Open Journal of Genetics
Vol.1 No.2(2011), Article ID:7309,2 pages DOI:10.4236/ojgen.2011.12003
Different denaturation rates between methylated and non-methylated genomic DNA can result in allele-specific PCR amplification
![]()
1Wessex Regional Genetics Laboratory, Salisbury District Hospital, Salisbury, UK;
2Department of Genetics, Kuala Lumpur Hospital, Kuala Lumpur, Malaysia;
3Human Genetics Division, Southampton University School of Medicine, Southampton, UK.
Email: Dave.Bunyan@salisbury.nhs.uk
Received 13 June 2011; revised 25 July 2011; accepted 20 August 2011.
Keywords: Differential Methylation; Allele-Specific PCR; Commercial Kit
ABSTRACT
We analysed a DNA sample from a father and child who were both heterozygous for a 7 base pair insertion in the MEST gene differentially-methylated promoter region, previously shown by PCR analysis of bisulphite-treated DNA to be on the methylated allele in the unaffected father and the unmethylated allele in the affected child. PCR from genomic DNA was then carried out using a commercial PCR kit with its recommended initial DNA denaturation step of 2 minutes. Subsequent sequence analysis showed that only the non-methylated allele had been amplified, the father appearing to be homozygous normal and the child appearing to have a homozygous 7 b.p. insertion. The PCR protocol was then modified in order to use a longer DNA denaturation stage prior to the addition of the polymerase enzyme. Upon doing so, both the methylated and non-methylated alleles were then identifiable by sequencing with the mutation appearing in its expected heterozygous form. These results highlight the fact that the methylation status of DNA can affect the denaturation rate prior to PCR and result in allele drop-out, showing that the standard protocols of commercial kits should be used with caution when working with methylated regions of DNA.
1. INTRODUCTION
Due to the high GC content of differentially-methylated regions (DMRs), the PCR amplification of these sites from untreated genomic DNA often involves the use of special polymerase enzymes or kits which often do not have “hotstart” capabilities. We regularly use the FailsafeTM system (Epicentre Biotechnologies, Madison, Wisconsin, USA) which has as its standard protocol an initial DNA denaturation step of 1 - 2 minutes following the addition of the polymerase enzyme.
Recently, we identified a two-generation family with a 7 b.p. insertion in the MEST gene differentially-methylated promoter region (c.1-99_1-93insdupGGGCTGC). This insertion was shown by previous PCR analysis of bisulphite-treated DNA to be on the methylated chromosome in the unaffected father and on the non-methylated chromosome in the affected child (data not shown). PCR from genomic DNA was then carried out on both individuals using the FailsafeTM system. However, subsequent sequencing analysis showed that the child appeared to be homozygous for the 7 b.p. insertion whilst the father only had the normal allele, indicating that only the non-methylated chromosome had been amplified during PCR in each case. The PCR protocol was then modified to include an extra DNA denaturation stage prior to the addition of the enzyme in order to also create a PCR product from the methylated chromosome.
2. MATERIALS AND METHODS
PCR analysis of the MEST DMR was initially carried out using the FailsafeTM PCR system with FailsafeTM Buffer G and the standard supplied protocol which has a suggested pre-PCR denaturation time of 1 - 2 minutes at 92˚C - 98˚C (we used 2 minutes at 94˚C). Subsequent PCR was performed using a modified protocol which added an extra 2 - 10 minutes of DNA denaturation to the start of the protocol, prior to the addition of the FailsafeTM polymerase enzyme. The primers used in the analysis were aaccagcacaccccggca (forward) and aatgcg-
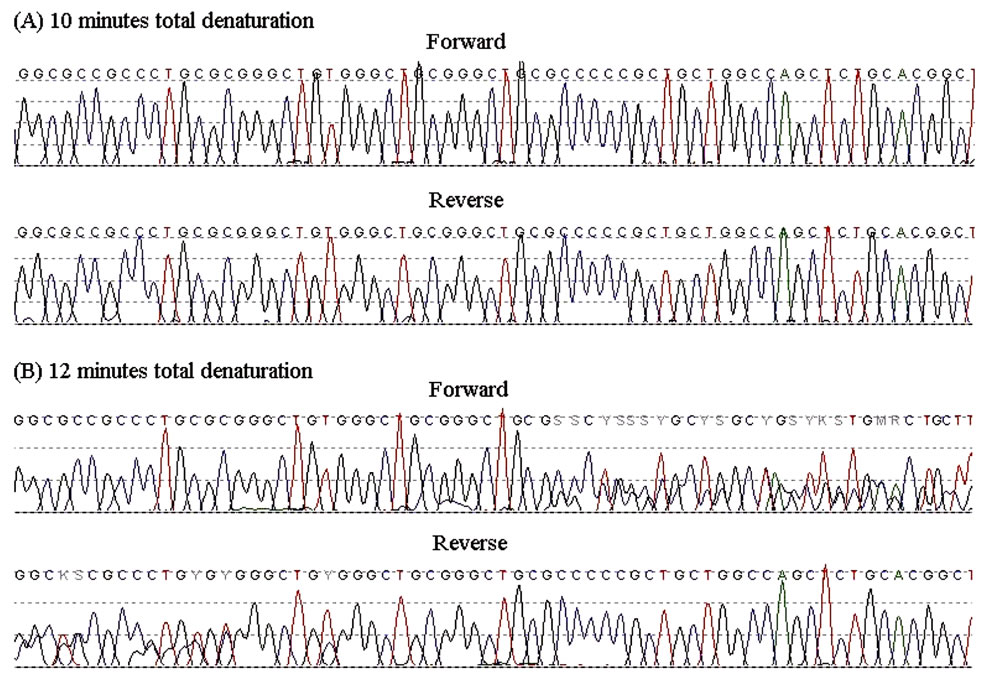
Figure 1. Sequence chromatograms of the PCR products from the father (forward and reverse) that had undergone an initial total denaturation time of 10 minutes (A) or 12 minutes (B).
tgccgcctgtcg (reverse) with a PCR annealing temperature of 60˚C.
3. RESULTS
After increasing the length of DNA denaturation prior to polymerase enzyme addition, the methylated allele from the MEST DMR was then identifiable at the sequencing stage, with the methylated allele starting to appear after a total of 12 minutes of DNA denaturation prior to the PCR cycles. Results from the father are shown in Figure 1.
4. DISCUSSION
The methylation status of genomic DNA has been previously shown to have an influence on DNA denaturation [1], but there are no previous reports of it having an affect on the PCR analysis of differentially methylated genomic regions. In the two individuals studied here, the preferential amplification of a single allele was clearly proven to be methylation-specific and not sequencespecific. These results were obtained by using a commercial kit and following its standard supplied protocol. It was only by adapting the standard protocol that we were able to amplify the methylated allele in the DMR of the MEST gene, an increase in the initial DNA denaturation step being necessary for the amplification of both non-methylated and methylated alleles.
The data presented here offer a cautionary tale by highlighting the fact that commercial PCR kits with short initial DNA denaturation protocols can be affected by the methylation status of genomic DNA, leading to potential allelic drop-out. Such commercial kits are ideal for GC-rich regions, but great care should be taken if working with known methylated regions. However, the phenomenon of DNA denaturation being influenced by its methylation status may allow selective amplification of the non-methylated homologue in differentially methylated regions if required, although the timing of the denaturation process would have to be highly optimised to suit the specific sequence and primers used.
REFERENCES
- Diede, S.J., Guenthoer, J., Geng, L.N., et al. (2010) DNA methylation of developmental genes in pediatric medulloblastomas identified by denaturation analysis of methylation differences. Proceedings of the National Academy of Sciences, 107, 234-239. doi:10.1073/pnas.0907606106