Advances in Biological Chemistry
Vol.04 No.05(2014), Article ID:48598,20 pages
10.4236/abc.2014.45036
Concluding Step in Cell Restitution Cycle: ER Transport Vesicles with Sphingolipids in the Outer Leaflet of the Membrane Restore Lysosomes
Amalia Slomiany, Bronislaw L. Slomiany
Biomedical and Health Sciences, School of Dental Medicine, Rutgers, The State University of New Jersey,
Newark, USA
Email: slomiaam@sdm.rutgers.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 25 July 22014; revised 26 July 2014; accepted 1 August 2014
ABSTRACT
Restitution of the cell organelles and the membrane implicates serine palmitoyltransferase (SPT) in signal-specific and selective assembly of the transport vesicles. Here, we reveal that SPT, embedded in the outer leaflet (OL) of endoplasmic reticulum (ER), is engaged in the synthesis of ER transport vesicles that recondition cell organelles, and the inner leaflet (IL) SPT in the restitution of the cell membrane. The OL SPT impacts assembly of sphingomyelinase (SMase)―susceptible ER vesicles but not the SMase-resistant and sphingolipid (SPhL) core―carrying vesicles that refurbish the cell membrane. The investigation of the SPT-initiated differences in the placement of SPhL in vesicular membranes by utilizing ER depleted of OL SPT, allows us to conclude that the restitution of endosomal and lysosomal membranes is achieved with the involvement of OL SPT, whereas the IL SPT is involved in formation of the lipid core for glycosphingolipids (GSL) and sphingomyelin (SM) of the apical and basolateral cell membrane. These findings along with our previously published report (Slomiany and Slomiany, Advances in Biological Chemistry, 2013, 3, 275-287), provide a clear distinction between the processes that renovate cell membrane and its organelles from that of the endocytotic cell debridement, and show that vesicles are navigated to the specific organelles and the cell membrane by the biomembrane constituents programmed in ER.
Keywords:
ER Transport Vesicles Specificity, Serine Palmitoyltransferase, Sphingomyelin, Lysosomes Restitution, Cell Debridement, Autophagocytes

1. Introduction
An activation of the particular sets of events is evoked in the nucleus by signals reporting changes in the environment that necessitates cell component restitution, and forecasts initiation of synchronized processes that at given time restore critical cell elements. Consequently, the episodes entail the simultaneous transcriptional activity that defines composition and dictates co-expression of closely associated functional elements that remain as whole from the time of transcription through translation and the final stage of their specific deposition within the cell. As envisioned in a plethora of signal-induced transcriptional activations, the processes occur in a discrete and controlled stages from the packaging of genes into chromatin and its localization within nucleus to the recruitment of multi-complexes whose conformation and activity result from specific protein-protein or protein-DNA interactions and translation of function- and placement-related mRNAs [1] - [5] . Unexplainably, once beyond the nucleus inner boundary, the events are not contemplated as inseparable entities undergoing conversion into distinct products of the discrete sites of endoplasmic reticulum (ER) [6] - [8] . The concept of the organized, judicious and error minimizing recreation of cell parts is overshadowed by the inherently scattered approach that favors techniques scrutinizing individual and random posttranscriptional events, which in our understanding might have skewed the assessment of the processes involved in cell restitution cycle [9] - [19] .
The most popular way to probe a protein’s destination and function, through knocking out genes, may create a unnoticeable central change that destroys destination marker of the transporter, or the modified by deletion complex may trigger elimination of misshapen product, or divert the product to an inappropriate cellular location [10] [11] [16] [20] - [23] . Hence, as the consequence of gene knockout, the cell architecture and function may be modified, the cell rejected or destroyed [24] - [26] . Similarly, the approach relying on point mutation technique may affect protein folding and its organization in the membrane, and thus disturb the vesicular structure and the fidelity of the restoration [14] [17] [21] - [23] . Just as risky is to focus on one specific posttranslational modification regulating cellular signaling that involves phosphorylation or ubiquitylation. The latter investigations uncover more than 20,000 phosphorylation sites and 19,000 ubiquitylation sites, but their site-spe- cific occurrence, and whether the presence of one modification influences the appearance of others, remain unknown [27] [28] . Collectively, the major transgression of such global approaches to integrate protein expression with posttranslational modification, protein activity, and its concentration is that the utilized systems require the destruction of cell compartmentalized arrangement, the order that prevents cellular elements from encountering and interacting with extraneous constituents [9] [21] [29] - [38] . The investigations embracing genetic manipulation, point mutation, and specific posttranslational modification are conducted and assessed under the conditions that minimize function of cell compartments membrane-specific lipids, or reduce their involvement to mere hydrophobic background for the protein folding. Consequently, the proteins released from their confined spaces are not in their native state, and that generates artificial cross talk, co-modified proteins, and results in erroneous explanations of the events. Yet, the tenuous schemes continue to dominate the investigation, and the singled-out events in the void of factors that in reality determine the coordinated and defect-free cell assembly and function are over interpreted.
The general problem summarized above is exemplified in the interpretation of the in vitro studies on the individual protein affinity to unspecific liposomes, where the assays cannot measure correct membrane integration and precise membrane lipid environment [37] [38] . The capability and promiscuity of proteins and peptides to bind to lipid bilayers with un-physiological topology create many incongruities diverging from specific cellular events [9] - [12] [21] [22] [24] - [26] . Subsequently, the results that demonstrate mere association of the purified protein with artificial lipid bilayer are accepted as the accurate functional engagement that is crucial in numerous membrane receptors, or in genuine protein association with outer or inner leaflet of the specific membrane. Collectively, the results of the studies embracing genetic manipulations, point mutations, highly specific antibodies assessing presence of protein in cells, or global approach to proteome analysis under conditions that detect and quantitate protein moiety in the vicinity of the organelle, but disregard the role of its intercalation in the cell-designed lipid environment defined for the organelle, are unable to differentiate between true or false artificial interactions. Afterward, the amalgamation of the undetected shortfalls in the general strategies that are used as master templates that determine the function of posttranslational switches regulating protein activity, placement and maintenance of homeostasis, builds indistinctness whether the investigated proteins are scattered throughout the cell, derived from endocytotic elimination of membranes destined for lysosomal degradation, or are ER-produced replacements delivering newly synthesized membrane fragments and organelle-specific cargo to the site-requiring restoration. In an essence, the outlined tactics generate highly confusing picture of the cell restitution and debridement cycle, which in nature ought to proceed in highly ordered fashion.
Dauntingly, ER is the home base for all newly synthesized components of the cell, those in form of vesicular transporters destined to assortment of the compartments, as well as those that remain as structural part of ER itself that are retained on its luminal or cytosolic site, and as whole, determine the quality of the anabolic replacements and precision with which the replacements are delivered to the cellular compartments [21] - [23] . In our interpretation of the early nuclear events, the translational units assemble the systems that then, en bloc, restore organellar and cell membrane elements. This is accomplished through generation of the ER-initiated transport vesicles of specific composition that regenerate organellar and cell membrane components and functions [39] - [41] . The vesicles for organelles’ restitution contain organelle-specific membrane lipids as well as co- translationally intercalated proteins that are permanently incorporated into the membrane and en bloc delivered to the precise site, where they remain in place until portion of the organelles undergo another cycle of restitution that evokes autophagocytic dissection [42] . As demonstrated earlier [43] , the process necessitates bifurcation of the ER proceedings producing mitochondria-directed transporters from those that facilitate synthesis of restitution vesicles that require Golgi-specific posttranslational modification instituted by Golgi integral components. In an essence, the process involves honing of sphingolipids (SPhL) to fit precisely their point of destination.
In expanding the basic concept of meticulous restitution of the cell elements, our investigation on the synchronized with translation membrane lipid synthesis was to elucidate the role of the de novo synthesized SPhL core, namely sphingosine (SphNH2) and ceramide (Cer), and their utilization to generate glycosphingolipids (GSLs) and sphingomyelin (SM) in Golgi vesicles. Based on the findings that ER integral enzyme responsible for the synthesis of SphNH2, the serine palmitoyltransferase (SPT), is associated with inner and outer leaflet of ER membrane [44] , we were able to demonstrate that the SPT of the outer, the cytosol-facing leaflet (OL) SPT, augments synthesis of endosome-directed transport vesicles with SM in the outer leaflet, whereas the inner leaflet (IL) SPT is providing SPhL core for the synthesis of GSL and SM in the vesicles responsible for the restitution of apical and basolateral membrane. Thus, following fusion with cell membrane, the vesicular inner leaflet lipids assembled in Golgi are facing the outer environment of the cell. The SM produced on the outer leaflet of Golgi, demarcates the transport vesicles destined to endosomes, and as is the case with other transporters, their membrane and cargo define endosome membrane composition and contents. With this, our findings allowed us to demonstrate that the cell membrane excised fragments, generated through endocytotic activity, do not construct endosome organelles, but reflect the process of cell debridement through generation of organelle and cell membrane-specific autophagocytes with the common affinity for lysosomal structures [42] [44] .
Our concept thus emphasizes the crucial role of coordinated ER events responsible for the retention of cell identity, the process that entails a role of specific lipids in ER-initiated assembly of transport vesicles with specific biomembrane configuration, that through en bloc fusion with destination site, cement the retention of the organellar and cellular membranes identity [39] - [50] . This holistic view of biogenesis of the intracellular transport entities and the identification of the biomembrane -specific lipids in the delivery and restitution of the individual cells elements contribute to visualization of the processes reflecting generation of autophagosomal vesicles carrying organelle-specific membrane resections destined for lysosomal degradation, and thus control cell debridement [42] . Cumulatively, the gradual validation of this concept allows us to conclude that the exact, destination-specific vesicular constructs, and the surgically precise autophagosomal resects, are crucial attributes of the faithful restitution and homeostatic repair of the cell [39] - [50] .
2. Materials and Methods
2.1. Preparation of Cells for Subcellular Fractionation
For the isolation of cell components, the initial step of fragmentation of the structure is crucial and a method is required that gives minimum breakage. Hence, in the method described herein we used the techniques which allowed us to purify cell organelles with minimum fragmentation of their structure, so that cell cytosol (CC) was not admixed with organellar components and that cell membranes were not admixed with intracellular membranes. In further preparation of organelles, we aimed to isolate the undamaged structures and use them for the incubation with CC and formation of the transport or autophagosomal vesicles.
The cells were prepared from rat gastric mucosa and the liver as described previously [41] [46] - [50] . The single cells that were separated from larger debris with the aid of specific cell size nylon mesh, were centrifuged at 50 xg for 2 min, washed twice with the enzyme-free medium, twice with the Minimum Essential Medium (MEM), and counted in hemocytometer. Thus prepared cells were then incubated in MEM for 3 hours with or without radiolabel, then where indicated incubated for 30 min with 100 nM cycloheximide (CHX), and then used for preparation of nuclei [51] , subcellular organelles, cell cytosol [41] [43] [46] [48] [52] and cellular membranes [42] [52] . In the experiments dedicated to the vesicles synthesis with translation active CC or RNase or CHX treated CC, the preparations of the organelles were additionally rinsed with phosphate buffered saline (PBS), or 0.5M NaCl or urea-PBS, in order to remove the associated residual cytosolic proteins that otherwise would remain on their membranes. The synthesis of phospholipids (PL) and protein was assessed using radiolabeled [3H]inositol, [3H]arachidonate, [14C]choline, [3H]serine, [3H]palmitate and [32P]ATP [40] [41] [46] - [48] [50] . The radiolabeled CHX incubated cells’ organelles were incubated with vesicles-depleted CHX CC and the formed products subjected to separation from organelles as described earlier for ER, Golgi and mitochondria- derived biosynthetic transport vesicles [40] -[43] [47] -[50] [52] [53] . The vesicles generated in the CHX-treated cells were subjected to fusion experiments with Golgi, ER, mitochondria and lysosomes in the medium containing cold, vesicles-free, CHX-derived CC at concentration of 15 mg protein/ml of incubation mixture enriched with 50 M ATP, 250 M CTP, 50 M GTP, 5 mM creatine phosphate, 8.0 IU/ml creatine kinase, and where indicated 100 nM CHX or 25 g/ml RNase, 10 M UDP-Glc and 10 M palmitoyl CoA [40] - [43] [47] - [50] [53] .
2.2. Preparation of the Cell Cytosol
The viable cells (normal, or CHX-treated) homogenized for 10 sec at 600 rpm in 3 volumes of buffer containing 0.25 M sucrose, 50 mM TRIS-HCl (pH 7.4), 25 mM magnesium acetate and 10 mM each of aprotinin, leupeptin, chemostatin, and 1 mM phenylmethylsulfonylfluoride were centrifuged at 5000 xg for 15 min. The supernatant, diluted with 2 volumes of homogenization buffer, was re-centrifuged at 10,000 xg for 20 min. The resulting supernatant was then subjected to centrifugation at 100,000xg for 1h. Thus obtained soluble fraction was adjusted to 15 - 18 mg protein/ml, admixed with an ATP generating system consisting of 40 mM ATP, 200 mM creatine phosphate, 2000 units/ml creatine phosphokinase, and referred to as active CC or CHX CC. Then, the vesicles remaining in the obtained cytosols were removed by centrifugation at 150,000 xg for 1 h, and the spun down material suspended in 55% sucrose, overlaid with 55% - 30% gradient and centrifuged at 150,000 xg for 16 h. The radiolabeled material was recovered from the gradient as reported earlier for isolation of transport vesicles from ER, Golgi and mitochondria [40] [41] [43] [47] [48] [50] [52] - [54] .
2.3. Preparation of Cellular Organelles and Membranes
The cell membranes and subcellular organelle fractions (mitochondria, ER, Golgi) were recovered from the cold or radiolabeled cells as described earlier [40] - [43] [46] [47] [54] - [58] . The ER and Golgi organelles sediment, remaining after separation of nuclei, mitochondria, endosomes and lysosomes, and cell cytosol, was suspended in the buffer containing 0.2 M PIPES (pH 6.9), 2.0 M glycerol, 1 mM EGTA and 1.0 mM magnesium acetate and applied on the top of discontinuous gradient of 2.0/1.5/1.3/1.0 M sucrose and centrifuged at 100,000 xg for 16 h. The cell membranes were recovered from 1.0 M sucrose, ER from 1.3 M and 1.5 M sucrose and Golgi from the top of the 2.0 M sucrose. Each sucrose-separated fraction was subjected to further purification. The cell membranes were washed with original PIPES buffer and centrifuged at 3,000 rpm for 2 min. To separate apical epithelial membranes, the buffer was adjusted with 0.2% Triton X-100 and the mixture incubated at 4˚C for 5 min [42] [43] [52] . This treatment resulted in breaking up the phospholipids-rich membranes into smaller segments and that allowed us to separate apical membranes containing cholesterol, glycosphingolipids and glycoproteins. The latter membranes were recovered by low speed centrifugation at 3000 rpm for 2 min.
The mitochondria and lysosomes were purified from 10,000 xg spun fraction [54] - [56] [58] . From the initial fraction, the fluffy layer of broken mitochondria and ER microsomes that covered the crude mitochondrial pellet was suctioned off, the pellet was gently resuspended in five volumes of medium consisting of 70 mM sucrose, 0.2 M mannitol, 0.1 mM disodium EDTA, and 1 mM TRIS, pH 7.2, and the suspension was spun for 10 min at low speed (500 xg), the supernatant was recovered and spun for 10 min at 9000 xg [43] [53] - [56] . This manipulation was repeated three times. Thus purified preparation of mitochondria contained only occasional broken mitochondria, ER membranes, or lysosomes. The lysosomes were recovered from the mitochondrial fraction following treatment that afforded swelling of mitochondria [54] - [56] . The swollen mitochondria were separated from lysosomes by means of isoosmotic gradient [56] , and the lysosomes used for incubation with vesicles recovered from CC, and the organelles subjected to incubation with vesicle-free CHX CC. Mitochondrial fraction recovered from the gradient was used for the preparation of mitochondrial lipids and thin layer chromatography [42] [43] . The crude endosomes were isolated from post-mitochondrial supernatant subjected to centrifugation for 10 min at 27,000 rpm. The pellet was suspended in 50 mM phosphate buffered saline containing 0.5 mM EDTA, pH 7.4, adjusted with 62% sucrose to 40.6% and 5 ml aliquots transferred to centrifuge tube which then was over-layered with 3.5 ml each of 35% and 8% sucrose and centrifuged in Beckman rotor 40 Ti at 27.8 K for 90 min. The pure endosomes were recovered from 35% - 8% interphase.
2.4. Isolation and Characterization SPT from Purified ER
The ER isolated from other cell organelles as described previously and here under “Preparation of cellular organelles and membranes was washed with 50 mM Tris-HCl, 25 mM KCl, 5 mM magnesium acetate, pH 7.5 (TK buffer) to remove sucrose, suspended in the same buffer with or without 2 M urea and incubated on ice for 1h [44] . The ER incubates were then centrifuged at 15,000 xg for 30 min, the supernatants dialyzed against TK buffer while the residual ER membranes were washed with the same buffer. The obtained fractions of ER containing 50 - 100 µg protein were suspended in 0.1 M HEPES, pH 7.5, 5 mM dithiotreitol (DTT), 1 mM EDTA, 10 µM pyridoxal phosphate, 5 mM L-serine containing [3H]serine with final specific activity of 2,500 cpm/nmol and 0.1 mM palmitoylCoA. Thus prepared reaction mixtures contained in 80 µl of assay buffer were incubated at 37˚C for 20 min and subjected to lipid extraction with chloroform/methanol/0.2 N NH4OH (3:2:4, v/v/v/). Following centrifugation at 3000 rpm for 10 min, the chloroform layer was recovered and washed 4 times with water saturated with chloroform. Then each chloroform-recovered aliquot was dried, reconstituted in 100 µl chloroform, of which 90 µl were applied to thin layer chromatography plate and subjected to separation using solvent system consisting of chloroform/methanol/2N NH4OH (40/10/1, v/v/v). To identify the SphNH2 products, the remaining 10 µl of the extract was run separately alongside and stained with ninhydrin. The rest of the plate was subjected to iodine vapors and, the corresponding to ninhydrin positive bands, were scraped and their radioactivity counted. After establishing SPT activity in the ER membranes and 2 M urea extracts, further characterization of the fraction containing enzymatic activity was performed following protein polyacrylamide gel electrophoretic (PAGE) separation, and electroblotting of the separated fraction [44] . The results of these experiments are shown in Figure 1, which demonstrate that 56 and 58 KDa protein bands are responsible for SPT activity resulting in synthesis of SphNH2.
2.5. Generation and Purification of the Vesicles
ER- and Golgi-derived transport vesicles were generated in the presence of radiolabeled precursors according to procedure described previously [39] - [41] [43] [47] - [50] [53] and where indicated, in the presence of cycloheximide (CHX) [43] [51] [57] - [60] . The ER or Golgi membranes, mixed with CC, ATP-generating system, UTP, CTP GTP, fatty acyl CoA and water soluble cold or radiolabeled lipids precursors, were incubated for 30 min at 37˚C, centrifuged over 0.3 M sucrose and treated with stripping buffer at 2˚C for 15 min followed by centrifugation at 10,000 xg for 10 min to separate transport vesicles from ER or Golgi membranes. The separated from maternal membranes transport vesicles were recovered from the supernatant resulting from centrifugation of the mixture at 150,000 xg for 1 h. The crude fraction of the transport vesicles was suspended in 55% sucrose, overlaid with 55% - 30% gradient and centrifuged at 150,000 xg for 16 h. The purified transport vesicles were recovered from the gradients as reported earlier [39] [42] [43] [45] [47] - [50] [53] [60] .
2.6. Incubation of Transport Vesicles with ER, Golgi, Mitochondria, Endosomes and Lysosomes
One volume of radiolabeled vesicles (1.3 - 1.5 mg protein/ml) was suspended in one volume of CC or CHX CC (15 mg protein/ml), and added to one volume of cold cell organelles (5 mg protein/ml). The reaction was allowed to proceed from 0 - 30 min at 4˚C (control) and at 37˚C in the presence of ATP regenerating system consisting of 40 mM ATP, 200 mM creatine phosphate, 2000 units/ml of creatine phosphokinase, or in the ATP depleting system containing 5 mM glucose and 500 units/ml hexokinase. After incubation, the respective organelles were recovered by centrifugation through three volumes of 0.5 M sucrose at 3000 rpm for 5 min. The ER vesicles recovered from the supernatant after incubation with Golgi were purified on 55% - 30% sucrose gradient

Figure 1. Polyacrylamide gel electrophoretic separation (I) and depiction of purified fractions (III) of the serine-palmitoyl transferase (SPT) isolated from ER urea extract and represented in OL lanes, and the SPT remaining in the extracted ER denoted as IL lanes. The molecular markers are depicted in MM lane. The section II represents ER’s SPT proteins activity in the PAGE separated and electroblotted to the PVDF membrane fractions. As demonstrated, over 99% of SPT activity was concentrated in section D containing 56 - 58 K proteins.
and used in fusion experiments with mitochondria and lysosomes [47] [54] [58] . In turn, the Golgi fraction recovered from the first cycle of fusion was isolated, introduced to medium generating transport vesicles and, the reaction products of the second cycle of vesicles synthesis, subjected to separation into Golgi maternal organelles and Golgi-derived vesicles. The Golgi-derived vesicles were than incubated with cold endosomes. One volume of the recovered vesicles (0.9 - 1.1 mg/ml) was suspended in one volume of CC (15 mg/ml) and added to one volume of purified endosomes (5 mg/ml). As described for the ER vesicles fusion with Golgi, each reaction was allowed to proceed for up to 30 min and under the same conditions. Finally, the endosomes and the Golgi-derived vesicles remaining in CC were recovered and their radiolabeled SM quantitated. At that point, the radiolabeled endosomes and the nonreactive with endosomes Golgi vesicles were subjected to sphingomyelinase (SMase) treatment followed by radiolabeled lipid analysis [59] . In each experiment the fusion of transport vesicles with acceptor organelle was followed by treatment with 2 M urea at 4˚C or 0.5 M NaCl in order to remove the vesicles which have not undergone en bloc fusion. The associated but not fused vesicles were combined with the free vesicles that remained in CC medium. Then, the recovered organelles were centrifuged through 0.5 M sucrose, washed and subjected to radiolabeled lipid analysis. The lipid analyses concentrated on identification of SM and Cer, the products of Golgi-specific SM synthase, and SMase degradation, respectively. Therefore, to show SM and Cer, the alkali-susceptible phosphoglycerides were eliminated by subjecting lipid extracts to alkaline methanolysis and then thin layer chromatography along with standards of SM and Cer. In experiments containing radiolabel derived from palmitate labeling, the alkaline degradation products were subjected to one dimensional thin layer chromatography but in two solvent system, first to separate Cer and free fatty acids using mixture of petroleum ethers/ethyl ethers/acetic acid (80:30:1, v/v/v), while the second solvent system consisting of chloroform/methanol/water (65:35:8, v/v/v) was used to identify and quantitate SM that after first chromatography remained at the origin.
3. Results
Our investigation of the mechanisms by which cell organelles are refurbished converged on the role of ER, the continuous membrane system which represents the nuclear envelope and peripheral membrane network that while accommodating cell restitution and secretion demands, remains intact. This mystery perplexed us and led to initial concept that synthetic activity in and at ER proceeds in synchrony with biomembrane assembly and henceforward generation of transport vesicles that deliver the secretory cargo from ER to cell organelles and cell membrane [39] [40] [42] [45] - [50] [60] . Further insight and the expansion of the working hypothesis were gained with identification of membrane lipid markers that foreshowed vesicles delivery to the precise site of the cell membrane and the organelles. At the outset, ER-initiated transport bifurcates to deliver nuclear translation products to mitochondria and to Golgi and the transporters destination is certified through their membrane lipid specificity, the integral membrane proteins, and the vesicular cargo [39] [43] [47] [50] [53] . We have found, that while mitochondria-specific transporters are marked by the presence of phosphatidylglycerol (PG), and absence of phosphatidylinositol (PI) and Cer, the Golgi-destined transporters contain the latter two [43] [45] [46] [50] [53] .
Based on these fundamental and distinctive dissimilarities, we reasoned that Golgi-directed transport must represent a mixture of vesicles marked for delivery of: (1) the materials that restore Golgi structure and function, (2) the constituents of cell membranes and cell secretory products and (3) the essential elements for endosomes to refurbish lysosomes and thus carry out intracellular catabolic tasks.
Within the assigned categories of the vesicles directed from ER to Golgi, our studies documented structure and contents of apical and basolateral transport vesicles which revealed that their membrane assembly is dependent on the SphNH2- and Cer-synthesizing ER -constitutive enzymes providing SPhL core for GSL and SM assembled in Golgi [45] [47] [50] [53] [60] . The challenge confronted here was to identify membrane markers that differentiate between SM-containing basolateral and endosomal vesicles that with other membrane constituents co-determine their divergent destination sites. Earlier, we have found that initial steps in synthesis of SM and GSL lipid core depend on ER constitutive enzyme, the SPT which we have shown to consist of two forms, one susceptible to extraction with 2 M urea, and one resistant to the treatment [44] . Here, we present results of the study pursuing the impact of SPT placement on the synthesis of transport vesicles destined for Golgi, and onward to apical and basolateral cell membranes, and to endosomes. As illustrated in Figure 1, both forms of SPT are represented by 56 - 58 K peptides, and the urea-released proteins remain as active as those that following extraction remain in the ER membranes. The urea freed SPT contained 49.6% (42.6 - 56.5 range) of the total enzyme activity (Figure 2), yet the intact ER membranes while partially depleted of SPT, generated transport vesicles.

Figure 2. SPT activity in the purified intact ER, and the urea extracted ER. As demonstrated, on an average 50% SPT activity remained in the urea- treated ER membranes.
With the role of SPT in mind, we next investigated the impact of OL SPT removal on the production of ER generated transport vesicles delivered to Golgi. As shown in Figure 3, and reflected in the SM production by the Golgi-destined, and in turn, Golgi-produced transport vesicles, the urea-extracted OL SPT-depleted ER delivered 69% less (N = 3) vesicles.
The depletion of the urea accessible and thus cytosol-facing SPT, disproportionally reduced production of ER-generated transport vesicles dedicated for endosome-specific transport, but it did not hinder the synthesis of GSL and SM containing vesicles delivering cargo to cell membrane.
As calculated from the results presented in Figure 3, which demonstrate the impact of the ER extraction on the delivery of the material to Golgi and formation of Golgi transport vesicles, the untreated ER produced 49.4% Golgi vesicles that reacted with endosomes, 41.2% without affinity for endosomes and 9.4% radiolabeled ER vesicular lipids that remained with Golgi membranes. The urea extracted ER produced 23.9% Golgi vesicles reacting with endosomes and 63.7% nonreactive with endosomes, while 12.4% vesicular radiolabel remained with Golgi membranes. Expectedly, the treatment with urea had an impact on ER biomembrane synthesis, as clearly demonstrated in the reduced synthesis of PS, but the disproportional impact of OL SPT removal on the production of the Golgi transport vesicles supplying endosomes was unforeseen. Accessibility to urea extraction

Figure 3. Characterization of the [3H]serine labeled lipid profiles of the Golgi-derived transport vesicles that originated from the serine labeled intact and urea extracted ER. Briefly, the serine labeled ERs were subjected to incubation resulting in generation of ER transport vesicles, followed by the incubation of the purified ER vesicles with Golgi and subsequently subjecting Golgi to treatment affording Golgi transport vesicles. Thus obtained Golgi vesicles were used in incubation with endosomes. Consequently, the experiment afforded the chase of radiolabeled lipids synthesized in ER, used for formation of ER-derived transport vesicles, transferred to Golgi, used in formation of Golgi transport vesicles and finally transferred to endosomes or remaining in reaction mixture and not showing affinity for endosomes. The aliquots of transport vesicles obtained at the described stages of the experiment, the endosomes and the residual Golgi membranes recovered after separation of Golgi transport vesicles were subjected to lipid extraction and serine-labeled lipid identification.
pointed to SPT exposure and its activity in the ER leaflet facing cytosol. Remarkably and unexpectedly, the results suggested that the removal of OL SPT was mainly associated with generation of Golgi vesicles reacting with endosomes (reduction from 54.5% to 27.3%), while the production of the vesicles not reacting with endosomes remained unaffected.
At this stage of the investigation the results of the experiments with partially depleted SPT from ER allowed us to presume that the partial removal of SPT disproportionally or exclusively impacted formation of Cer on ER leaflet facing cytosol and that impacted production of ER, and in sequence, Golgi vesicles that reacted with endosomes. The critical data that implicated ERs OL SPT in assembly of endosome-directed Golgi transport vesicles were obtained from experiments that involved treatment of Golgi transport vesicles with sphingomyelinase (SMase) (Figure 4). When subjected to SMase digestion (30 min incubation at 37˚C with 10 U SMase), 50% - 60% of SM in Golgi-derived vesicles generated from the untreated with urea ER was susceptible to enzymatic

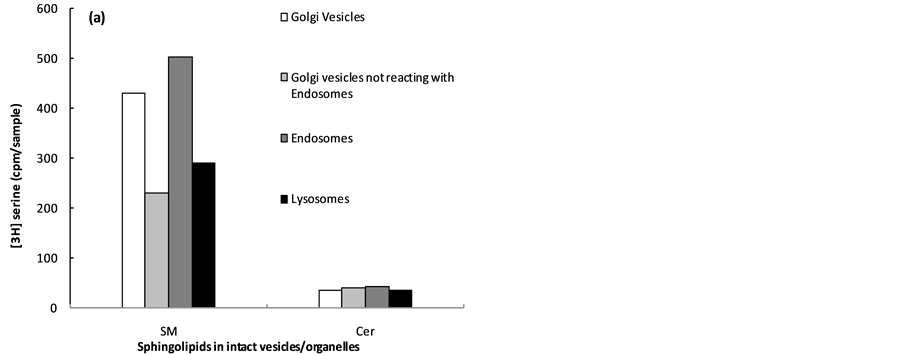

Figure 4. Sphingomyelinase (SMase) susceptibility of the SM of Golgi-derived transport vesicles, endosomes and lysosomes. The [3H]serine-labeled Golgi transport vesicles and the transport induced label deposited in endosomes and lysosomes were characterized for contents of SM and Cer before SMase treatment (a), and after enzymatic treatment (b). The % of SM susceptible to SMase degradation is shown in (c). As demonstrated, the serine labeled SM derived from labeled Cer synthesized in ER was partially susceptible to digestion in Golgi transport vesicles and not susceptible to SMase in Golgi vesicles not reacting with endosomes. In contrast, the SM transferred via Golgi transport vesicles to endosomes and lysosomes was entirely converted to Cer. In our interpretation, the SMase susceptibility of the SM-labeled transport vesicles reacting with endosomes and lysosomes demonstrates that SM of their membrane constitutes outer leaflet of the transporters fusing with endosomes and lysosomes.
degradation, whereas the Golgi vesicles not reacting with endosomes were not affected by SMase treatment. Moreover, the radiolabeled SM transferred by Golgi transport vesicles to endosomes was completely converted to radiolabeled Cer. An identical outcome of the experiment with SMase was attained in reaction of SM labeled endosomes with lysosomes. As demonstrated, the lipid extracts of the lysosomes incubated with SMase contained Cer. Thus, as became evident, the Golgi produced SM-containing transport vesicles susceptible to SMase digestion, and affected by the removal of OL SPT, was those destined for endosomes and lysosomes. Subsequent analysis of the Golgi-derived SMase resistant vesicles determined that this is the characteristic feature of the vesicles nonreactive with endosomes, while the vesicles that were not susceptible to SMase and retained intact SM were cell membrane-specific transporters.
To assert that following SMase treatment the recovered endosomal material while containing labeled Cer instead of SM remained intact, the containment of neutral and acidic endosomal SMase activity was investigated. As shown in Figure 5, the incubation of SM-depleted (Cer containing) endosomes in the medium with radiolabeled SM substrate has not generated the products that would suggest leakage of endosomal neutral or acidic SMase. However, with addition of Triton X-100, the enzymatic activity was released from the organelles and the radiolabeled water-soluble product derived from the SM substrate was detected (Figure 5). Thus, the experiment with endosomes containing Cer instead SM asserted that fusion of Golgi-transport vesicles with endosomes produces membranes containing SM in the outer leaflet that is susceptible to SMase, but the organelle retains the Cer and remains intact.

Figure 5. The impact of SMase treatment on the retention of lipolytic enzymes in endosomes. Intact and SMase treated and SM-depleted endosomes were subjected to treatment in the cytosol medium enriched with [14C]choline-labeled SM and incubated for up to 30 min. The release of [14C]choline was quantitated by measuring the release of water-soluble radiolabel ([14C]choline). As evident, the digestion of SM by the intact and SM-depleted endosomes remains unchanged throughout incubation period. However, addition of Triton X-100 to either SM-containing or SM-depleted endosomes releases endosomal SMase activity. The results of the experiments shown support the results demonstrating endosomes and lysosomes susceptibility to the externally provided SMase and their resistance to intra-organellar SMases transported to lysosomes via Golgi vesicles reacting with endosomes.
The characterization of the stage where Golgi-delivered SphNH2-radiolabeled material incorporated into the Golgi-derived vesicles and in turn into endosomes and lysosomes (Figures 4(a)-(c) and Figure 5), provided convincing evidence that ER vesicles labeled in SphNH2 moiety are introduced into the endosomal and lysosomal membrane in the same membrane alignment as OL SM vesicles produced by Golgi. Moreover, the fact that upon SMase treatment, the retained by endosomes and lysosomes SM-radiolabeled Golgi vesicles afforded radiolabeled Cer and the treated organelles remained intact and further degradation (presence of labeled SphNH2) was not detected, gave the support to our contention that (1) the vesicles directed to endosomes and in turn to lysosomes originated in ER and were assembled with the aide of SPT facing cytosolic environment, (2) they fuse and thus restore these organelles membranes, (3) the results confirmed that lysosomal membranes are rebuilt in the process of fusion with ER-derived vesicular membranes modified in Golgi, and (4) the products of fusion remained external SMase susceptible but were not exposed to internal endosomal/lysosomal SMases.
In contrast, the Golgi vesicles remaining in the medium after SMase treatment or reaction with endosomes (Figure 4(a), Figure 4(b)), whether from the experiments utilizing untreated ER or one extracted with urea, retained SM. This allowed us to conclude that the cell membrane-destined vesicles differ from those delegated to endosome, their SM and GSL are assembled in the inner leaflet of the transport vesicle membrane and therefore their SM is not affected by SMase. Consequently, their fusion with cell membrane exposes their SphLs (GSLs and SM) to the outer cell environment. Indeed, the GSL of gastric epithelial cells are exposed on the apical cellular surfaces, whereas the intact hepatocytes are susceptible to SMase treatment.
Collectively, the investigation of the ER without cytosol-exposed SPT allowed us to differentiate between its utilization in generation of vesicles transporting cargo to endosomes and those intended for restitution of cell membrane. As reflected in the data presented here, the differential placement of SM in the transport vesicles, made possible to recognize endosomes assembly and set them apart from the products of endocytotic activity, the vesicles that represent resected fragments of cell membrane. As we have shown in the prior communication [42] , the endocytotic vesicles are SMase resistant, and represent the cell membrane autophagosomes. As well known, endocytosis creates vesicles by the inversion of cellular membrane, and just as membrane of the synthetic transport vesicles that fuse with cell membrane, the reaction produces vesicles with the SM in the inner leaflet of the membrane. In our conclusion, based on the findings delineating cellular restitution cycle, the SM lipid core integration in the outer and the inner leaflet of ER-assembled vesicles is the major lipid marker that allows to separate the synthetic, cargo-delivering vesicles initiated in ER and marked in Golgi for endosomes and lysosomes, from the endocytosis-produced autophagosomal vesicles destined for lysosomal digestion.
The other critical function-oriented distinction between endosomes and the endocytosis-produced autophagosomal vesicles is that endosomes not only contain hydrolytic cargo but, as the part of the sequence of events, also fuse and rebuilt lysosomal membranes and provide lysosomes with the newly synthesized cargo. As lysosomes represent the final stage of cellular cycle, the process of renewal and degradation must be contained within the organelle. In our concept, supported by the data presented earlier [42] and in Figure 6, the organelle renewal and terminal degradation are observed during the catabolic processing of cell organelle-derived autophagosomes. The concomitant processing of autophagosomes and lysosomes is accomplished via lysosomal engulfment and internalization of the engulfed autophagosome together with engulfing portion of itself. As demonstrated in Figure 6(a), Figure 6(b), the incubation of cell membrane endocytosis-derived autophagosomes labeled through incorporation of serine or palmitate into PL and SPhL with cold lysosomes afforded SphNH2 (panel A) and SphNH2 and fatty acids (panel B). The lysosomal self degradation during endocytosis-originating cell membrane autophagosomes degradation, is reflected in the outcome of the reactions employing converse, the labeled lysosomes with serine (panel C) and palmitate (panel D) incubated with cold autophagosomes. In both sets of experiments terminal degradation of SphLs (that can originate only from cell membrane reflects degradation of endocytosis-derived autophagosomes) is observed as labeled SphNH2 and fatty acids (FA) are identified, while Cer is no longer detectible. Moreover, the labeled lysosomes retain fraction of labeled SM. These results clearly show, that endocytosis-produced vesicles i.e. the membrane-derived autophagosomes, in order to be digested to basic metabolites (SphNH2 and FA), must be first engulfed by the lysosomes, and then degraded with part of lysosomal membrane. The presence of labeled SM in experiments depicted in Figure 6(c), Figure 6(d), most likely represents the newly restored lysosomal membrane that is not internalized and hence not subjected to degradation. Such turn of events would support the concept that the reaction of endosomes with lysosomes generates SM-containing membrane that maintains and refurbishes lysosomes and the process is concomitant with lysosomal degradative processes. Moreover, the Golgi transport




Figure 6. Major differences between reactions demonstrating endocytosis-derived vesicles with lysosomes and endosomes reaction with lysosomes. Panel A and B illustrate outcome of the reaction between serine- and palmitate-labeled endocytosis products that is cell membrane autophagosomes containing labeled phoshoglyceride phospholipids/phosphatidylserine (PL/PS), SM, GSL, while Cer, SphNH2 and FA (Panel A) are in trace amount. Following incubation with lysosomes, the serine labeled (A) shows presence of SphNH2, and traces of PL/PS and SM that demonstrates that cell membrane autophagocytes were completely degraded. The results with palmitate labeled autophagosomes show degradation that affords generation of SphNH2 and FA, and thus illustrates terminal hydrolysis of the autophagocytes in lysosomes. In contrast, reaction of serine-labeled lysosomes and cold autophagosomes reflects partial susceptibility of the lysosomal membranes, partial reduction of PS/PL, SM, and some enrichment in SphNH2 (panel C) and SphNH2 and FA (panel D). In our interpretation, the results obtained with labeled lysosomes suggest that degradation of autophagosomes involves partial degradation of lysosomal membranes. Such outcome of the reaction, without release of lysosomal enzymes is only feasible if lysosomal membranes engulf autophagosomes and both are internalized and subjected to internal lysosomal hydrolysis.
vesicular pathway delivers newly synthesized membrane containing SM in the OL and just as in case of all organelles delivers cargo via fusion of ER-synthesized Golgi-modified vesicles with endosomes and lysosomes, and therefore the outer leaflet of lysosomes is SMase susceptible, but not accessible to inner lysosomal hydrolytic enzymes.
Consequently, at any point of active process of endocytic excision of the cell membrane and the active pro- cess of fusion of endosomes with lysosomes, and simultaneous cellular organelle-derived autophagosomes digestion in the lysosomes, the lysosomal membrane is being refurbished, intact, and its characteristics are retained because they are endowed through a sole process of ER-originated vesicular transport and restitution. The asset of the delineated process established through the investigation of lipids, concentrating in this communication on SM synthesis and degradation, is that it eliminates prospect of mixing, combining or interspacing various membranes components destined for engulfment by lysosomes and catabolic recycling with that of newly synthesized membrane replacements that afford restitution of the organelles. Clearly, the SM-containing endosomal membranes are sole suppliers that regenerate lysosomes, and the structural features of lysosomal membranes reflect such derivation.
Together, our results provide concrete evidence that every organelle within the cell and the cell membrane retain strictly guarded characteristics that otherwise, with indiscriminate incorporation of any lipid structure, would change into mixture of undefined and ill-functioning entities.
4. Discussion
Cells and their organelles are surrounded by the lipid-bilayer membranes that compartmentalize biochemical reactions and pathways, and provide setting for the membrane-specific proteins to perform their designated functions [61] - [67] . Whether the cell is in state of quiescence or is dividing to create new cells, it is central to tissue homeostasis that the processes of renewal and metabolic restitution are faithfully reproduced, the newly synthesized products take the place of the excised elements, and the lysosomal cell debridement ensues. Thus, from the outset of the coded metabolic conversions, the retention of the characteristic specificity of the cellular and organellar membranes defined by the protein and specific lipids, encrypt their paramount structural attri- butes that guard cells from untoward conversions [14] - [16] [20] [30] . The specific lipid environment that is repeatedly assembled for every integral membrane protein is not acquired at any time, or by chance, but reflects co-translational creation of the membrane for which the particular structure is essential [39] -[42] [45] -[50] [53] [60] . Bearing in mind the complexity of membrane receptors, their distinct positioning and multiple traversing in order to achieve competent form, the simplistic view of their incorporation into membrane after translation fails to explain the phenomenal fidelity of their placement within the specific membrane and delivery to the precisely assigned site [11] [23] - [25] [33] [38] [39] [61] [62] [67] [68] . Owing to vacuity of information on the significance of co-assembly of membrane lipid environment, which together with membrane protein marks destination of the ER-constructed entities, a series of thought-provoking scientific findings offers highly detailed, yet dim and distorted, view of cell renewal. The example of such void is the interpretation of Toll-like receptors signal transduction as “promiscuous” due to binding reactivity with phosphatidylinositol phosphates (PIPs) and phosphatidylinositol bisphosphates (PIP2s) but “beneficial means of diversifying the subcellular sites of innate immune signal transduction” [5] [69] - [72] . In our view, established through examination of the vesicular specificity of cellular transport, the results provide exquisite example of the vesicles transporting specific TLRs to the apical and basolateral membrane armed with their specific destination markers in form of PIPs (PI3P and PI4P), respectively [40] [41] [50] [53] [60] . Moreover, the vesicles named endosomes are most likely the cell membrane fragments (autophagosomes) undergoing excision and on their way for lysosomal degradation. The presence of “promiscuous” reactivity with PIP2s (PI3,5P and PI4,5P) reflects their apical and basolateral cell membrane derivation. As established, the PIP2s are generated in cell membrane, while Golgi to which newly assembled vesicles are delivered, including those directed to endosomes, provides them with PI3P and PI4P markers [48] - [50] [53] . Until then, the ER-derived transport vesicles destined to Golgi contain PI only [39] [40] [43] [45] [47] [49] [50] [53] .
Thus, while we share the idea that intracellular vesicular transport plays a significant role in the process of gene products delivery, our concept diverges from the widespread assumption that transport vesicles are mere inert containers delivering proteins [12] [14] [21] [25] [26] [29] [30] [32] [37] [38] [61] [62] [64] [70] - [72] . Our investigations of intracellular transport have shown that transport vesicles, at the inception of their synthesis in ER, acquire organelle and cell membrane specific proteins intercalated within the co-translationally produced specific lipids assembled into the membrane that provide the specific environment for the integrated proteins and, as such are delivered and en bloc fused with the site for which the replacement has been signaled [5] [39] [42] [46] - [48] [60] . Pursuing this concept, we have demonstrated that ER vesicles generated for transport via Golgi differ from those directed for the restitution of mitochondria [43] . The lipid environment produced for renewal of mitochondrial membranes is enriched in PG and devoid of PI and Cer. In contrast, the Cer- and PI-containing transport vesicles deliver ER cargo to Golgi, whereupon further transformations of their membrane lipids signal their delivery to endosomes, and the apical and basolateral membranes [39] [42] [45] [47] [48] [50] . While in the earlier obtained evidence we identified that PIPS (PI3P, PI4P, PI5P) marked other than apical membrane destination, but the quandary remained regarding the specific attributes of the vesicles containing PI4P and delegated to basolateral and/or endosomal membranes [46] [49] [50] . After all, the identified PI4P could not represent the exclusive factor that determined delivery of the specific cargo to both sites since this would lead to erroneous delivery and improper cell membrane restitution [23] [29] [30] . As well known, normally, such an event is not taking place because the endosomal hydrolytic cargo is not delivered through basolateral release or newly synthesized albumin delivered to lysosomes. The other specific membrane lipid, SM, assembled by Golgi SM synthases indicates further modifications of the endosome and basolateral vesicular membranes, but its presence on both transporters has not provided satisfactory explanation regarding its role in paving vesicles destination. The explanation in terms of existence of lipid flippases and floppases is offered [13] as the determining factor in SM placement, but as is the case of lipid transporting proteins [34] [35] their involvement in translocation is investigated using artificial liposomes made up of lipid mixture that is not reflecting cell membrane profiles. Thus, further doubts arise from experimental paradigms employing same liposomal composition for the processes in organelles and real cell membrane containing different lipid arrangements and orientations and from the omission of the role and the placement of the SPhL core and the Cer, synthesized in ER [13] [33] [34] [73] . It seems most likely that the appearance of lipid flipping results from experimental conditions that allow in vitro unphysiological interactions. Hence, in the view of the enumerated inaccuracies to reproduce cellular events, it is debatable whether observed flipping plays any function in custom tailoring of the inner and outer leaflet of Golgi-derived transport vesicles membranes in situ.
In our earlier study we demonstrated the significance of Cer in generation of ER transport vesicles delivering apomucin and albumin to Golgi for apical and basolateral cargo discharge, respectively [39] [45] [47] [50] [53] . In both instances, the Cer moiety synthesized in ER and as such transported within membrane of ER transport vesicles, was used for synthesis of GSL and SM, both assembled on the inner leaflet of Golgi, and after delivery to apical and basolateral membranes the GSL and SM faced outer cell environment [42] [47] [53] [59] [60] . Yet, the fraction of remodeled in Golgi vesicles that contained SM and PI4P, was not delivered to basolateral membranes but displayed affinity for endosomes. During our investigations of SphNH2 synthesis, it came into the view that constitutive enzyme of ER, the SPT, was localized to the inner and outer leaflet of the membrane [44] . Both, the cytosol-facing and intraluminal SPT were engaged in synthesis of SphNH2 and upon stripping the outer SPT activity; the resultant ER retained capacity to produce transport vesicles containing Cer and transport apomucin to Golgi. As we found out here, the fact that remained unnoticed in the earlier project concentrating on the apical and basolateral transport was that the release of cytosol-facing SPT weakened ER capacity to generate the endosome-specific transport vesicles. The diminished ability to assemble the endosome-directed vesicles was not resulting from inactivation of the SPT, since the urea extracted enzyme remained active, but rather from the fact that the urea-extractable SPT produced the SPhL core that was an essential component for the assembly of the specific vesicular membrane delivering cargo to endosomes. The reduced competence to assemble endosome-directed transporters was clearly established after the SPT-depleted ER labeled vesicles were chased through Golgi and thus Golgi-produced labeled transport vesicles directed to endosomes was estimated. As documented, the transport marked by the presence of labeled SPhL core in endosomes’ SM was drastically reduced, though the vesicles carrying apomucin and albumin cargo were not affected, all the while, the level of so-called flippases or other factors stipulated to impact organelle-specific transport was uniformly satisfied by utilizing in the experiments the fresh, active, and complete cytosol medium. This allowed us to voice the assertion that the ER’s outer leaflet SPT was determining factor in the assembly of endosome-specific transport vesicles membrane and that the SM created in Golgi from the cytosol facing SPhL core remained in membrane leaflet facing cytosol. As shown, the incubation of endosomes and endosome-directed transport vesicles with SMase, in the absence of the detergent, generated Cer. In contrast, the Golgi-assembled transport vesicles carrying apical and basolateral cargo must have used SPhL core integrated into the inner ER membrane and, therefore were not susceptible to SMase treatment. Only with addition of 0.2% Triton X-100, the apical and basolateral vesicles’ SM was digested and generated Cer. These results documented that endosome-directed Golgi transport vesicles are derived from the ER-preassembled membrane containing Cer in the outer leaflet and consequently their SM is formed in their outer, cytosol-facing membrane surface, whereas the apical and basolateral transport vesicles contain SM in their inner leaflet and are the product of SPT of the inner membrane surface of ER. Thus, the conclusion was reached that specific lipid marker of endosome assigned transport vesicles is SM present in their outer, cytosol-facing leaflet of their membranes, and just as in other vesicular transporters, the vesicles incorporate en bloc to the endosomes.
The attribute gained by endosomes from endosome-delegated transport vesicles, reflected in the SM placement, is that this feature provides their membrane with a characteristic that differentiate them from the endocytosis-released cell membrane-derived excisions. While the former carry newly synthesized cargo for the hydrolytic activity of lysosomes, the latter are the rejected fragments of cellular membrane destined for lysosomal hydrolysis and do not carry hydrolytic enzymes or share characteristic membrane features with endosomes. Indeed, without demonstration of the characteristic features of the outer and inner leaflet of the endocytic and endosome vesicles it is impossible to distinguish one from the other, and that appears to generate an assumption that the endocytic and endosomal vesicles commingle before lysosomal degradation [13] [35] [64] [74] [75] . Consequently, in the study presented here, we provided concrete evidence that endocytic vesicles carry their SM in the inner leaflet, while in Golgi-released endosome-directed transport vesicles SM is in the outer, cytosol facing leaflet. This distinction in SM placement in the endosomes is transferred onto lysosomes that by fusing with structures released from endosomes gain their characteristic membranes while they become refueled with newly synthesized hydrolytic cargo. On the other end, the endocytic vesicles do not fuse with lysosomes, instead they are engulfed by the lysosomal membranes and as we determined earlier, the engulfed endocytic vesicles and the lysosomal membrane involved in the engulfment process, are internalized and digested [42] . The results presented regarding susceptibility to SMase and the differences between hydrolytic products derived from SMase alone and from internal lysosomal digestion, provide sage evidence that confirms our view on the origin of endosomes and endocytic vesicles, and provide clear view of the cellular events that restore and cleanse cells in highly ordered fashion. With this in mind, we feel that interpretation of the endosome composition and function, as presently portrayed by others, is somewhat distorted by implying that cell membrane endocytotic products are partially incorporated into the organelle and as such that portion of excised membranes built endosomes. In the light of our findings, the endocytosis events, as described so far, are subjected to highly convoluted interpretation. It is highly unlikely that cell membrane-rejected fragments are reused to build endosomes before catabolic process is complete, or that singled out receptors reenter exact space in the cell membrane. Firstly, and as discussed earlier, the intercalation of membrane protein is too complex and cannot be reproduced in the similar fashion as dictated in ER, and secondly, the logical progression of cell repair must satisfy requirements of the cell surface and eliminate the excised membranes entirely. In our interpretation of the results that have shown systematic assembly of membrane components and their markers, the true endosomes are consisting of the newly assembled ER-derived membranes whose characteristics are reflecting the composition and membrane markers of transport vesicles containing SM in the outer cytosol facing leaflet and carrying enzymatic cargo for lysosomes. The specificity of the vesicles genesis allows deducing that endosome membrane components and the organelle contents reflect the composition of the synthetic transport vesicles but not the endocytotic vesicles destined for degradation.
At this stage of the cellular transport documentation underscoring the role of specific lipids in the ultimate environment for the protein spanning the membrane and branding vesicles surface to find the place in cell provide clear extension of our concept regarding cell-specific destination transport, restitution and retention of function, generates clear and genuine view of the processes in living cell. More so, our results demonstrate that placement of the enzyme on one or the other site of membrane leaflet determines the ER site for translation of the endosome destined cargo, as well as vesicular membrane protein and lipid components, without intervention of flippases or lipid transporting proteins [13] [34] [35] . And, in the same construal of the metabolic cycle, the products of endocytosis, distinguished by their SM placement, are reflecting the composition of apical or basolateral membranes and are recognizable as genuine cell membrane-derived autophagocytes which are destined for the engulfment by lysosomal membrane and intralysosomal degradation. In keeping with our results-driven conclusions that all cell membranes are in continuous cycle of synthesis, replacement and degradation, the speculation with regard of the restitution of the ER and Golgi, the organelles most heavily involved in synthesis, modification, and trafficking of protein, is eminent but not through reprocessing of transport vesicles [14] [15] [19] [21] [29] [31] [33] .
As found out in the case of ER, the part of membrane adjacent to nuclear contents provides the link between nuclear pores, and as demonstrated with nuclear turnover of phosphoinositides, is in a constant lateral motion transporting cytosolic proteins to the nucleus, while in the same membrane, the lipids’ synthesis-propelling motion delivers RNAs to cytosol [40] [41] . The reemerging from nucleus membrane must serve as the initial template that signals the translation of just produced and released mRNA and the integration of the product within co-synthesized organelle and/or membrane-specific lipids. Thus, during this process, the structural renewal of the ER and its functional ability in terms of transport vesicles production is accomplished. The structural components that define ER and provide identity and marker lipids for Golgi-directed transport are SPT and Cer synthase. Undoubtedly, the glycosyltransferases and the enzymes that are involved in posttranslational modifications of vesicles transiting through Golgi reflect Golgi structural make-up. Because of simultaneous synthesis of the vesicles refurbishing organelle membrane structure and function-related vesicles processing transported in and out of the organelles, the determination of a true organelle’s membrane lipid composition is not easily accomplished, and at this stage, the closest approximation of the organellar lipid composition can be achieved when cellular synthetic processes are halted [42] .
As we have demonstrated earlier, the best source of the information on the organelle structural components are their autophagosomes. Since the autophagosomes are the organelle and cell membrane fragments that are no longer involved in cargo processing, or in the modification of the transporters en route to their destination sites, their membranes reflect genuine composition of the organelle from which they are derived [42] . Misleadingly, numerous studies concentrated on the identification of common lipid and protein markers, and the investigations have been carried out on the totality of organelle and cell membrane-rejected fragments. Hence, the information gathered in many of these studies could not be brought to conclusive statement [19] [64] [68] [69] [76] - [82] . Consequently, the lipid-specific markers that could be utilized for the verification of autophagosomes derived from the individual organelles are lacking. In our judgment, just as the chase after common lipid markers in autophagosomes, the conclusions could not be reached from the investigations on proteins alone, as the techniques identifying protein are not distinguishing between newly synthesized proteins in the synthetic transport vesicles from those in autophagosomes en route to lysosomal degradation. The erroneous assumptions regarding commonality of the synthetic and autophagosomal vesicles composition seem to affect the concept of the so-called lipid rafts and generated multiplicity of explanations, and even denials of their existence [35] . In this regard, we believe that the identified cell membrane “rafts” reflect inserts, the fused patches of membranes of the transport vesicles delivered and fused to apical and/or basolateral membranes. The difficulties in reaching agreement on their presence stem from the fact that the majority of these studies are performed on cells that differ in their membrane composition. The example of such differences is in the case of gastric epithelial cells containing apical membrane enriched in GSLs, while hepatocytes although are epithelial cells contain substantially less GSLs, and the SM-containing transport vesicles are delivered to their basolateral membrane. Our investigations of lipid biomembrane synthesis and its sequential and organelle-specific transformations illuminate “rafts composition phenomenon” fitting gastrointestinal epithelial cells’ apical membrane that is highly enriched in GSL and glycoproteins’. The fact that application of the same criteria to hepatocytes would fail to detect the rafts, reflects that basolateral membranes rafts are not enriched in GSL, and other lipids and membrane receptors should be used for their identification. In our understanding, the appearance of the so-called rafts in cell membranes signifies frequent vesicular fusion with cell membrane, while secreting the cell specific product. Thus it is not a coincidence that the rafts are first identified as the structures specific to cells involved in secretion, while their presence in other cells are not discernable.
The gaps in the concept of signal transduction-induced cell restitution seem to originate from the singular concentration on gene products function in cell-disrupted environment. That maneuvers our understanding of cells restitution as explicit protein phenomenon. Undeniably, the gene products are of paramount significance, but their function is controlled by precisely determined by the cytosol- and organelle-governed environment. This premise, however, is not demonstrable with the sophisticated techniques used to detect protein transport only.
In our concept, the sequential execution of the cell restitution cycle, offers the complete picture of the exquisite processes that originate in ER membrane with the synthesis of the replacement elements and is completed in lysosomes where the cell replaced elements are catabolized. The steps in this process require complete fidelity of the “replacement parts” composition, and are balanced through the precise resections and debridement of the cell. The elements of cell organelle structure that allow us to differentiate between the above characterized processes are embedded in membrane lipid organization and their specific profiles predetermined and preassembled in ER.
References
- Kapp, L.D. and Lorsch, J.R. (2004) The Molecular Mechanics of Eukaryotic Translation. Annual Reviews of Biochemistry, 73, 657-704. http://dx.doi.org/10.1146/annurev.biochem.73.030403.080419
- Koga, H., Kaushik, S. and Cuervo, A.M. (2011) Protein Homeostasis and Aging: The Importance of Exquisite Quality Control. Ageing Research Review, 10, 205-215. http://dx.doi.org/10.1016/j.arr.2010.02.001
- Walter, P. and Ron, D. (2011) The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Sci- ence, 334, 1081-1086. http://dx.doi.org/10.1126/science.1209038
- Vincenz, L. and Hartl, U. (2014) Sugarcoating ER Stress. Cell, 156, 1125-1127. http://dx.doi.org/10.1016/j.cell.2014.02.035
- Jung, H., Gkogkas, C.G., Sonenberg, N. and Holt, C.H. (2014) Remote Control of Gene Function by Local Translation. Cell, 157, 26-40. http://dx.doi.org/10.1016/j.cell.2014.03.005
- Lusk, C.P., Blobel, G. and King, M.C. (2007) Highway to the Inner Nuclear Membrane: Rules for the Road. National Review of the Molecular Biology, 8, 414-420. http://dx.doi.org/10.1038/nrm2165
- Siniossoglou, S. (2009) Lipins, Lipids and Nuclear Envelope Structure. Traffic, 10, 1181-1187. http://dx.doi.org/10.1111/j.1600-0854.2009.00923.x
- Adams, R.L. and Wente, S.R. (2013) Uncovering Nuclear Pore Complexity with Innovation. Cell, 152, 1218-1221. http://dx.doi.org/10.1016/j.cell.2013.02.042
- Rothman, J.E. (1994) Mechanisms of Intracellular Protein Transport. Nature, 372, 55-63. http://dx.doi.org/10.1038/372055a0
- Bilder, D., Schober, M. and Perrimon, N. (2003) Integrated Activity of PDZ Complexes Regulates Epithelial Polarity. Nature Cell Biology, 5, 53-58. http://dx.doi.org/10.1038/ncb897
- Sharma, N., Low, S.H., Misra, S., Pallavi, B. and Weimbs, T. (2006) Apical Targeting of Synthaxin 3 Is Essential for Epithelial Cell Polarity. Journal of Cell Biology, 173, 937-948. http://dx.doi.org/10.1083/jcb.200603132
- Kawano, M., Kumagai, K., Nishijima, M. and Handa, K. (2006) Efficient Trafficking of Ceramide from Endoplasmic Reticulum to the Golgi Apparatus Requires VAMP-Associated Protein-Interacting FFAT Motif of CERT. The Journal of Biological Chemistry, 281, 30279-30288. http://dx.doi.org/10.1074/jbc.M605032200
- van Meer, G., Halter, D., Sprong, H., Somerharju, P. and Egmond, M.R. (2006) ABC Lipid Transporters: Extruders, Flippases, or Flopless Activators? FEBS Letters, 580, 1171-1177. http://dx.doi.org/10.1016/j.febslet.2005.12.019
- Fölch, H. (2008) Regulation of Membrane Trafficking in Polarized Epithelial Cells. Current Opinions in Cell Biology, 20, 208-213. http://dx.doi.org/10.1016/j.ceb.2008.01.003
- Pfeffer, S.R. (2007) Unsolved Mysteries of Membrane Traffic. Annual Review of Biochemistry, 76, 629-645. http://dx.doi.org/10.1146/annurev.biochem.76.061705.130002
- Gohil, V.M. and Greenberg, M.L. (2009) Mitochondrial Membrane Biogenesis: Phospholipids and Proteins Go Hand in Hand. Journal of Cell Biology, 184, 469-472.
- Südhof, T.C. and Rothman, J.E. (2009) Membrane Fusion: Grappling with SNARE and SM Proteins. Science, 323, 474-477. http://dx.doi.org/10.1126/science.1161748
- Beck, R., Prinz, S., Distellköter-Bechart, P., Röhling, S., Adolf, F., Hoehner, K., et al. (2011) Coatomer and Dimeric ADP Ribosylation Factor 1 Promote Distinct Steps in Membrane Scission. Journal of Cell Biology, 194, 765-777.
- Brodsky, J.L. (2012) Cleaning Up: ER-Associated Degradation to the Rescue. Cell, 151, 1163-1167. http://dx.doi.org/10.1016/j.cell.2012.11.012
- Keiser, M., Alfalah, M., Propsting, M.J., Castelletti, D. and Naim, H.Y. (2006) Altered Folding, Turnover, and Polarized Sorting Act in Concert to Define a Novel Pathomechanism of Congenital Sucrose-Isomaltase Deficiency. Journal of Biological Chemistry, 281, 14393-14399. http://dx.doi.org/10.1074/jbc.M513631200
- Smith, M.H., Ploegh, H.L. and Weissman, J.S. (2011) Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science, 334, 1086-1090. http://dx.doi.org/10.1126/science.1209235
- Marion, G., Madeo, F. and Kroemer, G. (2011) Autophagy for Tissue Homeostasis and Neuroprotection. Current Opi- nions in Cell Biology, 23, 198-206. http://dx.doi.org/10.1016/j.ceb.2010.10.001
- Lukacs, G.L. and Verkman, A.S. (2012) CFTR: Folding, Misfolding and Correcting the ΔF508 Conformational Defect. Trends in Molecular Medicine, 18, 81-91. http://dx.doi.org/10.1016/j.molmed.2011.10.003
- Ron, E., Shenkman, M., Groisman, B., Izenshtein, Y B., Leitman, J. and Lederkremer, G.Z. (2011) Bypyss of Glycan- Dependent Glycoprotein Delivery to ERAD by Up-Regulated EDEM1. Molecular Biology of the Cell, 22, 3945-3954. http://dx.doi.org/10.1091/mbc.E10-12-0944
- Rodriguez-Boulan, E. and Müsch, A. (2005) Protein Sorting in Golgi Complex: Shifting Paradigms. Biochimica et Biophysica Acta (BBA), Molecular Cell Research, 1744, 455-464. http://dx.doi.org/10.1016/j.bbamcr.2005.04.007
- De Matteis, M.A. and Luini, A. (2008) Exiting the Golgi Complex. Nature Reviews of Molecular Cell Biology, 9, 273- 284. http://dx.doi.org/10.1038/nrm2378
- Mertins, P., Qiao, J.W., Patel, J., Udeshi, N.D., Clauser, K.R., Mani, D.R., Burgess, M.W., Gillette M.A., Jaffe, J.D. and Carr, S.A. (2013) Integrated Proteomic Analysis of Post-Translational Modifications by Serial Enrichment. Nature Methods, 10, 634-637. http://dx.doi.org/10.1038/nmeth.2518
- Swaney, D.L., Beltrao, P., Starita, L., Guo, A., Rush, J., Fields, S., Krogan, N.J. and Villén, J. (2013) Global Analysis of Phosphorylation and Ubiquitylation Cross-Talk in Protein Degradation. Nature Methods, 10, 676-682. http://dx.doi.org/10.1038/nmeth.2519
- Toulmay, A. and Prinz, W.A. (2011) Lipid Transfer and Signaling at Organelle Contact Sites: The Tip of the Iceberg. Current Opinions in Cell Biology, 23, 458-463.
- Mellman, I. and Nelson, W.J. (2008) Coordinated Protein Sorting, Targeting and Distribution in Polarized Cells. Nature Reviews Molecular Cell Biology, 9, 833-855. http://dx.doi.org/10.1038/nrm2525
- Lee, M.C., Miller, E.A., Goldberg, J., Orci, L. and Schekman, R. (2004) Bi-Directional Protein Transport between the ER and Golgi. Annual Reviews of Cell Development and Biology, 20, 87-123. http://dx.doi.org/10.1146/annurev.cellbio.20.010403.105307
- Glick, B.S. and Nakano, A. (2009) Membrane Traffic within the Golgi Apparatus. Annual Review of Cell Development Biology, 25, 113-132. http://dx.doi.org/10.1146/annurev.cellbio.24.110707.175421
- Houtkooper, R.H. and Vaz, F.M. (2008) Cardiolipin, the Heart of Mitochondrial Metabolism. Cellular and Molecular Life Sciences, 65, 2493-2206. http://dx.doi.org/10.1007/s00018-008-8030-5
- Lemmon, M.A. (2008) Membrane Recognition by Phospholipids Binding Proteins. Nature Reviews of Molecular Cell Biology, 9, 99-111. http://dx.doi.org/10.1038/nrm2328
- Whitelegge, J. (2011) Up Close with Membrane Lipid-Protein Complexes. Science, 334, 320-321. http://dx.doi.org/10.1126/science.1214084
- Osman, C., Voelker, D.R. and Langer, T. (2011) Making Heads or Tails of Phospholipids in Mitochondria. Journal of Cell Biology, 192, 7-16.
- Rusinova, R., Hobart, E.A, Koeppe II, R.E. and Andersen, O.S. (2013) Phosphoinositides Alter Lipid Bilayer Properties. Journal of General Physiology, 141, 673-690. http://dx.doi.org/10.1085/jgp.201310960
- Park, S.H., Das, B.B., Casagrande, F., Tian, Y., Nothnagel, H.J., Chu, M., Kiefer, H., Maier, K., De Angelis, A.A., Marassi, F.M. and Opella, S.J. (2012) Structure of the Chemokine Receptor CXCR1 in Phospholipid Bilayers. Nature, 491, 779-783.
- Slomiany, A., Grzelinska, E., Kasinathan, C., Yamaki, K., Palecz, D., Slomiany, B.A., et al. (1992) Biogenesis of Endoplasmic Reticulum Transport Vesicles Transferring Gastric Apomucin from ER to Golgi. Experimental Cell Research, 201, 1669-1682. http://dx.doi.org/10.1016/0014-4827(92)90280-L
- Slomiany, A. and Slomiany, B.L. (2011) Transformations of Phosphatidylinositol Phosphates in the Outer and Inner Nuclear Membrane Are Linked to Synthesis and Restitution of Cellular Membranes. Health, 3, 187-199. http://dx.doi.org/10.4236/health.2011.34035
- Slomiany, A., Grabska, M. and Slomiany, B.L. (2006) Homeostatic Restitution of Cell Membranes. Nuclear Membrane Lipid Biogenesis and Transport of Protein from Cytosol to Intranuclear Spaces. International Journal of Biological Sciences, 2, 216-226. http://dx.doi.org/10.7150/ijbs.2.216
- Slomiany, A. and Slomiany, B.L. (2013) Homeostatic Cell Cycle and the Origin of Autophagosomal Vesicles. Advances in Biological Chemistry, 3, 275-287. http://dx.doi.org/10.4236/abc.2013.33031
- Slomiany, A. and Slomiany, B.L. (2012) Phosphatidylglycerol-Containing ER-Transport Vesicles Built and Restore Outer Mitochondrial Membrane and Deliver Nuclear DNA Translation Products to Generate Cardiolipin in the Inner Mitochondrial Membrane. Advances in Biological Chemistry, 2, 132-145. http://dx.doi.org/10.4236/abc.2012.22016
- Sano, S., Slomiany, B.L. and Slomiany, A. (1995) Isolation and Characterization of the First Enzyme in the Pathway of Sphingolipids Synthesis. FASEB Journal, 9, A2719.
- Slomiany, A., Grzelinska, E., Grabska, M. and Slomiany, B.L. (1992) Intracelular Processes Associated with Glycoprotein Transport and Processing. Archives of Biochemistry and Biophysics, 298, 167-175. http://dx.doi.org/10.1016/0003-9861(92)90108-9
- Slomiany, A., Grzelinska, E., Yamaki, K. and Slomiany, B.L. (1992) Function of Intracellular Phospholipase A2 in Vectorial Transport of Apoproteins from ER to Golgi. International Journal of Biochemistry, 24, 1397-1406. http://dx.doi.org/10.1016/0020-711X(92)90065-9
- Slomiany, A., Grabska, M., Piotrowski, E. and Slomiany, B.L. (1994) Intracellular Processes Associated with Vesicular Transport from Endoplasmic Reticulum to Golgi and Exocytosis. Archives of Biochemistry and Biophysics, 310, 247-255. http://dx.doi.org/10.1006/abbi.1994.1164
- Slomiany, A. and Slomiany, B.L. (2003) Lipidomic Processes in Homeostatic and LPS-Modified Cell Renewal Cycle. Role of Phosphatidylinositol 3-Kinase Pathway in Biomembrane Synthesis and Restitution of Apical Epithelial Membrane. Journal Physiology and Pharmacology, 54, 533-551.
- Slomiany, A., Sano, S., Grabska, M., Yamaki, K. and Slomiany, B.L. (2004) Gastric Mucosal Cell Homeostatic Physiome. Critical Role of ER-Initiated Membrane Restitution in the Fidelity of Cell Function Renewal. Journal of Physiology and Pharmacology, 55, 837-860.
- Slomiany, A. and Slomiany, B.L. (2010) Cell Membranes Composition Is Defined in ER and Their Restitution Pro- ceeds by en Bloc Fusion of ER Generated Transport Vesicles. Health, 2, 1444-1454. http://dx.doi.org/10.4236/health.2010.212214
- Obrig, T.G., Culp, W.J., McKeehan, W.L. and Hardesty, B. (1971) The Mechanism by Which Cycloheximide and Related Glutarimide Antibiotics Inhibit Peptide Synthesis on Reticulocyte Ribosomes. Journal of Biological Chemistry, 246, 174-181.
- Croze, E.M. and Morré, D.J. (1984) Isolation of Plasma Membrane, Golgi Apparatus, and Endoplasmic Reticulum from Single Homogenates of Mouse Liver. Journal of Cellular Physiology, 119, 46-57. http://dx.doi.org/10.1002/jcp.1041190109
- Slomiany, A., Nowak, P., Piotrowski, E. and Slomiany, B.L. (1998) Effect of Ethanol on Intracellular Vesicular Trans- port from Golgi to the Apical Membrane. Role of Phosphatidylinositol 3-Kinase and Phospholipase A2 in Golgi Trans- port Vesicles Association and Fusion with the Apical Membrane. Alcohol, Clinical and Experimental Research, 22, 167-175.
- Parsons, D.F., Willims, G.R. and Chance, B. (1966) Characteristics of Isolated and Purified Preparations of the Outer and Inner Membranes of Mitochondria. Annals of the New York Academy of Sciences, 137, 643-666. http://dx.doi.org/10.1111/j.1749-6632.1966.tb50188.x
- Tjelle, T.E., Brech, A., Juvet, L.K., Griffiths, G. and Berg, T. (1996) Isolation and Characterization of Early Endosomes, Late Endosomes, Late Endosomes and Terminal Lysosomes: Their Role in Protein Degradation. Journal of Cell Sciences, 109, 2905-2914.
- Rome, L.H. and Crain, L.R. (1981) Degradation of Mucopolysacharide in Intact Isolated Lysosomes. Journal of Biological Chemistry, 256, 10763-10768.
- Schneider-Poetsch, T., Ju, J., Eyler, D.E., Dang, Y., Bhat, S., Merrick, W.C., Green, R., Shen, B. and Liu, J.O. (2010) Inhibition of Eukaryotic Translation Elongation by Cycloheximide and Lactimidomycin. Nature Chemistry and Biology, 6, 209-217. http://dx.doi.org/10.1038/nchembio.304
- Kawashima, A., Sato, A., Kawashima, M., Nitta, K., Yamura, W., Sugino, N., Nihei, H. and Natori, Y. (1998) A Simple Procedure for the Isolation of Rat Kidney Lysosomes. Kidney International, 54, 275-278. http://dx.doi.org/10.1046/j.1523-1755.1998.00958.x
- Liu, P. and Anderson, R.G.W. (1995) Compartmentalized Production of Ceramide at the Cell Surface. Journal of Biological Chemistry, 270, 27179-27185. http://dx.doi.org/10.1074/jbc.270.45.27179
- Slomiany, A., Grabska, M., Slomiany, B.A., Grzelinska, E., Morita, M. and Slomiany, B.L. (1993) Intracellular Transport, Organelle Biogenesis and Establishment of Golgi Identity: Impact of Brefeldin A on the Activity of Lipid Synthesizing Enzymes. International Journal of Biochemistry, 25, 891-901. http://dx.doi.org/10.1016/0020-711X(93)90245-A
- Nunnari, J. and Walter, P. (1996) Regulation of Organelle Biogenesis. Cell, 84, 389-394. http://dx.doi.org/10.1016/S0092-8674(00)81283-0
- Burke, B. and Ellenberg, J. (2002) Remodeling the Walls of the Nucleus. Nature Reviews Molecular Cell Biology, 3, 487-497. http://dx.doi.org/10.1038/nrm860
- Akhtar, A. and Gasser, S.M. (2007) The Nuclear Envelope and Transcriptional Control. National Review Genetics, 8, 507-517. http://dx.doi.org/10.1038/nrg2122
- Saftig, P. and Klumperman, J. (2009) Lysosome Biogenesis and Lysosomal Membrane Proteins: Trafficking Meets Function. Nature Reviews Molecular Cell Biology, 10, 623-635. http://dx.doi.org/10.1038/nrm2745
- Slomiany, A. and Slomiany, B.L. (2011) Transformations of Phosphatidylinositol Phosphates in the Outer and Inner Nuclear Membrane Are Linked to Synthesis and Restitution of Cellular Membranes. Health, 4, 187-199.
- Lloyd, A.C. (2013) The Regulation of Cell Size. Cell, 154, 1194-1205. http://dx.doi.org/10.1016/j.cell.2013.08.053
- Atilla-Gokcumen, G.E., Muro, E., Relat-Goberna, J., Sasse, S., Bedigian, A., Coughlin, M., Garcia-Manyes S. and Eggert, U.S. (2014) Dividing Cells Regulate Their Lipid Composition and Localization. Cell, 156, 428-439. http://dx.doi.org/10.1016/j.cell.2013.12.015
- Mari, M., Tooze, S.E. and Reggiori, F. (2011) The Puzzling Origin of the Autophagosomal Membrane. F1000 Biology Reports, 3, 25-34.
- Maiuri, M.C., Zalckvar, E., Kimchi, A. and Kroemer, G. (2007) Self-Eating and Self-Killing: Cross Talk between Autophagy and Apoptosis. Nature Reviews Molecular Cell Biology, 8, 741-752. http://dx.doi.org/10.1038/nrm2239
- Karunakaren, V. and Wickner, W. (2013) Fusion Proteins and Select Lipids Cooperate as Membrane Receptors for the soluble N-Methylmaleimide-Sensitive Factor Attachment Protein Receptor (SNARE) Vam7p. The Journal of Biological Chemistry, 288, 28557-28566. http://dx.doi.org/10.1074/jbc.M113.484410
- Hetz, C. (2012) The Unfolded Protein Response: Controlling Cell Fate Decision under ER Stress and Beyond. National Reviews Molecular Cell Biology, 13, 89-102.
- Idone, V., Tam, C. and Andrews, N.W. (2008) Two-Way Traffic on the Road to Plasma Membrane Repair. Trends in Cellular Biology, 18, 552-559. http://dx.doi.org/10.1016/j.tcb.2008.09.001
- Neupert, H. and Herrmann, J.M. (2007) Translocation of Proteins into Mitochondria. Annual Review of Biochemistry, 76, 723-749. http://dx.doi.org/10.1146/annurev.biochem.76.052705.163409
- Kobayashi, T., Beuchat, M.H., Chevallier, J., Makino, A., Mayran, N., Escola, J.M., Lebrand, C., Cosson, P., Kobayashi, T. and Gruenberg, J. (2002) Separation and Characterization of Late Endosomal Membrane Domains. Journal of Biological Chemistry, 277, 32157-32164. http://dx.doi.org/10.1074/jbc.M202838200
- Hammond, G.R.V., Machner, M.P. and Balla, T. (2014) A Novel Probe for Phosphatidylinositol 4-Phosphate Reveals Multiple Pools beyond the Golgi. Journal of Cell Biology, 205, 113-126. http://dx.doi.org/10.1083/jcb.201312072
- Yoshimori, T. and Noda, T. (2008) Toward Unraveling Membrane Biogenesis in Mammalian Autophagy. Current Opinions in Cell Biology, 20, 401-407. http://dx.doi.org/10.1016/j.ceb.2008.03.010
- Hayashi-Nishino, M., Fujita, M., Noda, T., Yamaguchi, A., Yoshimori, T. and Yamamoto, A. (2009) A Subdomain of the Endoplasmic Reticulum Forms a Cradle for Autophagosome Formation. National Cell Biology, 5, 1180-1185.
- Tooze, S.E. and Yoshimuri, T. (2010) The Origin of the Autophagosomal Membrane. National Cell Biology, 12, 831- 835. http://dx.doi.org/10.1038/ncb0910-831
- Hailey, D.W., Rambold, A.S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P.K. and Lippincott-Schwartz, J. (2010) Mitochondra Supply Membranes for Autophagosomal Biogenesis during Starvation. Cell, 141, 656-667. http://dx.doi.org/10.1016/j.cell.2010.04.009
- van der Vaart, A. and Reggiori, F. (2010) The Golgi Complex as a Source for Yeast Autophagosomal Membranes. Autophagy, 6, 800-801. http://dx.doi.org/10.4161/auto.6.6.12575
- Ravikumar, B., Moreau, K., Jahreiss, L., Puri, C. and Rubinsztein, D.C. (2010) Plasma Membrane Contributes to the Formation of Pre-Autophagosomal Structures. National Cell Biology, 12, 747-757. http://dx.doi.org/10.1038/ncb2078
- Hurley, J.H. and Schulman, B.A. (2014) Atomistic Autophagy: The Structures of Cellular Self-Digestion. Cell, 157, 300-311. http://dx.doi.org/10.1016/j.cell.2014.01.070