases especially in a developing country such as Nigeria. This becomes imperative considering the suggestion that pathogen-mediated selection (PMS) is the driving force maintaining diversity at MHC loci.
Cite this paper
StevenB. Ugbo,AbdulmojeedYakubu,Jude N.Omeje,BwasehS. Bibinu,IbrahimS. Musa,JosephO. Egahi,N. I.Dim, (2015) Assessment of Genetic Relationship and Application of Computational Algorithm to Assess Functionality of Non-Synonymous Substitutions in DQA2 Gene of Cattle, Sheep and Goats. Open Journal of Genetics,05,145-158. doi: 10.4236/ojgen.2015.54011
References
- 1. Biek, R. and Real, L.A. (2010) The Landscape Genetics of Infectious Disease Emergence and Spread. Molecular Ecology, 19, 3515-3531.
http://dx.doi.org/10.1111/j.1365-294X.2010.04679.x - 2. Zhou, H., Hickford, J.G.H. and Fang, Q. (2005) Polymorphism of the DQA2 Gene in Goats. Journal of Animal Science, 83, 963-968.
- 3. Yakubu, A., Salako, A.E., De Donato, M., Takeet, M.I., Peters, S.O., Adefenwa, M.A., Okpeku, M., Wheto, M., Agaviezor, B.O., Sanni, T.M., Ajayi, O.O., Onasanya, G.O., Ekundayo, O.J., Ilori, B.M., Amusan, S.A. and Imumorin, I.G. (2013) Genetic Diversity in Exon 2 at the Major Histocompatibility Complex DQB1 Locus in Nigerian Indigenous Goats. Biochemical Genetics, 51, 954-966.
http://dx.doi.org/10.1007/s10528-013-9620-y - 4. Niranjan, S.K., Deb, S.M., Kumar, S., Mitra, A., Sharma, A., Sakaram, D., Naskar, S., Sharma, D. and Sharma, S.R. (2010) Allelic Diversity at MHC Class 11 DQ Loci in Buffalo (Bubalusbubalis): Evidence for Duplication. Veterinary Immunology and Immunopathology, 138, 206-212.
http://dx.doi.org/10.1016/j.vetimm.2010.07.014 - 5. Amills, M., Ramirez, O., Tomas, A., Obexer-Ruff, G. and Vidal, O. (2008) Positive Selection on Mammalian MHC-DQ Genes Revisited from a Multispecies Perspective. Genes and Immunity, 9, 651-658.
http://dx.doi.org/10.1038/gene.2008.62 - 6. Ballingall, K.T., Luyai, A. and McKeever, D.J. (1997) Analysis of Genetic Diversity at the DQA Loci in African Cattle: Evidence for a BoLA-DQA3 Locus. Immunogenetics, 46, 237-244.
http://dx.doi.org/10.1007/s002510050268 - 7. McKenzie, G.W., Abbott, A., Zhou, H., Fang, Q., Merrick, N., Forrest, R.H., Sedcole, J.R. and Hickford, J.G. (2012) Genetic Diversity of Selected Genes That Are Potentially Economically Important in Feral Sheep of New Zealand. Genetics Selection Evolution, 42, 43.
- 8. Vandre, R.K., Gowane, G.R., Sharma, A.K. and Tomar, S.S. (2014) Immune Responsive Role of MHC Class II DQA1 Gene in Livestock. Livestock Research International, 2, 1-7.
- 9. Vandre, R.K., Sharma, A.K., Gowane, G.R., Rajoriya, R., Rajoriya, S., Sinha, R.K., Kumar, A., Shivhare, M., Caser, D.D. and Meshram, S.K. (2014) Polymorphism and Disease Resistance Possessions of MHC Class II BoLA Genes. DHR International Journal of Biomedical and Life Sciences, 5.
http://www.doublehelixresearch.com/DHRIJBLS - 10. George, P.D.C., Rajasekaran, R., Sudandiradoss, C., Ramanathan, K., Purohit, R. and Sethumadhavan, R. (2008) A Novel Computational and Structural Analysis of nsSNPs in CFTR Gene. Genomic Medicine, 2, 23-32.
http://dx.doi.org/10.1007/s11568-008-9019-8 - 11. Liu, L. and Kumar, S. (2013) Evolutionary Balancing Is Critical for Correctly Forecasting Disease Associated Amino Acid Variants. Molecular Biology and Evolution, 30, 1252-1257.
http://dx.doi.org/10.1093/molbev/mst037 - 12. Zemla, D., Kostova, T., Gorchakov, R., Volkova, E., Beasley, D.W.C., Cardosa, J., Weaver, S.C., Vasilakis, N. and Naraghi-Arani, P. (2014) Genesv—An Approach to Help Characterize Possible Variations in Genomic and Protein Sequences. Bioinformatics and Biology Insights, 8, 1-16.
http://dx.doi.org/10.4137/BBI.S13076 - 13. Choi, Y., Sims, G.E., Murphy, S., Miller, J.R. and Chan, A.P. (2012) Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE, 7, e46688.
http://dx.doi.org/10.1371/journal.pone.0046688 - 14. Larkin, M.A., Blackshields G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J. and Higgins, D.G. (2007) Clustal W and Clustal X Version 2.0. Bioinformatics, 23, 2947-2948.
http://dx.doi.org/10.1093/bioinformatics/btm404 - 15. Bromberg, Y. and Rost, B. (2007) SNAP: Predicts Effect of Non-Synonymous Polymorphisms on Function. Nucleic Acids Research, 35, 3823-3835.
http://dx.doi.org/10.1093/nar/gkm238 - 16. Ng, P.C. and Henikoff, S. (2003) SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Research, 31, 3812-3814.
http://dx.doi.org/10.1093/nar/gkg509 - 17. Bairoch, A. and Apweiler, R. (2000) The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Research, 28, 45-48.
http://dx.doi.org/10.1093/nar/28.1.45 - 18. Bromberg, Y., Yachdav, G. and Rost, B. (2008) SNAP Predicts Effect on Protein Function. Bioinformatics Applications Note, 24, 2397-2398.
http://dx.doi.org/10.1093/bioinformatics/btn435 - 19. Felsenstein, J. (1985) Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution, 39, 783-791.
http://dx.doi.org/10.2307/2408678 - 20. Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. and Kumar, S. (2011) MEGA 5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution, 28, 2731-2739.
http://dx.doi.org/10.1093/molbev/msr121 - 21. Zhao, Y., Xu, H., Shi, L. and Zhang, J. (2011) Polymorphisms in Exon 2 of MHC Class II DRB3 Gene of 10 Domestic Goats in Southwest China. Asian-Australasian Journal of Animal Science, 24, 752-756.
http://dx.doi.org/10.5713/ajas.2011.10398 - 22. Amills, M. (2014) The Application of Genomic Technologies to Investigate the Inheritance of Economically Important Traits in Goats. Advances in Biology, 2014, Article ID: 904281.
http://dx.doi.org/10.1155/2014/904281 - 23. McManus, C., Paim, T.D.P., de Melo, C.B., Brasil, B.S.A.F. and Paiva, S.R. (2014) Selection Methods for Resistance to and Tolerance of Helminths in Livestock. Parasite, 21, Article No.: 56.
http://dx.doi.org/10.1051/parasite/2014055 - 24. Hickford, J.G.H., Forrest, R.H.J., Zhou, H., Fang, Q. and Frampton, C.M. (2011) Association between Variation in Faecal Egg Count for a Mixed Field-Challenge of Nematode Parasites and Ovine MHC-DQA2 Polymorphism. Veterinary Immunology and Immunopathology, 144, 312-320.
http://dx.doi.org/10.1016/j.vetimm.2011.08.014 - 25. Kemper, K.E., Emery, D.L., Bishop, S.C., et al. (2011) The Distribution of SNP Marker Effects for Faecal Worm Egg Count in Sheep, and the Feasibility of Using These Markers to Predict Genetic Merit for Resistance to Worm Infections. Genetics Research, 93, 203-219.
http://dx.doi.org/10.1017/S0016672311000097 - 26. Preston, S.J.M., Sandeman, M., Gonzalez, J. and Piedrafita, D. (2014) Current Status for Gastrointestinal Nematode Diagnosis in Small Ruminants: Where Are We and Where Are We Going? Journal of Immunology Research, 2014, Article ID: 210350.
http://dx.doi.org/10.1155/2014/210350 - 27. Ellis, S.A. and Hammond, J.A. (2014) The Functional Significance of Cattle Major Histocompatibility Complex Class I Genetic Diversity. Annual Review of Animal Biosciences, 2, 285-306.
http://dx.doi.org/10.1146/annurev-animal-022513-114234 - 28. Wuliji, T., Hickford, J.G.H., Lamberson, W.R., Shanks, B.C. and Azarpajouh, S. (2014) Ovine Footrot Gene Marker Screening in a Katahdin Sheep Flock. Proceedings of Joint Annual Meeting of ASAS, Kansas City, 20-24 July 2014.
- 29. Takeshima, S., Chen, S., Miki, M., Kado, M. and Aida, Y. (2008) Distribution and Origin of Bovine Major Histocompatibility Complex Class II DQA1 Genes in Japan. Tissue Antigens, 72, 195-205.
http://dx.doi.org/10.1111/j.1399-0039.2008.01092.x - 30. Sun, Y., Zhang, X., Xi, D., Li, G., Wang, L., Zheng, H., Du, M., Gu, Z., Yang, Y. and Yang, Y. (2015) Isolation and cDNA Characteristics of MHC-DRA Genes from Gayal (Bos frontalis) and Gaytle (Bos frontalis × Bostaurus). Biotechnology & Biotechnological Equipment, 29, 33-39.
http://dx.doi.org/10.1080/13102818.2014.986128 - 31. Reche, P.A. and Reinherz, E.L. (2003) Sequence Variability Analysis of Human Class I and Class II MHC Molecules: Functional and Structural Correlates of Amino Acid Polymorphisms. Journal of Molecular Biology, 331, 623-641.
http://dx.doi.org/10.1016/S0022-2836(03)00750-2
NOTES

*Corresponding authors.